
Abstract
During cell division, replication of the genomic DNA is performed by high-fidelity DNA polymerases but these error-free enzymes can not synthesize across damaged DNA. Specialized DNA polymerases, so called DNA translesion synthesis polymerases (TLS polymerases), can replicate damaged DNA thereby avoiding replication fork breakdown and subsequent chromosomal instability.
We focus on the involvement of mammalian TLS polymerases in DNA damage tolerance mechanisms. In detail, we review the discovery of TLS polymerases and describe the molecular features of all the mammalian TLS polymerases identified so far. We give a short overview of the mechanisms that regulate the selectivity and activity of TLS polymerases. In addition, we summarize the current knowledge how different types of DNA damage, relevant either for the induction or treatment of cancer, are bypassed by TLS polymerases. Finally, we elucidate the relevance of TLS polymerases in the context of cancer therapy.
Similar content being viewed by others

DNA damage response (DDR)
Genomic information is stored as deoxyribonucleic acid (DNA) in every living organism and needs to be protected and maintained to guarantee genomic integrity. Each of the 1013 cells of the human body contains 30'000-40'000 genes encoded by 3 × 109 base pairs of the DNA [1–3]. The integrity of the DNA is constantly threatened either by spontaneous decay or by damage induced by endogenous and environmental sources. In every single cell, tens of thousands of DNA lesions per day are formed due to spontaneous hydrolysis and the attack of reactive oxygen species (ROS) and other intracellular metabolites [4]. In the context of cancer research, prominent examples for environmental factors which induce DNA damage are ultraviolet (UV)-light inducing [6-4]pyrimidine-pyrimidone photoproducts ([6-4]PP) and cyclobutane pyrimidine dimers (CPDs), and cigarette smoke, which contains a variety of carcinogens, e.g. benzo(α)pyrene (BaP) [5]. Cancer treatment regimens are frequently based on DNA damage inducing agents. For instance, multimodality therapies of solid tumors are often based on cisplatin, a platinum analogue, which induces intra- and interstrand DNA crosslinks [6].
In addition, accurate DNA duplication is an essential step carried out by a complex DNA replication machinery but errors during this process can also compromise genomic integrity. For example, damaged DNA, which cannot be replicated by the high fidelity replicative DNA polymerases, can lead to stalled replication forks and subsequent replication fork breakdown results in chromosomal instability [7].
To counteract the constant loss or the modification of DNA bases, cells evolved a complex and interplaying system, the so-called DNA damage response (DDR) [8, 9]. During DDR, DNA lesions are detected, leading to the activation of a signal cascade resulting either in the repair or the tolerance of the DNA damage, thereby regulating the cellular outcome after genomic insult [4] (Figure 1).

DNA damage induced by spontaneous decay or endogenous and environmental sources can either be repaired or tolerated (Adapted from [245]). See text for details.
The cellular DNA repair machinery includes non-homologous end joining (NHEJ) and homologous recombination (HR) to repair double strand breaks (DSBs), base excision repair (BER) to counteract modification of the nitrogenous bases, nucleotide excision repair (NER) to excise bulky nucleotide alterations such as UV-induced [6-4]PPs, mismatch repair (MMR) to exchange mispaired nucleotides and direct damage repair for reversal of alkylated nucleotides (Figure 1) [10]. Although DNA repair processes are not as accurate as high-fidelity DNA replication, DNA repair is considered to be error-free. In eukaryotes, DNA damage tolerance involves a error-free pathway dependent on homologous recombination and a more mutagenic pathway based on TLS polymerases [11]. In this review we focus on the contribution of TLS polymerases to DNA damage tolerance and their relevance in cancer research.
Mammalian TLS polymerases: state-of the art
History and Discovery
In 1956, the group of Arthur Kornberg discovered and described an enzyme purified from Escherichia coli (E. coli), which is able to create an appropriate copy of its DNA substrate, i.e. DNA polymerase (Pol) I [12]. DNA Pol I was shown to generate a copy of the single-stranded DNA of the small bacterial virus ΦX174. The generated DNA kept the infectious activity, thereby confirming that DNA Pol I is able to generate genetically active DNA [13, 14]. DNA Pol II was discovered in 1970 [15, 16] and shortly afterwards DNA Pol III was discovered as the third DNA-replicating enzyme [17]. Miroslav Radman and coworkers published in 1974 the "SOS repair" model, proposing that the UV-induced mutations of both λ phage and host E. coli are due to a "mutation-prone" cellular replication mechanism [18]. Also in the 1970s, a screen in Saccharomyces cerevisiae (S. cerevisiae) for reversionless (rev) mutants unable to revert an auxotropic marker after UV irradiation led to the discovery of the first eukaryotic genes encoding error-prone TLS polymerases, i.e. REV1 (encoding the TLS Pol Rev1) and REV3 (encoding the catalytic subunit of TLS Pol ζ) [19]. A strategy similar to the one which led earlier to the discovery of the UmuDC genes (defects in these genes render cells non-mutable) in E. coli[20] resulted in the discovery of and REV7, the structural subunit of TLS Pol ζ, in S. cerevisiae[21]. The co-discovery of bacterial and eukaryotic TLS polymerases revealed the conservation of a cellular process that had until then been considered to be a bacterial-specific function. Subsequently, it was shown that DNA Pol II is part of the "SOS repair" [22, 23]. Although the DinB [24] and the UmuDC [20] genes of E. coli were discovered much earlier, it was shown only in the 1990s that these gene products constitute the error-prone DNA Pol IV [25] and Pol V, respectively [26, 27]. DNA Pol IV and Pol V are inducible by DNA damage and can efficiently bypass various forms of DNA lesions thereby generating most of the SOS-repair dependent mutations [28]. Other mammalian TLS polymerases such as Pol η (eta; hRAD30A/XPV), Pol ι (iota; hRAD30B), Pol κ (kappa; DINB1) and Pol θ (theta; POLQ) were identified by searches for homologues of genes of previously identified TLS polymerases [25, 29–32]. The TLS Pol μ (mu) [33, 34] and Pol λ (lambda) were discovered and described more recently [33, 35]. The most recently described TLS Pol ν (nu) was found due to homology with mus308[36].
The ability of eukaryotic TLS polymerases to bypass DNA lesions, was firstly described for the yeast TLS Pol ζ, mediating the bypass of UV-induced thymine-thymine cyclobutane pyrimidine dimers (TT-CPDs) [37]. The property of Rev1 to insert deoxycytidine monophosphate (dCMPs) opposite abasic sites was first described in yeast [38]. Subsequently, the role of human TLS Pol ζ to bypass DNA lesions [39] and the function of human Rev1 as dCMP transferase opposite abasic sites [40] were proposed. The UV lesion bypass activity of human TLS Pol η was discovered by the fact that xeroderma pigmentosum variant (XPV) patients show increased susceptibility to UV-induced skin cancer [41] and hypermutability [42] due to a defect of TLS Pol η. The human TLS Pol ι was shown to be able to incorporate deoxynucleotides opposite the 3' T of [6-4]PPs and abasic sites [43] and opposite N2-adducted guanines [44]. Human TLS Pol κ was identified and it was first shown that TLS Pol κ protect cells against the lethal and mutagenic effects of BaP [45]. Human TLS Pol θ was identified more recently and it was shown that TLS Pol θ is implicated in somatic hypermutation (SHM) [46–48]. Similarly, TLS Pol λ and TLS Pol μ are both implicated in V(d)J recombination during the immunoglobulin (IgG) diversification process [49, 50] whereas TLS Pol ν is able to bypass thymine glycols (Tg) [51]. It can not be excluded that additional TLS polymerases will be discovered in mammalian genomes.
Fidelity of TLS
TLS is defined as the incorporation of a nucleotide across DNA damage followed by extension of the potentially mispaired primer-template, which can be error-free or error-prone [52]. The basic necessity for the presence of TLS polymerases reflects a trade-off between the maintenance of genomic integrity by avoiding replication fork breakdown and subsequent chromosomal instability and the occurrence of mutations on the nucleotide level by the TLS polymerases mediated DNA damage bypass reaction.
Although the tertiary structure consisting of palm, thumb and fingers is conserved among the different polymerase families, the thumb and fingers of the TLS polymerases are smaller. Compared to the DNA replication polymerases where the fingers tightly bind the incoming dNTPs and make a conformational change upon correct Watson-Crick base pairing, the active site of TLS polymerases is more open and less constrained to reject wrong paired base pairs. Therefore, TLS polymerases are able to mediate the bypass reaction of non-coding DNA lesions. The additional little finger of the Y family TLS polymerases supports the stabilization of the template DNA and influences fidelity and activity [53].
The error rate of DNA replication polymerases of the families A, B and C including correct incorporation of the nucleotide and the proofreading activity is between 10-6 and 10-8. Auxiliary proteins such as proliferating cell nuclear antigen (PCNA) and replication protein A (RP-A) [54] and postreplicative MMR decrease the error rate to 10-8 and 10-10 . The error rate of the TLS polymerases ranges from 10-1 to 10-3 for replication of undamaged DNA [55, 56]. Due to the characteristic low fidelity DNA synthesis and the lack of an exonuclease proofreading activity, it was initially assumed that TLS is generally a mutagenic process. Recently it became clear that the use of specialized TLS polymerases at specific lesions can be error-free. The best example is the ability of TLS Pol η to bypass TT-CPDs, the main DNA lesion induced by both UVB and UVA radiations, in a non-mutagenic manner [57, 58].
In this context, it is important to address whether the accurate bypass of a particular lesion by a TLS Pol in vitro can be used as an indicator whether this polymerase also processes the corresponding lesion in vivo. In addition to the TT-CPDs in vitro bypass activity of TLS Pol η, it was also shown that inactivation and deletion of Pol η decreases UV survival of human [59, 60] and yeast cells [29, 61]. Similarly, TLS Pol θ is able to bypass oxidative DNA lesions, i.e. apurinic/apyrimidinic (AP) sites in vitro[62] and knockout of TLS Pol θ in the chicken DT40 B-cell line resulted in hypersensitivity to hydrogen peroxide (H2O2) [63]. Thus, in general, the in vivo sensitivity to a DNA damage inducing agent of cells deficient for a specific polymerase can be predicted by the in vitro ability of the Pol to bypass the induced DNA lesion. However, it became clear that the bypass reaction of most DNA adducts requires the concerted action of protein complexes containing several TLS polymerases. Inactivation or deletion of a TLS polymerase can disrupt protein-protein interactions essential for lesion bypass therefore indirectly affecting the in vivo sensitivity to the DNA lesion inducing agent. Thus, based on the in vivo sensitivity of an inactivation or deletion mutant, it can not be concluded that the induced DNA damage is processed by the modified TLS Pol activity. For example, although TLS Pol ζ is sensitive to UV-irradiation, other TLS polymerases perform the bypass reaction whereas Pol ζ mainly performs the subsequent extension step [64] (see also Table 1).
TLS polymerase families
The Y-family TLS polymerases η, ι, κ and REV1 [65] and the B-family TLS Pol ζ [66, 67] perform most of the TLS in mammalian cells and are well characterized. Less is known about the member of the A-family TLS polymerases, which were more recently identified. An involvement in DNA damage tolerance was also shown for the TLS polymerase members of the X-family although their involvement in DNA repair processes might be their main cellular function (Figure 2).

Overview of TLS polymerases (Adapted from [246]). See text for details.
Family A: TLS polymerases theta (θ) and nu (ν)
The A family polymerases consist of DNA Pol γ (gamma), which is the mitochondrial DNA replicase and the TLS Pol θ and TLS Pol ν.
TLS Pol θ
The POLQ gene encoding TLS Pol θ was mapped on chromosome 3q. The C-terminal region of TLS Pol θ includes the polymerase motifs A, B and C, which are typical for A family polymerases whereas the N-terminal region contains an ATP domain. The POLQ gene encodes a protein of 2592 amino acids (aa). The protein sequence shares homology to the Mus308 protein of Drosphila melanogaster[30, 68]. Further research estimated a protein of 290 kDa, with an N-terminal ATPase helicase domain although no helicase activity could be detected so far [68]. Additionally, TLS Pol θ has been shown to have 5' dRP lyase activity that is involved in short patch BER in vitro[69]. It was shown that TLS Pol θ is able to bypass AP sites and also thymine glycols, e.g. a DNA damage product of ionizing radiation and other oxidative mutagens. TLS Pol θ preferentially incorporates adenine (A) opposite an AP site, which allows DNA replication to continue by using the incorporated nucleotide as a primer (henceforth referred to as extension step) [62]. Additionally, TLS Pol θ can carry out the extension step from mismatches after error prone dNTP incorporation by human TLS Pol ι [70] or S. cerevisiae TLS Pol ζ opposite [6-4]PPs in vitro[43]. Due to the lacking 3' to 5' exonuclease proofreading activity [68], the fidelity of TLS Pol θ during dNTP incorporation is lower than usual for A family polymerases [62]. It is proposed that TLS Pol θ has a function in SHM of IgG diversification due to misincorporation of nucleotides opposite AP sites and the low fidelity during DNA replication of undamaged DNA [46–48]. Conversely, it was suggested that human TLS Pol θ from HeLa cells nuclear extracts synthesizes DNA with a high fidelity and possesses 3' to 5' exonuclease proofreading activity [71].
TLS Pol θ mutant mice show an increase of spontaneous and radiation-induced micronuclei formation [72, 73] and TLS Pol θ knockout chicken DT40 B-cell line shows hypersensitivity to hydrogen peroxide (H2O2) [63]. Furthermore, CH12 mouse B lymphoma cells containing a knockdown of TLS Pol θ showed elevated sensitivity to UV irradiation, the crosslinking agent mitomycin C and cisplatin, etoposide, ionizing irradiation and the alkylating agent methyl methanesulphonate (MMS) [74]. Thus, TLS Pol θ is involved in the tolerance of a broad range of DNA adducts, which indicates that TLS Pol θ mainly functions as an extender polymerase similar to TLS Pol ζ.
TLS Pol ν
The full length POLN gene comprises 24 exons with a length of 900 aa. The POLN gene is located on chromosome 4p16.2 and is deleted in approximately 50% of breast carcinomas [75]. The POLN gene encodes a protein with a size of 160 kDa. The C-terminal polymerase domain of TLS Pol ν consists of the typical A family polymerase motifs A, B and C and shares 29% identity with the C-terminus of TLS Pol θ. Neither a 3' to 5' nor a 5' to 3' nuclease domain were identified [36]. In vitro experiments showed the ability of TLS Pol ν to bypass thymine glycols [51]. Interestingly, it has been shown that TLS Pol ν is involved in cross link repair and homologous recombination. In detail, depletion of TLS Pol ν sensitizes HeLa cells to the DNA cross-linking agent mitomycin C but not to UV irradiation [76]. Depletion of TLS Pol ν in U2OS cells reduced the efficiency of homologous recombination in a GFP-based reporter assay and increased the sensitivity of HeLa cells to camptothecin-induced DSBs, i.e. a substrate for homologous recombination [77]. Contradictorily, deletion of POLN did not sensitize chicken DT40 cells to campothecin. However, the same study showed that TLS Pol ν in chicken DT40 cells has a dominant role in homologous recombination-dependent immunoglobulin gene conversion and in TLS-dependent immunoglobulin hypermutation [78].
Family B: TLS Pol zeta (ζ)
The family B includes the highly accurate DNA polymerases δ (delta), ε (epsilon), α (alpha), and the error-prone TLS Pol ζ [79, 80].
TLS Pol ζ
Unlike the replicative DNA polymerases δ and ε, TLS Pol ζ lacks the 3' to 5' exonuclease proofreading activity. The human TLS Pol ζ and its yeast homologue are heterodimeric proteins consisting of the catalytic subunit REV3 and the structural subunit REV7 [37, 81]. The human REV3 protein has two transcripts that have a length of 3052 and 3130 aa and the larger protein has a size of 353 kDa compared to 173 kDa of the yeast REV3. The discrepancy in size between the yeast REV3 and the human REV3 is due to the exon 13 with a length of 1388 aa. Human REV3 shows ~36% identity with the N-terminal region, ~29% identity with the central REV7 binding region and ~39% identity with the C-terminal DNA polymerase region of the yeast homologue. The C-terminal region consists of six B-family conserved DNA polymerase motifs and two zinc finger motifs [66, 67, 82]. Human REV3 is located on chromosome 6q21 and its mouse equivalent on chromosome 10 [83, 84]. Interestingly, the REV3 gene is located in the chromosomal region 6q21 within the fragile site FRA6F, which is known to be commonly deleted in several types of human leukemias and solid tumors [85]. The human REV3 contains an out-of-frame ATG in the 5' region that reduces the rate of correct transcripts. Moreover, a sequence upstream of the AUG initiator codon has the potential to form a stem-loop hairpin that lowers the rate of translation. It is suggested that the characteristic structural features in combination with the alternative splicing are responsible for the observed low REV3 expression levels [66, 83]. Indeed, the protein concentration of REV3 in Xenopus laevis egg extracts is much lower than those of other replication and repair proteins and does not change within the early embryonic development [86].
In contrast to viable REV3 null yeast mutants, the disruption of REV3 in mice causes embryonic lethality around midgestation [87–90]. It is known that during the early stages of embryogenesis checkpoints are actively silenced [91] to allow rapid cell division and it was proposed that REV3 is essential during this strict temporal program. The embryonic lethal effect could not be rescued by the absence of p53 suggesting a p53-independent pathway. However, mouse embryonic fibroblasts (MEFs) with a p53-deficient background could be generated [92, 93].
An in vitro study showed that REV3 expression levels are directly regulated by a p53 responding element in the REV3 promoter region. In addition, REV3 expression was increased after DNA damage induction in a p53-dependent manner [94]. An independent study also showed increased REV3 mRNA level after cisplatin treatment [95]. Over-expression of REV3 in yeast led to an elevated rate of UV-induced mutagenesis [96]. These findings together with the embryonic lethal effect after REV3 abrogation indicate that the level of REV3 has to be tightly regulated to maintain genomic integrity.
Within the exon 13 of REV3, serine 995 was shown to be phosphorylated by CHK2 [97]. In addition, REV3 shares an AT-hook domain with AHDC1, which has been proposed to be phosphorylated by either ATM and ATR upon DNA damage, indicating a putative regulation of REV3 by ATM and ATR [98].
The human TLS Pol ζ is thought to be the major contributor to the error-prone bypass of DNA lesions. Relevant for tumorigenesis, REV3 knockout in MEFs leads to increased chromosomal instability in a p53-deficient background [92]. Moreover, in mice with a conditional deletion of REV3, thymic lymphomas occurred with decreased latency and elevated incident in a p53-deficient background [99]. Relevant for cancer therapy, REV3 down-regulation in human foreskin fibroblasts revealed decreased mutation frequency after treatment with UV or BaP-diolepoxide [100]. Similarly, MEFs derived from mice expressing REV3 antisense revealed decreased mutagenic frequency after UV treatment [101]. Recent in vitro and in vivo studies revealed that inhibition of REV3 expression increased the sensitivity of lymphoma to cisplatin [102]. Similarly, REV3 depletion in combination with cisplatin treatment decreased the growth rate of a p53-deficient non small cell lung cancer cell line (NSCLC) transplanted into mice and prolonged the survival of the host. The same study showed that the frequency of 6-thioguanine resistant colonies after cisplatin treatment was reduced in REV3 deficient cells compared to the control [103]. Thus, although REV3 is involved in the maintenance of genomic integrity, inhibition of REV3 expression might enhance the anti-tumor activity of DNA-damage inducing agents as discussed below.
Recent findings propose TLS Pol ζ to have a function not only in TLS synthesis but also in DNA repair, e.g. REV3 deletion impairs HR [95] and ICL repair [104, 105]. Furthermore, SHM and/or class switch recombination (CSR) of IgG were affected by TLS Pol ζ ablation [106].
Although human REV3 contains a REV7 binding region no interaction between full-length REV3 and REV7 could be demonstrated so far. However, it was shown, that a human REV3 fragment interacts with full-length REV7 and a part of human REV7 interacts with human REV3 and REV1 [107]. The human REV7 protein has a length of 211 aa and a size of 24 kDa and shares ~23% identity with the yeast REV7. The human REV7 is located on the human chromosome 1p36. REV7 displays ~23% identity with the spindle checkpoint assembly protein MAD2 and it is therefore also known as MAD2B and MAD2L2 in higher eukaryotes. The REV7 contains a HORMA (Hop1/Rev7/Mad2) domain that is known to interact with chromatin [108]. Additionally, REV7 interacts with CDH1 and CDC20 of the anaphase-promoting complex/cyclosome (APC/C) [109] and the protein MAD2, a spindle checkpoint protein [81], indicating that REV7 is involved in the regulation of mitosis. Interestingly, the bacterial pathogen Shigella delivers the effector IpaB into epithelial cells to efficiently colonize the epithelium. It has been shown that IpaB interacts with MAD2L thereby inducing a cell cycle arrest [110].
Family X: DNA Pol beta (β), TLS Pol lambda (λ) and TLS Pol mu (μ)
The polymerases of the X family include DNA Pol β, terminal deoxynucleotidyl transferase (TdT), TLS Pol λ and TLS Pol μ. All the X family polymerases lack the 3' to 5' exonuclease proofreading activity.
DNA Pol β
DNA Pol β is a 39 kDa monomeric protein and the encoding gene POLB is located on chromosome 8 in both mice and human [111]. DNA Pol β consists of two protease resistant segments linked by a short protease sensitive segment indicating that DNA Pol β activity might be controlled by proteolytic activity. The 8 kDa N-terminal lyase domain shows a strong affinity to ssDNA [112], whereas the 31 kDa C-terminal polymerase domain specifically binds double-stranded nucleic acids [113]. The 31 kDa polymerase domain consists of three subdomains. The catalytic subdomain (palm) coordinates two metal-ions and mediates the nucleotidyltransferase reaction [114] and the other subdomains mediate the binding of duplex DNA (thumb) and nascent base pairs (fingers) [115]. It has been shown that DNA Pol β is able to fill short gaps in double stranded DNA [116]. The 8 kDa lyase domain was shown to direct DNA Pol β to phosphorylated 5' side of a DNA gap for its bypass [117] and recently to mediate the removal of 5' dRP from the AP site via β-elimination after the incision step by the AP endonuclease [118]. Beside the role of DNA Pol β in short patch BER, a function in long patch BER was proposed. Both short- and long-patch repair are impaired after DNA Pol β ablation [119–121]. Additionally, DNA Pol β has a role in bypassing DNA lesions such as cisplatin-DNA adducts [122]. It was shown that DNA Pol β is not involved in the diversification of IgG [123]. DNA Pol β knockout mice are not viable reflecting the important role of Pol β during embryonic development [124].
Down-regulation of DNA Pol β sensitized mouse fibroblasts to cisplatin, UV-irradiation, oxidizing- and methylating agents [125–128]. Ectopic expression of DNA Pol β leads to aneuploidy, aberrant localization of the centrosome-localized γ-tubulin protein during mitosis, checkpoints defects in vitro and tumour induction in vivo[129]. In addition, DNA Pol β expression is upregulated in chronic myelogenous leukemia (CML) patients [130]. Thus, a strict regulation of DNA Pol β activity is essential to maintain genomic integrity.
TLS Pol λ
TLS Pol λ has a size of approximately 69 kDa and its gene POLL is located on the chromosome 10 in human and on the chromosome 19 in mice [33, 35]. The human TLS Pol λ consists of 575 aa and shares 32% residue identity with Pol β, comprising a C-terminal Pol domain including palm, thumb and fingers and the 8 kDa 5' dRP lyase domain. Additionally, TLS Pol λ contains an N-terminal BRCT (BRCA1 C-terminus) domain followed by a serine/proline rich region that is absent in Pol β [131]. Tandem BRCT domains mediate binding to phosphorylated proteins and are widely found in proteins involved in DDR [132]. TLS Pol λ shows terminal TdT activity [131], which prefers the incorporation of pyrimidine nucleotides [133]. The TLS Pol λ has been shown to be less accurate for base substitutions and much less accurate for single-base deletions [134]. Further, TLS Pol λ is unable to differentiate between matched and mismatched primer termini during the extension step, therefore suggesting TLS Pol λ as a candidate for NHEJ and as mismatch extender during TLS [134, 135]. Additional in vitro studies showed that TLS by Pol λ requires the BRCT domain and is physically and functionally dependent on Ku during NHEJ [136]. Recently, it has been shown, that a TLS Pol λ variant containing of a single nucleotide polymorphism (SNP), a cytosine/thymine variation, leads to increased mutation frequency, chromosomal aberration and defects in NHEJ [137].
TLS Pol λ is also discussed to participate in BER. Uracil-containing DNA was efficiently repaired in an in vitro reconstituted BER reaction by the 5' dRP lyase activity of TLS Pol λ, in coordination with its polymerization activity [138]. TLS Pol λ null mice are viable and fertile, but shortening of the heavy chain coding joints was reported [49].
TLS Pol μ
TLS Pol μ has a size of 55 kDa and its gene POLM is located on the human chromosome 7. It consists of 492 aa and shares 42% identity with TdT [33, 35]. TLS Pol μ, as TLS Pol λ, contains a polymerase domain and a BRTC domain. In contrast to Pol β and λ, the TLS Pol μ lacks a 5' dRP lyase activity [138]. Isolated TLS Pol μ is highly error-prone for frameshifts during DNA synthesis. Interestingly, TLS Pol μ is able to extend from mismatches by frameshift synthesis mechanism and thereby promoting microhomology search and microhomology pairing between the primer and the DNA template [139]. Moreover, TLS Pol μ shows template-independent polymerase activity under physiological conditions (Mg2+ present) preferring the incorporation of pyrimidines and thereby generating terminal microhomology, which can be ligated by the XRCC4-DNA ligase IV [140]. All these findings suggest TLS Pol μ as a candidate for NHEJ of DSBs. It has been shown that TLS Pol μ, as TLS Pol λ, interacts with the Ku-DNA complex through its BRCT domain [136].
Interestingly, TLS Pol μ null mice are viable and fertile, but they show impaired V(D)J recombination due to shortening of the light chain coding ends, but not of the heavy chain coding ends [49, 50].
Family Y: Rev1, TLS Pol eta (η), kappa (k) and iota (ι)
The human TLS Pol members of the Y family include REV1, TLS Pol η, TLS Pol k and TLS Pol ι [65]. All the Y family members lack the 3' to 5' exonuclease proofreading activity [56] and share a general conserved N-terminal polymerase domain for the catalytic activity and a non-conserved C-terminus, which, at least for the human TLS Pol ι [141] and Pol κ [142], is responsible for the regulation of the activity. The conserved N-terminus of the DNA polymerase domain includes five motifs (I to V) corresponding to the catalytic core complex. Motif I and II form the catalytic epicentre (palm) with its three acidic residues harbouring the two metal ions mediating the nucleotide transfer. Despite sequence differences, the palm domain with its nucleotide transfer function is widely conserved between Y-family TLS, A- and B-family replicative DNA polymerases. The motifs III and IV belong to the finger and thumb domain, respectively. They bind the triphosphate of the nascent incoming dNTP and mediate the incorporation of the nascent nucleotide whereas the additional motif V binds the primer strand. The C-terminus of the motif V resides either the so called little finger (LF), polymerase associated domain (PAD) or the wrist that supports the DNA synthesis activity and is conserved and unique among the Y-family TLS polymerases [53, 143, 144].
Rev1
The 1251 aa human REV1 protein has a size of 138 kDa and is encoded by the gene REV1, located on chromosome 2. Beneath the typical Y-family conserved domains, a BRCT domain is located at the N-terminus [132]. At the C-terminal end, there are two ubiquitin binding motifs (UBM) [145] followed by a polymerase interaction region [146].
The polymerase activity of Rev1 is restricted to the incorporation of C over G and DNA lesions such as AP sites [147]. It has been proposed that the nucleotide insertion activity is not the main function of REV1 but that REV1 helps to coordinate the polymerase switch between the normal- and the substituting TLS Pol upon PCNA monoubiquitination. Murine Rev1 binds ubiquitin through its UBMs and thereby mediating its localization to DNA damage foci. UBM mutants showed increased mutational aberrations after UV irradiation and elevated sensitivity to UV irradiation and cisplatin, which was further increased in UBM and BRCT double-mutants [148]. Although it was shown that murine REV1 binds monoubiquitinated PCNA via its UBMs, it is assumed that the polymerase switch function of REV1 might also be dependent on the other protein interaction domains of REV1, e.g. the BRCT and the polymerase interaction region. In detail, the C-terminal end of human Rev1 is able to interact with several TLS polymerases including Pol η, Pol ι, Pol κ and Pol ζ, thus supporting the assumption that Rev1 acts as a scaffold protein for several TLS polymerases [107, 146, 149]. Additionally, murine REV1 binds PCNA through its BRCT domain and the monoubiquitination of PCNA enhances this reaction [150].
Rev1 ablation sensitizes DT40 chicken cells to various DNA damaging agents including cisplatin, UV irradiation and MMS. Additionally, Rev1 is required for the maintenance of chromosomal stability after UV irradiation [150]. Rev1 deletion in chicken DT40 cells did not affect basal and damage induced sister chromatid exchange and immunoglobulin gene conversion indicating that homologous recombination repair is likely to be intact [151]. However, the same study showed that knockout of Rev1 in chicken DT40 cells reduced the level of non-templated immunoglobulin gene mutations indicating a defect in translesion bypass of DNA replication blocking lesions. An in vivo mouse model showed that REV1 inhibition in B-cell lymphoma reduces cisplatin and cyclophosphamide induced mutagenesis and prolongs survival of mice upon cyclophosphamide treatment [102].
Recently, it has been shown that Rev1 silencing impairs the replication of G-quadruplex (G4) structures thereby, on one side, limiting the recycling of histones and, on the other side, favouring the incorporation of newly synthesized histones resulting in changes of the epigenetic pattern, e.g. gene silencing [152].
TLS Pol η
Loss of TLS Pol η activity in human results in a cancer-prone syndrome known as xeroderma pigmentosum variant (XPV), which is characterized by an increased incidence of skin cancers and sensitivity to sunlight [153]. Human TLS Pol η consists of 713 aa and is encoded by the POLH (Xeroderma pigmentosum variant, XPV) gene, localized on chromosome 6. TLS Pol η has a size of 78 kDa. Additional to the N-terminal conserved polymerase domain, TLS Pol η consists of a Rev1-interacting region (RIR), an ubiquitin binding zinc finger (UBZ), a nuclear localization domain (NLD) and two PIPs [154]. XPV cells are sensitive to UV irradiation and show an increased mutagenic rate despite functional NER indicating that TLS Pol η bypasses specific UV lesions in a non-mutagenic manner [41, 57, 155]. It is proposed that in the absence of TLS Pol η, TLS Pol ι serves as the error-prone polymerase which bypasses the UV-induced lesions [156]. Also Rev1 is suggested to have a regulatory role in TLS of UV-induced lesions [157]. The PIP and the UBZ domain of TLS Pol η are essential for binding the monoubiquitinated PCNA and its TLS activity. Mutation in either the PIP or the UBZ domain increases the UV sensitivity [145].
Interestingly, it has been shown that loss of TLS Pol η in mice leads to decrease in adenine/thymine mutations during SHM of IgG indicating that TLS Pol η bypasses adenine and thymine in an error prone manner [158].
TLS Pol κ
The human TLS Pol κ is encoded by POLK gene and has a length of 870 aa. The TLS Pol κ has a size of 99 kDa and is located on chromosome 5. The N-terminal part consists of the conserved polymerase domain whereas the variable C-terminus consists of a RIR, two UBZ and a PIP. It has been shown that TLS Pol κ co-localizes to a lesser extend with PCNA at replication foci after UV irradiation, hydroxyurea or BaP treatment compared to TLS Pol η [159]. It has been shown that embryonic stem (ES) cells deficient in the TLS Pol κ gene are more sensitive and acquire more mutations after treatment with BaP and that TLS Pol κ bypasses BaP-G accurately and efficiently in vivo[45]. Additionally, XPV cells treated with siRNA against TLS Pol κ reveal increased UV sensitivity [160] indicating that TLS Pol κ is able to bypass UV-induced DNA lesions. Recent findings suggest that TLS Pol κ has a function during NER and that its activity is dependent on RAD18 and monoubiquitinated PCNA [161].
A role of TLS Pol κ in promoting tumorigenesis has been discussed since ectopic expression of Pol κ leads to DSBs, aneuploidy and tumorigenesis in nude mice [162].
TLS Pol ι
Human TLS Pol ι is encoded by the POLI gene consists of 715 aa. The TLS Pol ι has a size of 80 kDa and is localized on chromosome 18. TLS Pol ι shares with the other Y family members the N terminal conserved polymerase domain and at the variable C-terminal a RIR, two UBMs and the PIP. Interestingly, TLS Pol ι possesses a 5' dRP lyase activity [163] that is located within a NLD [164].
As in the case of TLS Pol η, the PIP and the UBM domains of TLS Pol ι are important for localization to the replication fork by binding monoubiquitinated PCNA [145]. The localization and accumulation of TLS Pol ι to stalled replication forks is dependent on physical interaction with TLS Pol η [165]. Recently, it has been shown that BER activity is decreased in human fibroblasts in which TLS Pol ι is stably downregulated resulting in increased sensitivity to the oxidizing agents H2O2 and menadione [164]. Additionally, after H2O2 treatment, TLS Pol ι binds to chromatin and interacts with the BER factor XRCC1, suggesting a role of TLS Pol ι not only in TLS but also in the repair process of oxidative DNA damage [164].
Activation of TLS and polymerase switch reaction
In response to DNA damage, it is proposed that activation of the DNA damage tolerance mechanisms is mainly mediated by modifications of PCNA [166, 167]. Monoubiquitination of PCNA by the RAD6-RAD18 complex triggers DNA damage tolerance by TLS. Y-family TLS polymerases can bind to monoubiquitinated PCNA through ubiquitin-binding domains such as the UBM, the UBZ and a PCNA interacting peptide box (PIP), thereby initiating TLS [145, 148].
In S. cerevisiae, monoubiquitinated PCNA is subsequently polyubiquitinated by RAD5/Ubc13/Mms2, which triggers the error-free DNA damage tolerance response carried out by template switching including fork reversal or recombination past the lesion [168, 169]. There are two Rad5 orthologs in humans, HLTF and SHPRH, which are both capable of polyubiquitinating PCNA in vitro[170, 171]. An interplaying mechanism was proposed between the deubiquitinating enzyme USP1 and ubiquitination factors including RAD6 and RAD18. USP1 acts as a negative regulator, thus removing the ubiquitin residue from the monoubiquitinated PCNA to reduce the mutagenic effect of TLS polymerases [172, 173].
Studies in yeast indicate that other factors than PCNA might be involved in the recruitment of TLS polymerases to damaged sites and the subsequent polymerase exchange reaction. Similar to PCNA, the Fanconia anaemia (FA)-ID complex might also promote the exchange of replicative DNA polymerases by TLS polymerases after replication fork blockage [174–176]. The yeast Rad9-Rad1-Hus1 (9-1-1) checkpoint clamp, which is loaded by the RAD24-replication factor C (RFC) clamp loader, physically interacts with Pol ζ indicating a putative role of the (9-1-1) checkpoint clamp in the recruitment of TLS polymerases after checkpoint activation [177].
It was proposed that the major function of TLS polymerases is to allow replication to continue in the presence of DNA damage during S-phase [178]. However, subsequent studies in yeast revealed that TLS polymerases also have a function in post-replicative gap filling, the so called post replicative repair (PRR) [179]. PRR is not necessarily temporally separated from S-phase. Yeast REV1, which interacts and thereby regulates the activity of several TLS polymerases, is highly expressed in late S and early G2 phase [180]. Additionally, studies in mammalian U2OS cells revealed that human REV3 accumulates in G1-phase and at the G2/M transition [97]. In addition, it was show that replication-dependent TLS across UV-adducts is not affected in REV3-deficient MEFs. However, although a significant fraction of CPDs were still bypassed, post-replicative repair of [6-4]PPs was completely abolished in REV3-deficient MEFs [181]. Recently, it was shown in chicken DT40 cells that REV1 is essential to maintain normal replication fork progression in the presence of replication-blocking DNA lesions whereas PCNA is required for post-replicative gap filling [182].
Taken together, the activation of the DNA damage tolerance pathway is regulated by the mono- or polyubiquitination of PCNA, REV1, and the FA-ID complex, and might also dependent on the stage of the cell cycle. The regulation of TLS and post-replicative repair recently got into the limelight but more studies are needed to fully elucidate how TLS is regulated during DDR.
One- and two-polymerase mechanism
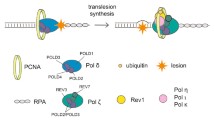
Some TLS polymerases are able to autonomously replicate over a DNA lesion by both incorporating nucleotides opposite the damaged DNA and by extending from the inserted nucleotides. This so called one- polymerase mechanism has been shown to be performed by TLS Pol κ replicating over AP sites in vitro and by TLS Pol η bypassing UV-induced CPDs in vivo. Interestingly, CPDs, the main DNA lesions induced by both UVB and UVA radiation, are bypassed by Pol η in an error-free manner (Figure 3).

One- polymerase error-free bypass of a UV-induced TT-CPD carried out by TLS Pol η (adapted from [247]). See text for details.
However, other lesions such as BaP-G or cisplatin-DNA adducts are mainly bypassed in a process requiring the continuous processing by two TLS polymerases, a so called two-polymerase mechanism. The first TLS polymerase incorporates the nucleotides opposite the DNA lesion and a second TLS polymerase subsequently extends from the inserted nucleotide. Depending on the type of DNA lesion, different pairs of TLS polymerases interact together to replicate the DNA lesion resulting in either an error-free or error-prone bypass (Figure 4).

Two-polymerase mechanisms for bypassing BaP-G (top) or cisplatin (cisPt) (bottom) DNA adducts. The insertion step is performed by one or a combination of several TLS polymerases incorporating nucleotides in an error-free or error-prone manner opposite the adduct whereas the extension step is mainly carried out by TLS Pol ζ (adapted from [247]). See text for details.
DNA damage specific TLS
A variety of different DNA damages can result in the arrest of DNA replication, subsequently requiring TLS to avoid the conversion of a stalled replication fork or post-replication gap into a genotoxic DNA double strand break. Thus, TLS is essential to maintain the genomic integrity and it is therefore not surprising that the system is redundant, i.e. every kind of DNA damage can be bypassed by various combinations of one- or two TLS polymerases (Table 1).
DNA apurinic/apyrimidinic (AP) sites
Nonenzymatic hydrolysis of the base-sugar bonds in DNA and the accumulation of BER-intermediates results in the formation of an estimated 10'000 AP sites per human cell per day [4]. Ionizing radiation and bleomycin, which both are used to treat various types of cancers, not only induce cytotoxic DNA DSBs but also AP sites [183]. AP sites are processed by BER and Pol β is the primary enzyme used for gap filling DNA synthesis during BER. If not repaired, AP sites can be bypassed by TLS polymerases. However, TLS across an AP site is highly error-prone since the sequence information of an AP site is missing. It was shown that TLS Pol β can bypass an AP site resulting in deletions and base substituting errors [184, 185]. A one-polymerase mechanism has been proposed based on an in vitro assay where the TLS Pol θ preferentially incorporates an adenine opposite the AP site followed by a guanine (G) and cytosine/thymine (C/T) [62]. An A opposite the AP site is also the best primer for the extension step by TLS Pol θ [62]. In addition, it has been shown in vitro that TLS Pol η is able to incorporate nucleotides opposite AP sites preferentially A and G and extend from the incorporated nucleotide favouring an A [186]. In vitro studies showed that isolated TLS Pol λ from calf thymus is able to replicate a DNA template containing an AP site in vitro[187]. Similarly, isolated human TLS Pol λ is able to synthesize over an AP site and this bypass is stimulated by PCNA in vitro[188]. TLS Pol κ is able to autonomously bypass AP sites in vitro[189]. Similarly, TLS Pol μ is capable to incorporate nucleotides opposite AP sites in vitro although deletions are frequently generated due to primer realignment [190]. Additionally, The replicative DNA Pol δ in the presence of PCNA preferentially inserted A across AP sites and is also able to extend from the lesion [191].
Alternatively, AP sites might also be bypassed by a two-polymerase mechanism. TLS Pol ι and REV1 are able to incorporate one nucleotide opposite an AP site but the extending polymerase was not identified [191].
7, 8-dihydro-8-oxoguanine (8-oxo-G)
Oxidative stress can lead to the generation of reactive oxygen species (ROS), which induce base modifications such as 8-oxo-G and thymine glycol [192, 193]. Studies have shown an increased level of oxidative DNA damage in cancerous tissue, e.g. a 9-fold increase of 8-oxo-G in tissue from breast cancer compared to surrounding normal tissue [194]. 8-oxo-G is generated by oxidative stress and leads to frequent misincorporation (10-75%) of adenine by human replicative DNA polymerases generating a G:C to T:A transversion [195]. In addition, TLS Pol κ also inserts mainly adenine opposite 8-oxo-G [196]. In vitro experiments revealed that TLS Pol ι is able to bypass 8-oxo-G with low efficiency in a generally non-mutagenic manner thereby preferring the incorporation of C over 8-oxo-G followed by A and G [197]. It has been shown that 8-oxo-G lesion in vitro can be bypassed by TLS Pol μ resulting in a -1 deletion due to primer realignment during TLS [190]. More recently, it has been shown in vitro that 8-oxo-G lesions are mainly bypassed by TLS Pol λ and TLS Pol η and that the presence of PCNA and RP-A increases the fidelity of correct C incorporation over the incorrect A incorporation opposite the 8-oxo-G 1200-fold for TLS Pol λ and 68-fold for TLS Pol η [198]. Additionally, PCNA and RP-A inhibited error-prone TLS opposite an 8-oxoG by DNA polyerase β [199].
Thymine glycol
Thymine glycol is the most common thymine lesion induced by reactive oxygen species (ROS). In vitro studies have shown, that TLS Pol θ is able to incorporate nucleotides opposite both 5R- and 5S-diastereoisomers of thymine glycol with similar efficiency but fails to process the extension step [62]. TLS Pol ν is able to bypass 5S-thymine glycol in an error-free manner whereas the bypass of 5R-thymine glycol was less accurate [51]. TLS Pol β and TLS Pol λ have been shown to bypass thymine glycols in gapped DNA structures. Additionally, dependent on the size of the gap, TLS Pol λ is able to perform the extension step. The bypass fidelity of TLS Pol λ is increased by the presence of PCNA [200]. Recently, a two-polymerase mechanism for error-free thymine glycol bypass including TLS Pol κ as nucleotide inserter and TLS Pol ζ as extender was proposed [201].
[6-4]pyrimidine-pyrimidone photoproduct ([6-4]PP)
The most abundant environmental source of DNA damage is UV-light, which induces nucleotide dimerization, e.g. CPDs and [6-4]PPs at a 3:1 ratio [202]. One hour at the beach results in the induction of approximately 1 × 105 UV-adducts per exposed cell [203].
The induction of [6-4]PPs induces a bend of 44° in the DNA helix, which triggers the efficient recognition and repair of [6-4]PPs by NER [204]. In a primer extension assay, only TLS Pol η was able to autonomously insert nucleotides opposite an [6-4]PP although with a significant lower efficiency than opposite CPDs and without detectable extension [51]. Replication-dependent bypass of UV-adducts was not delayed in REV3-deficient MEFs. However, post-replicative repair of [6-4]PPs was completely dependent on REV3, i.e. TLS Pol ζ [181]. Based on a in vivo plasmid assay, alternative two-polymerase mechanism models have been proposed for the bypass of [6-4]PPs. In the first model, TLS Pol ι and TLS Pol η alternatively incorporate nucleotides opposite a [6-4]PP in a process, which is error-free or error-prone at the 3' thymine and 3' cytosine, respectively, and which results in the subsequent extension by a yet unknown DNA TLS polymerase. In the second model, a yet unknown polymerase incorporates nucleotides opposite a [6-4]PP in a process, which is error-free at the 3' thymine or 3'cytosine. Subsequent extension is carried out by TLS Pol ζ [205]. An additional two-step model was proposed where TLS Pol ι incorporates nucleotides opposite a [6-4]PP in an error-prone manner and TLS Pol θ carries out the subsequent extension step from the mismatched primer terminus [70].
Cyclobutane pyrimidine dimer (CPD)
CPDs are not as much DNA helix distorting (9°) as [6-4]PPs and are therefore not an ideal substrate for NER [206]. Hence, CPDs persist longer after UV-irradiation than [6-4]PPs thereby blocking DNA replication more frequently thus rendering their tolerance more dependent on functional TLS. Depletion of TLS Pol η renders human cells sensitive to UV-irradiation, especially to CPD induction [41, 160]. TLS Pol η is able to replicate error-free over CPDs in vitro[207], with a higher error rate at the 3'T than at the 5'T [208].It has been shown in vivo that TLS was reduced and mutagenicity increased in cells lacking TLS Pol η using a quantitative TLS assay measuring TLS across CPDs. Also in vivo, most of the mutations were found opposite the 3'T of the CPD [209].
Subsequently, it has been proposed that CPDs in XPV cells, e.g. in the absence of TLS Pol η, are replicated by the two-polymerase mechanism in an error-prone manner. The two step model includes either TLS Pol ι, TLS Pol κ or a yet undefined polymerase or their combined action for the first and the second pyrimidine nucleotide incorporation opposite a CPD, followed by the extension step achieved by TLS Pol ζ and to a minor extend by TLS Pol κ [160]. Additionally, in vitro experiments revealed that TLS Pol μ can autonomously bypass CPDs in a mainly error-free manner and that the subsequent extension was further enhanced by TLS Pol ζ [190].
Benzo[α]pyrene-guanine (BaP-G)
BaP is a major compound of tobacco smoke and forms, upon metabolic activation, a covalent BaP-G DNA adduct, which is associated with the development of lung cancer [210]. BaP-G frequently mispairs during DNA replication with A therefore leading to G:T transversions [210]. In vitro and in vivo studies showed efficient bypass of BaP-G by TLS Pol κ using a gapped plasmid containing a BaP-G lesion [211]. Additionally, BaP-G has been shown to be bypassed by a two-polymerase mechanism in vivo. TLS Pol κ inserts nucleotides opposite a BaP-G error-free whereas insertion by TLS Pol η is error-prone. Subsequent extension is performed by TLS Pol ζ. Since the combined inhibition of TLS Pol κ and TLS Pol η did not decrease TLS to a similar extend than depletion of TLS Pol ζ, it was suggested that a third unknown TLS polymerase might be involved in insertion opposite a BaP-G DNA adduct [64]. Indeed, it was shown that TLS Pol μ was able to bypass bulky DNA lesions including BaP-G DNA adducts [190].
Intrastrand-crosslinks
The chemical agent cisplatin is used for therapeutical treatment of most solid tumors including lung cancer and malignant pleural mesothelioma and forms DNA intra- and interstrand-crosslinks (ICLs), which can lead to a blockage of the DNA replication machinery [6]. Intrastrand-crosslinks are the most prevalent form of cisplatin-induced DNA adducts (> 90%) [6] and are bypassed by one- or the two-polymerase mechanisms. In vitro experiments revealed that Pol β can bypass various cisplatin intrastrand adducts [122, 212]. It has been reported that TLS Pol η in vitro is able to bypass a d(GpG)-cisplatin intrastrand adduct thereby preferentially incorporating C opposite the d(GpG)-cisplatin intrastrand adduct [186, 213]. TLS Pol ζ can also bypass cisplatin intrastrand adducts although with low efficiency [214]. It was subsequently shown in vitro that TLS Pol μ is less efficient than pol η in catalyzing translesion synthesis past platinum intrastrand adducts but appears to be significantly more efficient than Pol β or Pol ζ [215]. Recently, it has been shown in vivo, that either TLS Pol η or TLS Pol κ incorporate the correct or incorrect, respectively, nucleotide opposite the d(GpG)-cisplatin intrastrand adduct and TLS Pol ζ carries out the extension step [64]. Similarly, in vivo experiments proposed a model where RAD18/RAD6 dependent monoubiquitination of PCNA activates the bypass of the d(GpG)-cisplatin intrastrand adduct by TLS Pol η and activated TLS Pol ζ performs the subsequent extension in a REV1-dependent manner [105].
Interstrand-crosslink (ICL)
An excellent review of ICL repair and cancer has been published recently [216]. In contrast to the DNA damages described above, ICLs cannot be bypassed since, as the name implies, both DNA strands are covalently linked and therefore no template for DNA synthesis is available. It was proposed that ICLs kill cells due to 1.) Blockage of DNA replication 2.) Stalling of transcription or 3.) Distortion of the chromatin thereby preventing the access of DNA-interacting proteins (reviewed in [216]). Thus, a complex ICL repair machinery evolved and it was shown that TLS polymerases are involved in this process.
Lipid peroxidation can occur inside the body or in foods before they are eaten resulting in the production of by-products capable of crosslinking DNA, e.g. β-unsaturated aldehydes [217]. Recent biochemical and cellular studies implicated that dialdehydes formed by lipid peroxidation induce minor groove ICLs, which are repaired by a TLS Pol κ dependent mechanism [218]. In cancer therapy, cisplatin is the most widely used crosslinking drug but only approximately 10% of the total DNA adducts induced by cisplatin are ICLs [6]. XPV cells, which are deficient for TLS Pol η, are hypersensitive to cisplatin [219, 220].
During the G1 phase of the cell cycle, ICL are repaired in a recombination-independent pathway including NER, TLS Pol ζ and Rev1 [221]. Experiments with X. laevis egg extracts revealed that during replication-dependent ICL repair, the replicative DNA polymerase stalls about 24 nucleotides before the crosslink [222]. At this site, the replicative DNA polymerase is replaced by a TLS polymerase, most probably TLS Pol ν [76], which extends the nascent strand to within 1 base pair of the ICL. Subsequently, DNA nucleases unhook the ICL and REV1 inserts a cytosine opposite the unhooked ICL [223, 224]. Several of the TLS polymerases may extend DNA synthesis beyond the ICL but only deletion of TLS Pol ζ abrogates TLS extension in X. laevis extracts and is was suggested that only Pol ν, REV1 and Pol ζ are essential for ICL repair (reviewed in [216]). Indeed, a recent study showed in human cells that only REV1 and TLS Pol ζ are required for the repair of ICLs whereas RAD18, TLS Pol η, REV1 and TLS Pol ζ are all necessary for replicative bypass of cisplatin intrastrand DNA crosslinks [175].
Relevance of TLS polymerases in cancer therapy
Targeting the error-prone Pol ζ by deletion of REV3 in yeast results in a reduced spontaneous mutation rate [225]. On the other hand, selective deletion of REV3 in mature B cells impaired proliferation and genomic stability [106]. Thus, as indicated in Figure 5, a reduction of the mutation rate by inhibition of TLS is inversely correlated with an increase in gross chromosomal instability. Since genomic instability is a hallmark of cancer, an important question is how carcinogenesis is influenced by changes in the expression or activity of TLS polymerases. XPV patients who are deficient for TLS Pol η activity, suffer from a very high cancer incidence since TT-CPDs, which are bypassed error-free by Pol η, are bypassed in the absence of Pol η by alternative TLS polymerases in an error-prone manner [160]. In addition, somatic mutations of Pol β were identified in adenocarcinoma of the colon [226] and mutations in the gene enconding Pol ι are associated with increased susceptibility to lung cancer in mice [10, 227, 228] and humans [229]. Single nucleotide polymorphisms in the human REV1 gene are associated with increased lung cancer risk [229]. Changes in expression and mutations in the genes encoding Pol ι and Pol κ have been found in human tumors [230–232]. Gene expression of TLS Pol θ is upregulated in two cohorts of patients with untreated primary breast cancers, which correlates with poor clinical outcome [233]. In summary, there is a growing body of literature indicating that increased as well as decreased TLS activity is associated with increased and/or accelerated tumorigenesis.

Trade-off between the accumulation of mutations due to DNA lesion bypass by TLS and the accumulation of genomic instability in the absence of TLS.
However, transient inhibition of TLS activity might be beneficial for cancer treatment. In detail, the concept of "synthetic lethality" is applied as a therapeutic approach where defects in two pathways alone can be tolerated but become lethal when combined. DDR is often abrogated in cancer cells and it was proposed to develop cancer treatments taking advantage of cancer-specific DDR alterations [234]. The principle of synthetic lethality was successfully applied in cancer therapy of patients carrying mutations in BRCA1 or BRCA2, a specific DNA-repair defect. Inhibition of poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP) resulted in synergistic antitumor activity in the treatment of hereditary ovarian- and breast cancer of patients with BRCA mutations [235].
In analogy, there are indications that targeting TLS polymerases per se, i.e. without additional chemotherapy, might be applicable for cancer therapy. For example, based on the overlap in function of mismatch repair and DNA polymerase proofreading activity, it was recently shown that inhibition of Pol β or γ induces synthetic sickness/lethality in MSH2- or MLH1-deficient human cancer cells, respectively [236]. Similarly, a recent study from our laboratory revealed that inhibition of REV3 expression per se resulted in decreased colony formation and accumulation of persistent DNA damage in cancer cell lines of different origin whereas cell growth of control cell lines was less affected [237].
In addition, it was proposed that targeting TLS should enhance the therapeutic effect and reduce the resistance formation of DNA-damaging chemotherapeutics [238]. Indeed, inhibition of REV3 expression sensitized human fibroblasts to cisplatin and decreased the formation of cisplatin resistant cells in vitro[95]. Recent findings in a transplantable mouse xenograft model showed that suppression of REV3 expression increased the sensitivity of chemoresistant adenocarcinomas to cisplatin and reduced occurrence of cisplatin-induced resistance [103]. Similarly, using a preclinical mouse model of Burkitt's lymphoma, it was shown that suppression of both REV1 and REV3 expression, sensitized lymphomas to cisplatin [102]. The same study also showed that REV1 suppression in lymphoma cells inhibited resistance formation after cyclophosphamide treatment in vitro and improved cyclophosphamide-based chemotherapy of lymphomas in vivo.
Interestingly, studies in yeast showed that resistance formation after hydroxyurea treatment, which inhibits the ribonucleotide reductase and therefore reduces/imbalances the nucleotide pool, is also suppressed by REV3 deletion [239]. Thus, suppression of TLS polymerases might also reduce resistance formation of drugs whose therapeutic effect is also based on the reduction/imbalance of the nucleotide pool, e.g. gemcitabine and pemetrexed. In summary, inhibition of the expression/activity of TLS activity may increase responsiveness to genotoxic treatments and improve the clinical outcome.
Although the effects of TLS inhibition on cancer cells are well investigated, less is known how inhibition of TLS affects normal cells. No deficiency in cell growth/survival was mentioned after antisense-based inhibition of REV3 expression in human non-tumor cell lines [66, 100, 237]. In contrast, it was shown by different groups that REV3 knockout reduced cell growth of MEFs [92, 93]. Thus, it will be crucial to carefully evaluate how normal cells are affected by any cancer therapy based on TLS inhibition. To minimize the risk of malignant transformation of normal cells due to the induction of genomic instability by TLS inhibition during cancer therapy, transient inhibition would be favourable over long-term inhibition, e.g. specific small molecules inhibitors would be favourable over a lentiviral-based system constitutively delivering a TLS polymerase-targeting siRNA.
To date, no specific inhibitors for Y family TLS polymerases are available except for the pyrene nucleotide analogs oxetanocin (OXT)-GTP and -ATP, which are able to inhibit TLS Pol η [240]. Additionally, some natural inhibitors are known to have inhibitory effects such as Petasiphenol, which is a specific inhibitor of TLS Pol λ in vitro[241]. It has been shown that Petasiphenol has antiangiogenic activity [242, 243]. Tormetic acid is another inhibitor of TLS Pol λ and β but also of replicative DNA polymerases, e.g. Pol α. Tormetic acid showed an antitumorigenic activity in vivo[243].
Conclusions
The existence of error-prone TLS polymerases reflects a trade-off between avoiding gross chromosomal instability due to replication fork breakdown and the occurrence of mutations on the nucleotide level (Figure 5).
It was proposed that the evolution of long lived and large animals such as vertebrates necessarily entailed the acquisition of potent tumor suppressive mechanisms [244]. Thus, compared to lower organisms such as bacteria and yeast, it can be speculated that the increased number of TLS polymerases evolved in higher organisms as a tumor suppressive adaptation. Alternatively, research carried out in the last years showed that mammalian TLS polymerases are not only involved in bypassing and repair of DNA lesions but also in the diversification processes of IgG and the maintenance of epigenetic modifications. In summary, TLS polymerases have a function beyond the maintenance of the genomic integrity and it will be interesting tofurther elucidate the involvement of TLS in the delicate balance between cancer suppression and longevity.
References
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W: Initial sequencing and analysis of the human genome. Nature. 2001, 409 (6822): 860-921.
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA: The sequence of the human genome. Science. 2001, 291 (5507): 1304-1351.
Lindahl T, Barnes DE: Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000, 65: 127-133.
Barnes DE, Lindahl T: Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004, 38: 445-476.
Hecht SS: Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999, 91 (14): 1194-1210.
Zamble DB, Lippard SJ: Cisplatin and DNA repair in cancer chemotherapy. Trends Biochem Sci. 1995, 20 (10): 435-439.
Aguilera A, Gomez-Gonzalez B: Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008, 9 (3): 204-217.
Chuang PT, Kawcak T, McMahon AP: Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev. 2003, 17 (3): 342-347.
van der Wilt CL, Backus HH, Smid K, Comijn L, Veerman G, Wouters D, Voorn DA, Priest DG, Bunni MA, Mitchell F: Modulation of both endogenous folates and thymidine enhance the therapeutic efficacy of thymidylate synthase inhibitors. Cancer Res. 2001, 61 (9): 3675-3681.
Lee GH, Nishimori H, Sasaki Y, Matsushita H, Kitagawa T, Tokino T: Analysis of lung tumorigenesis in chimeric mice indicates the Pulmonary adenoma resistance 2 (Par2) locus to operate in the tumor-initiation stage in a cell-autonomous manner: detection of polymorphisms in the Poli gene as a candidate for Par2. Oncogene. 2003, 22 (15): 2374-2382.
Broomfield S, Hryciw T, Xiao W: DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae. Mutation research. 2001, 486 (3): 167-184.
Bessman MJ, Kornberg A, Lehman IR, Simms ES: Enzymic synthesis of deoxyribonucleic acid. Biochimica et biophysica acta. 1956, 21 (1): 197-198.
Goulian M, Kornberg A: Enzymatic synthesis of DNA. 23. Synthesis of circular replicative form of phage phi-X174 DNA. Proc Natl Acad Sci USA. 1967, 58 (4): 1723-1730.
Goulian M, Kornberg A, Sinsheimer RL: Enzymatic synthesis of DNA, XXIV. Synthesis of infectious phage phi-X174 DNA. Proc Natl Acad Sci USA. 1967, 58 (6): 2321-2328.
Kornberg T, Gefter ML: DNA synthesis in cell-free extracts of a DNA polymerase-defective mutant. Biochem Biophys Res Commun. 1970, 40 (6): 1348-1355.
Kornberg T, Gefter ML: Purification and DNA synthesis in cell-free extracts: properties of DNA polymerase II. Proc Natl Acad Sci USA. 1971, 68 (4): 761-764.
Kornberg T, Gefter ML: Deoxyribonucleic acid synthesis in cell-free extracts. IV. Purification and catalytic properties of deoxyribonucleic acid polymerase III. J Biol Chem. 1972, 247 (17): 5369-5375.
George J, Devoret R, Radman M: Indirect ultraviolet-reactivation of phage lambda. Proc Natl Acad Sci USA. 1974, 71 (1): 144-147.
Lemontt JF: Mutants of yeast defective in mutation induced by ultraviolet light. Genetics. 1971, 68 (1): 21-33.
Kato T, Shinoura Y: Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol Gen Genet. 1977, 156 (2): 121-131.
Lawrence CW, Das G, Christensen RB: REV7, a new gene concerned with UV mutagenesis in yeast. Mol Gen Genet. 1985, 200 (1): 80-85.
Bonner CA, Hays S, McEntee K, Goodman MF: DNA polymerase II is encoded by the DNA damage-inducible dinA gene of Escherichia coli. Proc Natl Acad Sci USA. 1990, 87 (19): 7663-7667.
Iwasaki H, Nakata A, Walker GC, Shinagawa H: The Escherichia coli polB gene, which encodes DNA polymerase II, is regulated by the SOS system. J Bacteriol. 1990, 172 (11): 6268-6273.
Kenyon CJ, Walker GC: DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proc Natl Acad Sci USA. 1980, 77 (5): 2819-2823.
Wagner J, Gruz P, Kim SR, Yamada M, Matsui K, Fuchs RP, Nohmi T: The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Molecular cell. 1999, 4 (2): 281-286.
Tang M, Shen X, Frank EG, O'Donnell M, Woodgate R, Goodman MF: UmuD'(2)C is an error-prone DNA polymerase, Escherichia coli pol V. Proc Natl Acad Sci USA. 1999, 96 (16): 8919-8924.
Reuven NB, Arad G, Maor-Shoshani A, Livneh Z: The mutagenesis protein UmuC is a DNA polymerase activated by UmuD', RecA, and SSB and is specialized for translesion replication. J Biol Chem. 1999, 274 (45): 31763-31766.
Tang M, Pham P, Shen X, Taylor JS, O'Donnell M, Woodgate R, Goodman MF: Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000, 404 (6781): 1014-1018.
McDonald JP, Levine AS, Woodgate R: The Saccharomyces cerevisiae RAD30 gene, a homologue of Escherichia coli dinB and umuC, is DNA damage inducible and functions in a novel error-free postreplication repair mechanism. Genetics. 1997, 147 (4): 1557-1568.
Sharief FS, Vojta PJ, Ropp PA, Copeland WC: Cloning and chromosomal mapping of the human DNA polymerase theta (POLQ), the eighth human DNA polymerase. Genomics. 1999, 59 (1): 90-96.
Gerlach VL, Aravind L, Gotway G, Schultz RA, Koonin EV, Friedberg EC: Human and mouse homologs of Escherichia coli DinB (DNA polymerase IV), members of the UmuC/DinB superfamily. Proc Natl Acad Sci USA. 1999, 96 (21): 11922-11927.
McDonald JP, Rapic-Otrin V, Epstein JA, Broughton BC, Wang X, Lehmann AR, Wolgemuth DJ, Woodgate R: Novel human and mouse homologs of Saccharomyces cerevisiae DNA polymerase eta. Genomics. 1999, 60 (1): 20-30.
Aoufouchi S, Flatter E, Dahan A, Faili A, Bertocci B, Storck S, Delbos F, Cocea L, Gupta N, Weill JC: Two novel human and mouse DNA polymerases of the polX family. Nucleic acids research. 2000, 28 (18): 3684-3693.
Dominguez O, Ruiz JF, Lain de Lera T, Garcia-Diaz M, Gonzalez MA, Kirchhoff T, Martinez AC, Bernad A, Blanco L: DNA polymerase mu (Pol mu), homologous to TdT, could act as a DNA mutator in eukaryotic cells. The EMBO journal. 2000, 19 (7): 1731-1742.
Garcia-Diaz M, Dominguez O, Lopez-Fernandez LA, de Lera LT, Saniger ML, Ruiz JF, Parraga M, Garcia-Ortiz MJ, Kirchhoff T, del Mazo J: DNA polymerase lambda (Pol lambda), a novel eukaryotic DNA polymerase with a potential role in meiosis. J Mol Biol. 2000, 301 (4): 851-867.
Marini F, Kim N, Schuffert A, Wood RD: POLN, a nuclear PolA family DNA polymerase homologous to the DNA cross-link sensitivity protein Mus308. J Biol Chem. 2003, 278 (34): 32014-32019.
Nelson JR, Lawrence CW, Hinkle DC: Thymine-thymine dimer bypass by yeast DNA polymerase zeta. Science. 1996, 272 (5268): 1646-1649.
Nelson JR, Lawrence CW, Hinkle DC: Deoxycytidyl transferase activity of yeast REV1 protein. Nature. 1996, 382 (6593): 729-731.
Lawrence CW: Cellular roles of DNA polymerase zeta and Rev1 protein. DNA repair. 2002, 1 (6): 425-435.
Lin W, Xin H, Zhang Y, Wu X, Yuan F, Wang Z: The human REV1 gene codes for a DNA template-dependent dCMP transferase. Nucleic acids research. 1999, 27 (22): 4468-4475.
Kannouche P, Stary A: Xeroderma pigmentosum variant and error-prone DNA polymerases. Biochimie. 2003, 85 (11): 1123-1132.
Maher VM, Ouellette LM, Curren RD, McCormick JJ: Frequency of ultraviolet light-induced mutations is higher in xeroderma pigmentosum variant cells than in normal human cells. Nature. 1976, 261 (5561): 593-595.
Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L: Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature. 2000, 406 (6799): 1015-1019.
Washington MT, Minko IG, Johnson RE, Wolfle WT, Harris TM, Lloyd RS, Prakash S, Prakash L: Efficient and error-free replication past a minor-groove DNA adduct by the sequential action of human DNA polymerases iota and kappa. Mol Cell Biol. 2004, 24 (13): 5687-5693.
Ogi T, Shinkai Y, Tanaka K, Ohmori H: Polkappa protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proc Natl Acad Sci USA. 2002, 99 (24): 15548-15553.
Arana ME, Seki M, Wood RD, Rogozin IB, Kunkel TA: Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic acids research. 2008, 36 (11): 3847-3856.
Masuda K, Ouchida R, Takeuchi A, Saito T, Koseki H, Kawamura K, Tagawa M, Tokuhisa T, Azuma T, J OW: DNA polymerase theta contributes to the generation of C/G mutations during somatic hypermutation of Ig genes. Proc Natl Acad Sci USA. 2005, 102 (39): 13986-13991.
Zan H, Shima N, Xu Z, Al-Qahtani A, Evinger Iii AJ, Zhong Y, Schimenti JC, Casali P: The translesion DNA polymerase theta plays a dominant role in immunoglobulin gene somatic hypermutation. The EMBO journal. 2005, 24 (21): 3757-3769.
Bertocci B, De Smet A, Weill JC, Reynaud CA: Nonoverlapping functions of DNA polymerases mu, lambda, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity. 2006, 25 (1): 31-41.
Bertocci B, De Smet A, Berek C, Weill JC, Reynaud CA: Immunoglobulin kappa light chain gene rearrangement is impaired in mice deficient for DNA polymerase mu. Immunity. 2003, 19 (2): 203-211.
Takata K, Shimizu T, Iwai S, Wood RD: Human DNA polymerase N (POLN) is a low fidelity enzyme capable of error-free bypass of 5S-thymine glycol. J Biol Chem. 2006, 281 (33): 23445-23455.
Stallons LJ, McGregor WG: Translesion synthesis polymerases in the prevention and promotion of carcinogenesis. J Nucleic Acids. 2010, 2010:
Boudsocq F, Kokoska RJ, Plosky BS, Vaisman A, Ling H, Kunkel TA, Yang W, Woodgate R: Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J Biol Chem. 2004, 279 (31): 32932-32940.
Fortune JM, Stith CM, Kissling GE, Burgers PM, Kunkel TA: RPA and PCNA suppress formation of large deletion errors by yeast DNA polymerase delta. Nucleic acids research. 2006, 34 (16): 4335-4341.
Kunkel TA: DNA replication fidelity. J Biol Chem. 2004, 279 (17): 16895-16898.
McCulloch SD, Kunkel TA: The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008, 18 (1): 148-161.
Gibbs PE, McDonald J, Woodgate R, Lawrence CW: The relative roles in vivo of Saccharomyces cerevisiae Pol eta, Pol zeta, Rev1 protein and Pol32 in the bypass and mutation induction of an abasic site, T-T (6-4) photoadduct and T-T cis-syn cyclobutane dimer. Genetics. 2005, 169 (2): 575-582.
Washington MT, Johnson RE, Prakash S, Prakash L: Accuracy of thymine-thymine dimer bypass by Saccharomyces cerevisiae DNA polymerase eta. Proc Natl Acad Sci USA. 2000, 97 (7): 3094-3099.
Cordonnier AM, Fuchs RP: Replication of damaged DNA: molecular defect in xeroderma pigmentosum variant cells. Mutation research. 1999, 435 (2): 111-119.
Yamada A, Masutani C, Iwai S, Hanaoka F: Complementation of defective translesion synthesis and UV light sensitivity in xeroderma pigmentosum variant cells by human and mouse DNA polymerase eta. Nucleic acids research. 2000, 28 (13): 2473-2480.
Roush AA, Suarez M, Friedberg EC, Radman M, Siede W: Deletion of the Saccharomyces cerevisiae gene RAD30 encoding an Escherichia coli DinB homolog confers UV radiation sensitivity and altered mutability. Mol Gen Genet. 1998, 257 (6): 686-692.
Seki M, Masutani C, Yang LW, Schuffert A, Iwai S, Bahar I, Wood RD: High-efficiency bypass of DNA damage by human DNA polymerase Q. The EMBO journal. 2004, 23 (22): 4484-4494.
Yoshimura M, Kohzaki M, Nakamura J, Asagoshi K, Sonoda E, Hou E, Prasad R, Wilson SH, Tano K, Yasui A: Vertebrate POLQ and POLbeta cooperate in base excision repair of oxidative DNA damage. Molecular cell. 2006, 24 (1): 115-125.
Shachar S, Ziv O, Avkin S, Adar S, Wittschieben J, Reissner T, Chaney S, Friedberg EC, Wang Z, Carell T: Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. The EMBO journal. 2009, 28 (4): 383-393.
Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T: The Y-family of DNA polymerases. Molecular cell. 2001, 8 (1): 7-8.
Gibbs PE, McGregor WG, Maher VM, Nisson P, Lawrence CW: A human homolog of the Saccharomyces cerevisiae REV3 gene, which encodes the catalytic subunit of DNA polymerase zeta. Proc Natl Acad Sci USA. 1998, 95 (12): 6876-6880.
Lin W, Wu X, Wang Z: A full-length cDNA of hREV3 is predicted to encode DNA polymerase zeta for damage-induced mutagenesis in humans. Mutation research. 1999, 433 (2): 89-98.
Seki M, Marini F, Wood RD: POLQ (Pol theta), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic acids research. 2003, 31 (21): 6117-6126.
Prasad R, Longley MJ, Sharief FS, Hou EW, Copeland WC, Wilson SH: Human DNA polymerase theta possesses 5'-dRP lyase activity and functions in single-nucleotide base excision repair in vitro. Nucleic acids research. 2009, 37 (6): 1868-1877.
Seki M, Wood RD: DNA polymerase theta (POLQ) can extend from mismatches and from bases opposite a (6-4) photoproduct. DNA repair. 2008, 7 (1): 119-127.
Maga G, Shevelev I, Ramadan K, Spadari S, Hubscher U: DNA polymerase theta purified from human cells is a high-fidelity enzyme. J Mol Biol. 2002, 319 (2): 359-369.
Shima N, Munroe RJ, Schimenti JC: The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol Cell Biol. 2004, 24 (23): 10381-10389.
Shima N, Hartford SA, Duffy T, Wilson LA, Schimenti KJ, Schimenti JC: Phenotype-based identification of mouse chromosome instability mutants. Genetics. 2003, 163 (3): 1031-1040.
Ukai A, Maruyama T, Mochizuki S, Ouchida R, Masuda K, Kawamura K, Tagawa M, Kinoshita K, Sakamoto A, Tokuhisa T: Role of DNA polymerase theta in tolerance of endogenous and exogenous DNA damage in mouse B cells. Genes Cells. 2006, 11 (2): 111-121.
Shivapurkar N, Sood S, Wistuba II, Virmani AK, Maitra A, Milchgrub S, Minna JD, Gazdar AF: Multiple regions of chromosome 4 demonstrating allelic losses in breast carcinomas. Cancer Res. 1999, 59 (15): 3576-3580.
Zietlow L, Smith LA, Bessho M, Bessho T: Evidence for the involvement of human DNA polymerase N in the repair of DNA interstrand cross-links. Biochemistry. 2009, 48 (49): 11817-11824.
Moldovan GL, Madhavan MV, Mirchandani KD, McCaffrey RM, Vinciguerra P, D'Andrea AD: DNA polymerase POLN participates in cross-link repair and homologous recombination. Mol Cell Biol. 2010, 30 (4): 1088-1096.
Kohzaki M, Nishihara K, Hirota K, Sonoda E, Yoshimura M, Ekino S, Butler JE, Watanabe M, Halazonetis TD, Takeda S: DNA polymerases nu and theta are required for efficient immunoglobulin V gene diversification in chicken. The Journal of cell biology. 2010, 189 (7): 1117-1127.
Morrison A, Christensen RB, Alley J, Beck AK, Bernstine EG, Lemontt JF, Lawrence CW: REV3, a Saccharomyces cerevisiae gene whose function is required for induced mutagenesis, is predicted to encode a nonessential DNA polymerase. J Bacteriol. 1989, 171 (10): 5659-5667.
Braithwaite DK, Ito J: Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic acids research. 1993, 21 (4): 787-802.
Murakumo Y, Roth T, Ishii H, Rasio D, Numata S, Croce CM, Fishel R: A human REV7 homolog that interacts with the polymerase zeta catalytic subunit hREV3 and the spindle assembly checkpoint protein hMAD2. J Biol Chem. 2000, 275 (6): 4391-4397.
Murakumo Y: The property of DNA polymerase zeta: REV7 is a putative protein involved in translesion DNA synthesis and cell cycle control. Mutation research. 2002, 510 (1-2): 37-44.
Morelli C, Mungall AJ, Negrini M, Barbanti-Brodano G, Croce CM: Alternative splicing, genomic structure, and fine chromosome localization of REV3L. Cytogenet Cell Genet. 1998, 83 (1-2): 18-20.
Van Sloun PP, Romeijn RJ, Eeken JC: Molecular cloning, expression and chromosomal localisation of the mouse Rev3l gene, encoding the catalytic subunit of polymerase zeta. Mutation research. 1999, 433 (2): 109-116.
Morelli C, Karayianni E, Magnanini C, Mungall AJ, Thorland E, Negrini M, Smith DI, Barbanti-Brodano G: Cloning and characterization of the common fragile site FRA6F harboring a replicative senescence gene and frequently deleted in human tumors. Oncogene. 2002, 21 (47): 7266-7276.
Ogawara D, Muroya T, Yamauchi K, Iwamoto TA, Yagi Y, Yamashita Y, Waga S, Akiyama M, Maki H: Near-full-length REV3L appears to be a scarce maternal factor in Xenopus laevis eggs that changes qualitatively in early embryonic development. DNA repair. 9 (1): 90-95.
J OW, Kajiwara K, Kawamura K, Kimura M, Miyagishima H, Koseki H, Tagawa M: An essential role for REV3 in mammalian cell survival: absence of REV3 induces p53-independent embryonic death. Biochem Biophys Res Commun. 2002, 293 (3): 1132-1137.
Esposito G, Godindagger I, Klein U, Yaspo ML, Cumano A, Rajewsky K: Disruption of the Rev3l-encoded catalytic subunit of polymerase zeta in mice results in early embryonic lethality. Curr Biol. 2000, 10 (19): 1221-1224.
Wittschieben J, Shivji MK, Lalani E, Jacobs MA, Marini F, Gearhart PJ, Rosewell I, Stamp G, Wood RD: Disruption of the developmentally regulated Rev3l gene causes embryonic lethality. Curr Biol. 2000, 10 (19): 1217-1220.
Bemark M, Khamlichi AA, Davies SL, Neuberger MS: Disruption of mouse polymerase zeta (Rev3) leads to embryonic lethality and impairs blastocyst development in vitro. Curr Biol. 2000, 10 (19): 1213-1216.
Holway AH, Kim SH, La Volpe A, Michael WM: Checkpoint silencing during the DNA damage response in Caenorhabditis elegans embryos. The Journal of cell biology. 2006, 172 (7): 999-1008.
Wittschieben JP, Reshmi SC, Gollin SM, Wood RD: Loss of DNA polymerase zeta causes chromosomal instability in mammalian cells. Cancer Res. 2006, 66 (1): 134-142.
Zander L, Bemark M: Immortalized mouse cell lines that lack a functional Rev3 gene are hypersensitive to UV irradiation and cisplatin treatment. DNA repair. 2004, 3 (7): 743-752.
Krieg AJ, Hammond EM, Giaccia AJ: Functional analysis of p53 binding under differential stresses. Mol Cell Biol. 2006, 26 (19): 7030-7045.
Wu F, Lin X, Okuda T, Howell SB: DNA polymerase zeta regulates cisplatin cytotoxicity, mutagenicity, and the rate of development of cisplatin resistance. Cancer Res. 2004, 64 (21): 8029-8035.
Rajpal DK, Wu X, Wang Z: Alteration of ultraviolet-induced mutagenesis in yeast through molecular modulation of the REV3 and REV7 gene expression. Mutation research. 2000, 461 (2): 133-143.
Brondello JM, Pillaire MJ, Rodriguez C, Gourraud PA, Selves J, Cazaux C, Piette J: Novel evidences for a tumor suppressor role of Rev3, the catalytic subunit of Pol zeta. Oncogene. 2008, 27 (47): 6093-6101.
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y: ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007, 316 (5828): 1160-1166.
Wittschieben JP, Patil V, Glushets V, Robinson LJ, Kusewitt DF, Wood RD: Loss of DNA Polymerase {zeta} Enhances Spontaneous Tumorigenesis. Cancer Res.
Li Z, Zhang H, McManus TP, McCormick JJ, Lawrence CW, Maher VM: hREV3 is essential for error-prone translesion synthesis past UV or benzo[a]pyrene diol epoxide-induced DNA lesions in human fibroblasts. Mutation research. 2002, 510 (1-2): 71-80.
Diaz M, Watson NB, Turkington G, Verkoczy LK, Klinman NR, McGregor WG: Decreased frequency and highly aberrant spectrum of ultraviolet-induced mutations in the hprt gene of mouse fibroblasts expressing antisense RNA to DNA polymerase zeta. Mol Cancer Res. 2003, 1 (11): 836-847.
Xie K, Doles J, Hemann MT, Walker GC: Error-prone translesion synthesis mediates acquired chemoresistance. Proc Natl Acad Sci USA. 2010, 107 (48): 20792-20797.
Doles J, Oliver TG, Cameron ER, Hsu G, Jacks T, Walker GC, Hemann MT: Suppression of Rev3, the catalytic subunit of Pol{zeta}, sensitizes drug-resistant lung tumors to chemotherapy. Proc Natl Acad Sci USA. 2010, 107 (48): 20786-20791.
Zhang N, Liu X, Li L, Legerski R: Double-strand breaks induce homologous recombinational repair of interstrand cross-links via cooperation of MSH2, ERCC1-XPF, REV3, and the Fanconi anemia pathway. DNA repair. 2007, 6 (11): 1670-1678.
Hicks JK, Chute CL, Paulsen MT, Ragland RL, Howlett NG, Gueranger Q, Glover TW, Canman CE: Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol Cell Biol. 2010, 30 (5): 1217-1230.
Schenten D, Kracker S, Esposito G, Franco S, Klein U, Murphy M, Alt FW, Rajewsky K: Pol zeta ablation in B cells impairs the germinal center reaction, class switch recombination, DNA break repair, and genome stability. J Exp Med. 2009, 206 (2): 477-490.
Murakumo Y, Ogura Y, Ishii H, Numata S, Ichihara M, Croce CM, Fishel R, Takahashi M: Interactions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7. J Biol Chem. 2001, 276 (38): 35644-35651.
Aravind L, Koonin EV: The HORMA domain: a common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem Sci. 1998, 23 (8): 284-286.
Pfleger CM, Salic A, Lee E, Kirschner MW: Inhibition of Cdh1-APC by the MAD2-related protein MAD2L2: a novel mechanism for regulating Cdh1. Genes Dev. 2001, 15 (14): 1759-1764.
Iwai H, Kim M, Yoshikawa Y, Ashida H, Ogawa M, Fujita Y, Muller D, Kirikae T, Jackson PK, Kotani S: A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell. 2007, 130 (4): 611-623.
McBride OW, Kozak CA, Wilson SH: Mapping of the gene for DNA polymerase beta to mouse chromosome 8. Cytogenet Cell Genet. 1990, 53 (2-3): 108-111.
Kumar A, Widen SG, Williams KR, Kedar P, Karpel RL, Wilson SH: Studies of the domain structure of mammalian DNA polymerase beta. Identification of a discrete template binding domain. J Biol Chem. 1990, 265 (4): 2124-2131.
Casas-Finet JR, Kumar A, Morris G, Wilson SH, Karpel RL: Spectroscopic studies of the structural domains of mammalian DNA beta-polymerase. J Biol Chem. 1991, 266 (29): 19618-19625.
Pelletier H, Sawaya MR, Kumar A, Wilson SH, Kraut J: Structures of ternary complexes of rat DNA polymerase beta, a DNA template-primer, and ddCTP. Science. 1994, 264 (5167): 1891-1903.
Beard WA, Shock DD, Yang XP, DeLauder SF, Wilson SH: Loss of DNA polymerase beta stacking interactions with templating purines, but not pyrimidines, alters catalytic efficiency and fidelity. J Biol Chem. 2002, 277 (10): 8235-8242.
Singhal RK, Wilson SH: Short gap-filling synthesis by DNA polymerase beta is processive. J Biol Chem. 1993, 268 (21): 15906-15911.
Prasad R, Beard WA, Wilson SH: Studies of gapped DNA substrate binding by mammalian DNA polymerase beta. Dependence on 5'-phosphate group. J Biol Chem. 1994, 269 (27): 18096-18101.
Matsumoto Y, Kim K: Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science. 1995, 269 (5224): 699-702.
Dianov GL, Prasad R, Wilson SH, Bohr VA: Role of DNA polymerase beta in the excision step of long patch mammalian base excision repair. J Biol Chem. 1999, 274 (20): 13741-13743.
Biade S, Sobol RW, Wilson SH, Matsumoto Y: Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J Biol Chem. 1998, 273 (2): 898-902.
Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH: Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. 1996, 379 (6561): 183-186.
Hoffmann JS, Pillaire MJ, Maga G, Podust V, Hubscher U, Villani G: DNA polymerase beta bypasses in vitro a single d(GpG)-cisplatin adduct placed on codon 13 of the HRAS gene. Proc Natl Acad Sci USA. 1995, 92 (12): 5356-5360.
Esposito G, Texido G, Betz UA, Gu H, Muller W, Klein U, Rajewsky K: Mice reconstituted with DNA polymerase beta-deficient fetal liver cells are able to mount a T cell-dependent immune response and mutate their Ig genes normally. Proc Natl Acad Sci USA. 2000, 97 (3): 1166-1171.
Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K: Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994, 265 (5168): 103-106.
Horton JK, Srivastava DK, Zmudzka BZ, Wilson SH: Strategic down-regulation of DNA polymerase beta by antisense RNA sensitizes mammalian cells to specific DNA damaging agents. Nucleic acids research. 1995, 23 (19): 3810-3815.
Horton JK, Baker A, Berg BJ, Sobol RW, Wilson SH: Involvement of DNA polymerase beta in protection against the cytotoxicity of oxidative DNA damage. DNA repair. 2002, 1 (4): 317-333.
Sobol RW, Watson DE, Nakamura J, Yakes FM, Hou E, Horton JK, Ladapo J, Van Houten B, Swenberg JA, Tindall KR: Mutations associated with base excision repair deficiency and methylation-induced genotoxic stress. Proc Natl Acad Sci USA. 2002, 99 (10): 6860-6865.
Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH: The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000, 405 (6788): 807-810.
Bergoglio V, Pillaire MJ, Lacroix-Triki M, Raynaud-Messina B, Canitrot Y, Bieth A, Gares M, Wright M, Delsol G, Loeb LA: Deregulated DNA polymerase beta induces chromosome instability and tumorigenesis. Cancer Res. 2002, 62 (12): 3511-3514.
Canitrot Y, Laurent G, Astarie-Dequeker C, Bordier C, Cazaux C, Hoffmann JS: Enhanced expression and activity of DNA polymerase beta in chronic myelogenous leukemia. Anticancer Res. 2006, 26 (1B): 523-525.
Garcia-Diaz M, Bebenek K, Sabariegos R, Dominguez O, Rodriguez J, Kirchhoff T, Garcia-Palomero E, Picher AJ, Juarez R, Ruiz JF: DNA polymerase lambda, a novel DNA repair enzyme in human cells. J Biol Chem. 2002, 277 (15): 13184-13191.
Yu X, Chini CC, He M, Mer G, Chen J: The BRCT domain is a phospho-protein binding domain. Science. 2003, 302 (5645): 639-642.
Ramadan K, Maga G, Shevelev IV, Villani G, Blanco L, Hubscher U: Human DNA polymerase lambda possesses terminal deoxyribonucleotidyl transferase activity and can elongate RNA primers: implications for novel functions. J Mol Biol. 2003, 328 (1): 63-72.
Bebenek K, Garcia-Diaz M, Blanco L, Kunkel TA: The frameshift infidelity of human DNA polymerase lambda. Implications for function. J Biol Chem. 2003, 278 (36): 34685-34690.
Picher AJ, Garcia-Diaz M, Bebenek K, Pedersen LC, Kunkel TA, Blanco L: Promiscuous mismatch extension by human DNA polymerase lambda. Nucleic acids research. 2006, 34 (11): 3259-3266.
Ma Y, Lu H, Tippin B, Goodman MF, Shimazaki N, Koiwai O, Hsieh CL, Schwarz K, Lieber MR: A biochemically defined system for mammalian nonhomologous DNA end joining. Molecular cell. 2004, 16 (5): 701-713.
Terrados G, Capp JP, Canitrot Y, Garcia-Diaz M, Bebenek K, Kirchhoff T, Villanueva A, Boudsocq F, Bergoglio V, Cazaux C: Characterization of a natural mutator variant of human DNA polymerase lambda which promotes chromosomal instability by compromising NHEJ. PLoS One. 2009, 4 (10): e7290-
Garcia-Diaz M, Bebenek K, Kunkel TA, Blanco L: Identification of an intrinsic 5'-deoxyribose-5-phosphate lyase activity in human DNA polymerase lambda: a possible role in base excision repair. J Biol Chem. 2001, 276 (37): 34659-34663.
Zhang Y, Wu X, Yuan F, Xie Z, Wang Z: Highly frequent frameshift DNA synthesis by human DNA polymerase mu. Mol Cell Biol. 2001, 21 (23): 7995-8006.
Gu J, Lu H, Tippin B, Shimazaki N, Goodman MF, Lieber MR: XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. The EMBO journal. 2007, 26 (4): 1010-1023.
Nair DT, Johnson RE, Prakash S, Prakash L, Aggarwal AK: Replication by human DNA polymerase-iota occurs by Hoogsteen base-pairing. Nature. 2004, 430 (6997): 377-380.
Uljon SN, Johnson RE, Edwards TA, Prakash S, Prakash L, Aggarwal AK: Crystal structure of the catalytic core of human DNA polymerase kappa. Structure. 2004, 12 (8): 1395-1404.
Silvian LF, Toth EA, Pham P, Goodman MF, Ellenberger T: Crystal structure of a DinB family error-prone DNA polymerase from Sulfolobus solfataricus. Nat Struct Biol. 2001, 8 (11): 984-989.
Trincao J, Johnson RE, Escalante CR, Prakash S, Prakash L, Aggarwal AK: Structure of the catalytic core of S. cerevisiae DNA polymerase eta: implications for translesion DNA synthesis. Molecular cell. 2001, 8 (2): 417-426.
Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR: Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005, 310 (5755): 1821-1824.
Ohashi E, Murakumo Y, Kanjo N, Akagi J, Masutani C, Hanaoka F, Ohmori H: Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells. 2004, 9 (6): 523-531.
Lawrence CW: Cellular functions of DNA polymerase zeta and Rev1 protein. Adv Protein Chem. 2004, 69: 167-203.
Guo C, Tang TS, Bienko M, Parker JL, Bielen AB, Sonoda E, Takeda S, Ulrich HD, Dikic I, Friedberg EC: Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol Cell Biol. 2006, 26 (23): 8892-8900.
Tissier A, Kannouche P, Reck MP, Lehmann AR, Fuchs RP, Cordonnier A: Co-localization in replication foci and interaction of human Y-family members, DNA polymerase pol eta and REVl protein. DNA repair. 2004, 3 (11): 1503-1514.
Guo C, Sonoda E, Tang TS, Parker JL, Bielen AB, Takeda S, Ulrich HD, Friedberg EC: REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Molecular cell. 2006, 23 (2): 265-271.
Simpson LJ, Sale JE: Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. The EMBO journal. 2003, 22 (7): 1654-1664.
Sarkies P, Reams C, Simpson LJ, Sale JE: Epigenetic Instability due to Defective Replication of Structured DNA. Molecular cell. 2010, 40 (5): 703-713.
Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F: The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999, 399 (6737): 700-704.
Acharya N, Yoon JH, Gali H, Unk I, Haracska L, Johnson RE, Hurwitz J, Prakash L, Prakash S: Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase eta in translesion DNA synthesis. Proc Natl Acad Sci USA. 2008, 105 (46): 17724-17729.
Yagi Y, Ogawara D, Iwai S, Hanaoka F, Akiyama M, Maki H: DNA polymerases eta and kappa are responsible for error-free translesion DNA synthesis activity over a cis-syn thymine dimer in Xenopus laevis oocyte extracts. DNA repair. 2005, 4 (11): 1252-1269.
Wang Y, Woodgate R, McManus TP, Mead S, McCormick JJ, Maher VM: Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase eta, DNA polymerase iota causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007, 67 (7): 3018-3026.
Jansen JG, Tsaalbi-Shtylik A, Langerak P, Calleja F, Meijers CM, Jacobs H, de Wind N: The BRCT domain of mammalian Rev1 is involved in regulating DNA translesion synthesis. Nucleic acids research. 2005, 33 (1): 356-365.
Delbos F, De Smet A, Faili A, Aoufouchi S, Weill JC, Reynaud CA: Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse. J Exp Med. 2005, 201 (8): 1191-1196.
Ogi T, Kannouche P, Lehmann AR: Localisation of human Y-family DNA polymerase kappa: relationship to PCNA foci. J Cell Sci. 2005, 118 (Pt 1): 129-136.
Ziv O, Geacintov N, Nakajima S, Yasui A, Livneh Z: DNA polymerase zeta cooperates with polymerases kappa and iota in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients. Proc Natl Acad Sci USA. 2009, 106 (28): 11552-11557.
Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG: Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Molecular cell. 2010, 37 (5): 714-727.
Bavoux C, Leopoldino AM, Bergoglio V, J OW, Ogi T, Bieth A, Judde JG, Pena SD, Poupon MF, Helleday T: Up-regulation of the error-prone DNA polymerase {kappa} promotes pleiotropic genetic alterations and tumorigenesis. Cancer Res. 2005, 65 (1): 325-330.
Bebenek K, Tissier A, Frank EG, McDonald JP, Prasad R, Wilson SH, Woodgate R, Kunkel TA: 5'-Deoxyribose phosphate lyase activity of human DNA polymerase iota in vitro. Science. 2001, 291 (5511): 2156-2159.
Petta TB, Nakajima S, Zlatanou A, Despras E, Couve-Privat S, Ishchenko A, Sarasin A, Yasui A, Kannouche P: Human DNA polymerase iota protects cells against oxidative stress. The EMBO journal. 2008, 27 (21): 2883-2895.
Kannouche P, Fernandez de Henestrosa AR, Coull B, Vidal AE, Gray C, Zicha D, Woodgate R, Lehmann AR: Localization of DNA polymerases eta and iota to the replication machinery is tightly co-ordinated in human cells. The EMBO journal. 2003, 22 (5): 1223-1233.
Kannouche PL, Wing J, Lehmann AR: Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Molecular cell. 2004, 14 (4): 491-500.
Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M: Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. The EMBO journal. 2004, 23 (19): 3886-3896.
Andersen PL, Xu F, Xiao W: Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res. 2008, 18 (1): 162-173.
Chang DJ, Cimprich KA: DNA damage tolerance: when it's OK to make mistakes. Nat Chem Biol. 2009, 5 (2): 82-90.
Unk I, Hajdu I, Fatyol K, Szakal B, Blastyak A, Bermudez V, Hurwitz J, Prakash L, Prakash S, Haracska L: Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc Natl Acad Sci USA. 2006, 103 (48): 18107-18112.
Unk I, Hajdu I, Fatyol K, Hurwitz J, Yoon JH, Prakash L, Prakash S, Haracska L: Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc Natl Acad Sci USA. 2008, 105 (10): 3768-3773.
Ulrich HD: Deubiquitinating PCNA: a downside to DNA damage tolerance. Nat Cell Biol. 2006, 8 (4): 303-305.
Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD: Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006, 8 (4): 339-347.
Wang W: Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007, 8 (10): 735-748.
Hicks JK, Chute CL, Paulsen MT, Ragland RL, Howlett NG, Gueranger Q, Glover TW, Canman CE: Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol Cell Biol. 30 (5): 1217-1230.
Mirchandani KD, McCaffrey RM, D'Andrea AD: The Fanconi anemia core complex is required for efficient point mutagenesis and Rev1 foci assembly. DNA repair. 2008, 7 (6): 902-911.
Sabbioneda S, Minesinger BK, Giannattasio M, Plevani P, Muzi-Falconi M, Jinks-Robertson S: The 9-1-1 checkpoint clamp physically interacts with polzeta and is partially required for spontaneous polzeta-dependent mutagenesis in Saccharomyces cerevisiae. J Biol Chem. 2005, 280 (46): 38657-38665.
Friedberg EC, Lehmann AR, Fuchs RP: Trading places: how do DNA polymerases switch during translesion DNA synthesis?. Molecular cell. 2005, 18 (5): 499-505.
Lopes M, Foiani M, Sogo JM: Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Molecular cell. 2006, 21 (1): 15-27.
Waters LS, Walker GC: The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc Natl Acad Sci USA. 2006, 103 (24): 8971-8976.
Jansen JG, Tsaalbi-Shtylik A, Hendriks G, Verspuy J, Gali H, Haracska L, de Wind N: Mammalian polymerase zeta is essential for post-replication repair of UV-induced DNA lesions. DNA repair. 2009, 8 (12): 1444-1451.
Edmunds CE, Simpson LJ, Sale JE: PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Molecular cell. 2008, 30 (4): 519-529.
Fung H, Demple B: Distinct roles of Ape1 protein in the repair of DNA damage induced by ionizing radiation or bleomycin. J Biol Chem. 2011, 286 (7): 4968-4977.
Blanca G, Villani G, Shevelev I, Ramadan K, Spadari S, Hubscher U, Maga G: Human DNA polymerases lambda and beta show different efficiencies of translesion DNA synthesis past abasic sites and alternative mechanisms for frameshift generation. Biochemistry. 2004, 43 (36): 11605-11615.
Efrati E, Tocco G, Eritja R, Wilson SH, Goodman MF: Abasic translesion synthesis by DNA polymerase beta violates the "A-rule". Novel types of nucleotide incorporation by human DNA polymerase beta at an abasic lesion in different sequence contexts. J Biol Chem. 1997, 272 (4): 2559-2569.
Masutani C, Kusumoto R, Iwai S, Hanaoka F: Mechanisms of accurate translesion synthesis by human DNA polymerase eta. The EMBO journal. 2000, 19 (12): 3100-3109.
Ramadan K, Shevelev IV, Maga G, Hubscher U: DNA polymerase lambda from calf thymus preferentially replicates damaged DNA. J Biol Chem. 2002, 277 (21): 18454-18458.
Maga G, Villani G, Ramadan K, Shevelev I, Tanguy Le Gac N, Blanco L, Blanca G, Spadari S, Hubscher U: Human DNA polymerase lambda functionally and physically interacts with proliferating cell nuclear antigen in normal and translesion DNA synthesis. J Biol Chem. 2002, 277 (50): 48434-48440.
Ohashi E, Ogi T, Kusumoto R, Iwai S, Masutani C, Hanaoka F, Ohmori H: Error-prone bypass of certain DNA lesions by the human DNA polymerase kappa. Genes Dev. 2000, 14 (13): 1589-1594.
Zhang Y, Wu X, Guo D, Rechkoblit O, Taylor JS, Geacintov NE, Wang Z: Lesion bypass activities of human DNA polymerase mu. J Biol Chem. 2002, 277 (46): 44582-44587.
Choi JY, Lim S, Kim EJ, Jo A, Guengerich FP: Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases alpha, delta, eta, iota, kappa, and REV1. J Mol Biol. 2010, 404 (1): 34-44.
Croteau DL, Bohr VA: Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J Biol Chem. 1997, 272 (41): 25409-25412.
Lindahl T: Instability and decay of the primary structure of DNA. Nature. 1993, 362 (6422): 709-715.
Olinski R, Zastawny T, Budzbon J, Skokowski J, Zegarski W, Dizdaroglu M: DNA base modifications in chromatin of human cancerous tissues. FEBS Lett. 1992, 309 (2): 193-198.
Avkin S, Livneh Z: Efficiency, specificity and DNA polymerase-dependence of translesion replication across the oxidative DNA lesion 8-oxoguanine in human cells. Mutation research. 2002, 510 (1-2): 81-90.
Irimia A, Eoff RL, Guengerich FP, Egli M: Structural and functional elucidation of the mechanism promoting error-prone synthesis by human DNA polymerase kappa opposite the 7, 8-dihydro-8-oxo-2'-deoxyguanosine adduct. J Biol Chem. 2009, 284 (33): 22467-22480.
Zhang Y, Yuan F, Wu X, Taylor JS, Wang Z: Response of human DNA polymerase iota to DNA lesions. Nucleic acids research. 2001, 29 (4): 928-935.
Maga G, Villani G, Crespan E, Wimmer U, Ferrari E, Bertocci B, Hubscher U: 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007, 447 (7144): 606-608.
Maga G, Crespan E, Wimmer U, van Loon B, Amoroso A, Mondello C, Belgiovine C, Ferrari E, Locatelli G, Villani G: Replication protein A and proliferating cell nuclear antigen coordinate DNA polymerase selection in 8-oxo-guanine repair. Proc Natl Acad Sci USA. 2008, 105 (52): 20689-20694.
Belousova EA, Maga G, Fan Y, Kubareva EA, Romanova EA, Lebedeva NA, Oretskaya TS, Lavrik OI: DNA polymerases beta and lambda bypass thymine glycol in gapped DNA structures. Biochemistry. 2010, 49 (22): 4695-4704.
Yoon JH, Bhatia G, Prakash S, Prakash L: Error-free replicative bypass of thymine glycol by the combined action of DNA polymerases kappa and zeta in human cells. Proc Natl Acad Sci USA. 2010, 107 (32): 14116-14121.
Lindahl T, Wood RD: Quality control by DNA repair. Science. 1999, 286 (5446): 1897-1905.
Hoeijmakers JH: DNA damage, aging, and cancer. N Engl J Med. 2009, 361 (15): 1475-1485.
Kim JK, Patel D, Choi BS: Contrasting structural impacts induced by cis-syn cyclobutane dimer and (6-4) adduct in DNA duplex decamers: implication in mutagenesis and repair activity. Photochem Photobiol. 1995, 62 (1): 44-50.
Yoon JH, Prakash L, Prakash S: Error-free replicative bypass of (6-4) photoproducts by DNA polymerase zeta in mouse and human cells. Genes Dev. 2010, 24 (2): 123-128.
Park H, Zhang K, Ren Y, Nadji S, Sinha N, Taylor JS, Kang C: Crystal structure of a DNA decamer containing a cis-syn thymine dimer. Proc Natl Acad Sci USA. 2002, 99 (25): 15965-15970.
Johnson RE, Washington MT, Prakash S, Prakash L: Fidelity of human DNA polymerase eta. J Biol Chem. 2000, 275 (11): 7447-7450.
McCulloch SD, Kokoska RJ, Masutani C, Iwai S, Hanaoka F, Kunkel TA: Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature. 2004, 428 (6978): 97-100.
Hendel A, Ziv O, Gueranger Q, Geacintov N, Livneh Z: Reduced efficiency and increased mutagenicity of translesion DNA synthesis across a TT cyclobutane pyrimidine dimer, but not a TT 6-4 photoproduct, in human cells lacking DNA polymerase eta. DNA repair. 2008, 7 (10): 1636-1646.
Denissenko MF, Pao A, Tang M, Pfeifer GP: Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science. 1996, 274 (5286): 430-432.
Avkin S, Goldsmith M, Velasco-Miguel S, Geacintov N, Friedberg EC, Livneh Z: Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: the role of DNA polymerase kappa. J Biol Chem. 2004, 279 (51): 53298-53305.
Vaisman A, Chaney SG: The efficiency and fidelity of translesion synthesis past cisplatin and oxaliplatin GpG adducts by human DNA polymerase beta. J Biol Chem. 2000, 275 (17): 13017-13025.
Vaisman A, Masutani C, Hanaoka F, Chaney SG: Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase eta. Biochemistry. 2000, 39 (16): 4575-4580.
Vaisman A, Lim SE, Patrick SM, Copeland WC, Hinkle DC, Turchi JJ, Chaney SG: Effect of DNA polymerases and high mobility group protein 1 on the carrier ligand specificity for translesion synthesis past platinum-DNA adducts. Biochemistry. 1999, 38 (34): 11026-11039.
Havener JM, Nick McElhinny SA, Bassett E, Gauger M, Ramsden DA, Chaney SG: Translesion synthesis past platinum DNA adducts by human DNA polymerase mu. Biochemistry. 2003, 42 (6): 1777-1788.
Deans AJ, West SC: DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011, 11 (7): 467-480.
Stone MP, Cho YJ, Huang H, Kim HY, Kozekov ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris TM: Interstrand DNA cross-links induced by alpha, beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc Chem Res. 2008, 41 (7): 793-804.
Minko IG, Harbut MB, Kozekov ID, Kozekova A, Jakobs PM, Olson SB, Moses RE, Harris TM, Rizzo CJ, Lloyd RS: Role for DNA polymerase kappa in the processing of N2-N2-guanine interstrand cross-links. J Biol Chem. 2008, 283 (25): 17075-17082.
Albertella MR, Green CM, Lehmann AR, O'Connor MJ: A role for polymerase eta in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005, 65 (21): 9799-9806.
Chen YW, Cleaver JE, Hanaoka F, Chang CF, Chou KM: A novel role of DNA polymerase eta in modulating cellular sensitivity to chemotherapeutic agents. Mol Cancer Res. 2006, 4 (4): 257-265.
Shen X, Jun S, O'Neal LE, Sonoda E, Bemark M, Sale JE, Li L: REV3 and REV1 play major roles in recombination-independent repair of DNA interstrand cross-links mediated by monoubiquitinated proliferating cell nuclear antigen (PCNA). J Biol Chem. 2006, 281 (20): 13869-13872.
Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC: Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008, 134 (6): 969-980.
Masuda Y, Ohmae M, Masuda K, Kamiya K: Structure and enzymatic properties of a stable complex of the human REV1 and REV7 proteins. J Biol Chem. 2003, 278 (14): 12356-12360.
Hlavin EM, Smeaton MB, Noronha AM, Wilds CJ, Miller PS: Cross-link structure affects replication-independent DNA interstrand cross-link repair in mammalian cells. Biochemistry. 2010, 49 (18): 3977-3988.
Quah SK, von Borstel RC, Hastings PJ: The origin of spontaneous mutation in Saccharomyces cerevisiae. Genetics. 1980, 96 (4): 819-839.
Starcevic D, Dalal S, Sweasy JB: Is there a link between DNA polymerase beta and cancer?. Cell Cycle. 2004, 3 (8): 998-1001.
Lee GH, Matsushita H: Genetic linkage between Pol iota deficiency and increased susceptibility to lung tumors in mice. Cancer Sci. 2005, 96 (5): 256-259.
Wang M, Devereux TR, Vikis HG, McCulloch SD, Holliday W, Anna C, Wang Y, Bebenek K, Kunkel TA, Guan K: Pol iota is a candidate for the mouse pulmonary adenoma resistance 2 locus, a major modifier of chemically induced lung neoplasia. Cancer Res. 2004, 64 (6): 1924-1931.
Sakiyama T, Kohno T, Mimaki S, Ohta T, Yanagitani N, Sobue T, Kunitoh H, Saito R, Shimizu K, Hirama C: Association of amino acid substitution polymorphisms in DNA repair genes TP53, POLI, REV1 and LIG4 with lung cancer risk. Int J Cancer. 2005, 114 (5): 730-737.
Albertella MR, Lau A, O'Connor MJ: The overexpression of specialized DNA polymerases in cancer. DNA repair. 2005, 4 (5): 583-593.
J OW, Kawamura K, Tada Y, Ohmori H, Kimura H, Sakiyama S, Tagawa M: DNA polymerase kappa, implicated in spontaneous and DNA damage-induced mutagenesis, is overexpressed in lung cancer. Cancer Res. 2001, 61 (14): 5366-5369.
Yang J, Chen Z, Liu Y, Hickey RJ, Malkas LH: Altered DNA polymerase iota expression in breast cancer cells leads to a reduction in DNA replication fidelity and a higher rate of mutagenesis. Cancer Res. 2004, 64 (16): 5597-5607.
Lemee F, Bergoglio V, Fernandez-Vidal A, Machado-Silva A, Pillaire MJ, Bieth A, Gentil C, Baker L, Martin AL, Leduc C: DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc Natl Acad Sci USA. 2010, 107 (30): 13390-13395.
Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C: DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005, 434 (7035): 864-870.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009, 361 (2): 123-134.
Martin SA, McCabe N, Mullarkey M, Cummins R, Burgess DJ, Nakabeppu Y, Oka S, Kay E, Lord CJ, Ashworth A: DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer cell. 2010, 17 (3): 235-248.
Knobel PA, Kotov IN, Felley-Bosco E, Stahel RA, Marti TM: Inhibition of REV3 expression induces persistent DNA damage and growth arrest in cancer cells. Neoplasia (New York, NY. 2011, 13 (10): 961-970.
Wang Z: DNA damage-induced mutagenesis: a novel target for cancer prevention. Mol Interv. 2001, 1 (5): 269-281.
Northam MR, Robinson HA, Kochenova OV, Shcherbakova PV: Participation of DNA polymerase zeta in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics. 2010, 184 (1): 27-42.
Izuta S: Inhibition of DNA polymerase eta by oxetanocin derivatives. Nucleic Acids Symp Ser (Oxf). 2006, 269-270. 50
Mizushina Y, Kamisuki S, Kasai N, Ishidoh T, Shimazaki N, Takemura M, Asahara H, Linn S, Yoshida S, Koiwai O: Petasiphenol: a DNA polymerase lambda inhibitor. Biochemistry. 2002, 41 (49): 14463-14471.
Matsubara K, Mori M, Mizushina Y: Petasiphenol which inhibits DNA polymerase lambda activity is an inhibitor of in vitro angiogenesis. Oncol Rep. 2004, 11 (2): 447-451.
Maga G, Hubscher U: Repair and translesion DNA polymerases as anticancer drug targets. Anticancer Agents Med Chem. 2008, 8 (4): 431-447.
Degregori J: Evolved tumor suppression: why are we so good at not getting cancer?. Cancer Res. 2011, 71 (11): 3739-3744.
Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC: Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009, 73 (1): 134-154.
Lange SS, Takata K, Wood RD: DNA polymerases and cancer. Nat Rev Cancer. 2011, 11 (2): 96-110.
Livneh Z, Ziv O, Shachar S: Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle. 2010, 9 (4): 729-735.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PAK and TMM were both involved in writing and editing the manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Knobel, P.A., Marti, T.M. Translesion DNA synthesis in the context of cancer research. Cancer Cell Int 11, 39 (2011). https://doi.org/10.1186/1475-2867-11-39
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2867-11-39