
Abstract
Saccharomyces cerevisiae CEN.PK 113-7D is widely used for metabolic engineering and systems biology research in industry and academia. We sequenced, assembled, annotated and analyzed its genome. Single-nucleotide variations (SNV), insertions/deletions (indels) and differences in genome organization compared to the reference strain S. cerevisiae S288C were analyzed. In addition to a few large deletions and duplications, nearly 3000 indels were identified in the CEN.PK113-7D genome relative to S288C. These differences were overrepresented in genes whose functions are related to transcriptional regulation and chromatin remodelling. Some of these variations were caused by unstable tandem repeats, suggesting an innate evolvability of the corresponding genes. Besides a previously characterized mutation in adenylate cyclase, the CEN.PK113-7D genome sequence revealed a significant enrichment of non-synonymous mutations in genes encoding for components of the cAMP signalling pathway. Some phenotypic characteristics of the CEN.PK113-7D strains were explained by the presence of additional specific metabolic genes relative to S288C. In particular, the presence of the BIO1 and BIO6 genes correlated with a biotin prototrophy of CEN.PK113-7D. Furthermore, the copy number, chromosomal location and sequences of the MAL loci were resolved. The assembled sequence reveals that CEN.PK113-7D has a mosaic genome that combines characteristics of laboratory strains and wild-industrial strains.
Similar content being viewed by others

Background
The 1000-dollar genome, an iconic goal in human genomics, is already a reality for the yeast Saccharomyces cerevisiae (based on September 2011 quotes from several sequencing companies for sequencing a 12 Mb genome via paired-end short-read sequencing, at over 40-fold coverage).
Although a high quality reference genome of the laboratory strain S. cerevisiae S288C has been available since 1996 [1], there are four main reasons to (re)sequence the genomes of other S. cerevisiae strains. First, the considerable sequence divergence among S. cerevisiae species may cause practical complications, for example, the design of oligonucleotide arrays and cassettes for gene disruption in non-S288C strains. The discovery of > 250,000 polymorphisms in 71 S. cerevisiae strains sequenced at low coverage [2] illustrates that this is not a trivial problem. Secondly, although the genomes of S. cerevisiae strains appears to be much more strongly conserved than those of other organisms, such as E. coli[3], S. cerevisiae strains do show physiologically relevant differences in their gene complement. For example, the absence of a functional MALx3 gene in S. cerevisiae S288C leads to a maltose-negative phenotype, while an atypical ENA gene complement renders the laboratory strain CEN.PK113-7D more sensitive to lithium ions [4]. The possible importance of strain-specific genes is illustrated by the identification of a probable horizontal gene transfer event in the S. cerevisiae wine strain EC1118, that led to the acquisition of genes from the spoilage yeast Zygosaccharomyces bailii[5]. Third, in addition to the presence or absence of coding regions, differences can occur in non-coding regions, such as promoter regions. Knowledge of such differences is essential for the analysis and modelling of regulatory networks in systems biology [6]. Finally, laboratory evolution is rapidly gaining popularity as a tool to analyse genome function and to select for yeast strains with industrially relevant properties [7–11]. Genome comparisons based on mapping short-read data to a distant relative may overlook structural changes. Hence availability of a well-annotated, high-quality reference genome is essential to interpret the changes that occur during laboratory evolution.
Several wild and domestic yeast strains have been sequenced. At the moment, forty-seven genome projects for S. cerevisiae have been registered at GenBank from which twenty-eight contain a de novo assembled (draft) genome [1, 5, 12–20].
The isogenic family of CEN.PK strains was developed by crossing of different laboratory strains of S. cerevisiae in the 1990's by a consortium of German yeast researchers [21]. A subsequent multi-laboratory study in which four S. cerevisiae strains were compared, confirmed that the CEN.PK strains combine good accessibility to classical and molecular genetics techniques with excellent growth characteristics under controlled, industrially relevant conditions [22]. These strains, and in particular the haploid MATa strain CEN.PK113-7D, have since become extremely popular for studies in systems biology [23, 24]. Moreover, the excellent growth characteristics of the CEN.PK strains have resulted in their broad application in metabolic and evolutionary engineering studies, for example for the fermentation of pentose sugars [25–28], production of ethanol [29, 30] and spirits [31] production of lactate and pyruvate [32, 33], production of C4-dicarboxylic acids [34], isoprenoids [35, 36], and fungal polyketide (6-methylsalicylic acid) [37].
Genomic differences between S. cerevisiae CEN.PK113-7D and the S288C strain have been the subject of several studies. Daran-Lapujade and co-workers [38] performed a comparative genotyping of the two strains by hybridization of genomic DNA to oligonucleotide gene-expression arrays. This work led to the identification of several genes that were absent in CEN.PK113-7D, but present in S288C. Schacherer and co-workers [39] employed an oligonucleotide tiling microarray (Affymetrix S. cerevisiae Tiling 1.0R array) based on the S288C genome to detect locations of single nucleotide variation (SNV) in order to narrow down the amount of sequencing needed using traditional sequencing approaches and to find genes absent in CEN.PK113-7D such as RDS1 and EHD3. SNVs in CEN.PK113-7D compared to S288C have previously been characterized by mapping next-generation DNA sequencing data to the S288C reference genome followed by SNV calling [35]. The use of short read (35-bp) sequences and a limited coverage, prohibited detection of insertions and deletions (indels), unique CEN.PK113-7D sequences and structural variations.
The goal of the present study was to make a high-quality assembled and annotated reference genome of S. cerevisiae CEN.PK113-7D sequence available to the academic and industrial yeast research communities. Additionally, we aim to compare the CEN.PK113-7D sequence to that of strain S288C and other previously sequenced S. cerevisiae strains. To this end, we performed high-coverage sequencing, de novo genome assembly, scaffolding and annotation of S. cerevisiae CEN.PK113-7D strain. We explored differences with the S288C genome, including single nucleotide variations, small insertions and deletions (indels) and larger structural variation, copy number variation (CNV) and strain-specific sequences and ORFs.
Results and discussion
Genome assembly, scaffolding and annotation
The genome assembly of the CEN.PK113-7D strain sequence was performed by combining Illumina (36 M reads, 51 bp, paired-end) and 454 (0.6 M reads, mean length 280 bp) sequencing datasets (see Methods and Additional file 1: Supplementary methods) that together represented more than 150-fold coverage of the genome. A hybrid assembly strategy followed by scaffolding using paired-end read information resulted in 565 scaffolds with a total size of 11.6 Mbp (GenBank BioProject PRJNA52955; http://cenpk.bt.tudelft.nl) (Table 1), which were subsequently placed into chromosomal scaffolds based on homology to S288C. Genes in the CEN.PK113-7D genome were predicted using a combination of ab initio and alignment based gene predictors. Combination of predictions by Jigsaw [40] resulted in a total of 5472 ORFs that were predicted with high confidence, comparable to the 5538 genes annotated in S288C [41]. The difference could be attributed to imperfect gene predictions, to missing sequence in the CEN.PK113-7D genome mostly due to repetitive sequences and to genomic content missing from the sequence data due to sequencing bias (e.g. due to nucleotide composition of specific regions). Analysis of the 0.5 Mbp present in S288C but absent in the CEN.PK113-7D draft genome showed that ~26% was in genomic regions with low read mapping probably caused by extreme GC content. Most missing sequence was due to sequence repeats. Out of the 0.5 Mbp absent in the CEN.PK assembly more than ~90% was in repetitive regions. Moreover, about 24 kbp in the CEN.PK assembly was found absent in the S288C genome. This additional sequence will be discussed below.
Reassembly and analysis of replicated sequences
A major challenge of de novo genome assembly with the current read length of 'next generation sequencing' methods is still the identification and reconstruction of repetitive regions. Regions with sequence similarity within the genome are therefore often left unresolved and cause assembly fragmentation. The approximately 38 Long Terminal Repeat (LTR) transposons in the CEN.PK113-7D genome with a length of about 6 kbp each, as well as the whole genome duplication that occurred in the evolutionary history of S. cerevisiae[43] contribute to the presence of repetitive sequences throughout the genome.
S. cerevisiae has five retrotransposon families (Ty1-y5) (Additional file 2: Table S1). Each Ty element is flanked by LTR sequences [44]. A total of 50 LTR flanked retrotransposons have been characterized in the S288C strain [1], whereas only 17 retrotransposons were identified in the strain YJM789, a strain isolated from the lung of an AIDS patient with pneumonia [12]. Of the 50 retrotransposons found in S288C [1], evidence of the presence of 40 was obtained in CEN.PK113-7D genome assembly. We found that 39 retrotransposon sequences were indeed located on contig breaks, which suggests an intact transposon or a remnant of one (Additional file 3: Figure S1). A 40th transposon, YCLWTy5-1, was fully de novo assembled. YCLWTy5-1 is the only Ty5-type retrotransposon in S. cerevisiae (SGD). Analysis of the depth-of-coverage ratio between S288C and CEN.PK113-7D gave a rough estimate of the transposon content and suggested that CEN.PK113-7D had 38 Ty retrotransposons, two fewer than the 40 possible retrotransposons derived from the assembly. This difference might be caused by two contig break locations not containing full-length transposons, possibly due to Ty excision by internal recombination, leaving remnant LTRs.
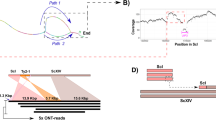
In addition to transposons, paralogs resulting from duplication events form a class of sequences with high similarities that are difficult to assemble de novo. To identify duplicated genes, read depths of the CEN.PK113-7D and S288C genomes were compared. Mapping sequencing reads of both S288C and CEN.PK113-7D to the S288C genome allowed the calculation of a depth of coverage ratio at every location on the genome (Figure 1A), which corrects for systematic biases in mapping depth of individual read sets, such as those caused by differences in GC content. This analysis enabled the identification of six replicated regions in the strain CEN.PK113-7D relative to S288C (Table 2). Five groups of genes comprised these regions including i) BIO2, IMA1, ii) RDL1, RDL2, iii) MAL33, MAL31, MAL32, YBR298C-A, YBR300C, iv) LEU2, NFS1 and v) PHO12, IMD2. To confirm the sequencing results, electrophoretic karyotypes and Southern blots with BIO2, IMA1, MAL32, RDL1 and PHO12 probes were carried out (Figure 1C and Additional file 4: Figure S2). Each probing experiment confirmed the duplication of the tested genes. The hybridization pattern with the RDL1 probe confirmed the presence of an extra copy on chromosome VI (CHRVI) in addition to the expected copy on CHRXV (Additional file 4: Figure S2). The PHO12 hybridization pattern in S288C revealed three chromosomes (I, II and VIII) of which only CHRVIII corresponds to PHO12 (Additional file 4: Figure S2). Hybridization with chromosomes I and II resulted from cross-hybridization with PHO11 (CHRI), PHO13 and PHO5 (both on CHRII). In addition to the three bands in S288C, CEN.PK113-7D exhibited a fourth hybridization on CHRXI (Additional file 4: Figure S2), which confirms the extra copy found in CEN.PK113-7D sequencing data.

Overview of the reconstruction of the MAL2 locus in S. cerevisiae CEN.PK113-7D. A) Depth of coverage analysis of MAL loci. Sequencing reads from the CEN.PK113-7D and S288C have been mapped onto the S288C genome sequence. Log2-ratio's of the average sequencing depth in 414-bp windows of CEN.PK113-7D over S288C have been plotted versus genomic position. Log2-ratio's were capped at -4. B) Schematic representation of the paired-end linkage analysis that showed anomalous read pairs (red arrows) on the end of chromosome III that mapped to BIO2 on CHRVII. C) Southern blots and karyotypes, denoted with 'b' and 'k', respectively, of S288C and CEN.PK113-7D, denoted with 'S' anc 'C', respectively. Probes for IMA1, BIO2 and MAL32 were amplified with the primers listed in Additional file 5: Table S8. Square boxes indicate hybridized chromosomes. Chromosomes marked with an asterisk (*) depicts cross-hybridization of the IMA1 probe to paralogs of IMA1 (see text). D) Schematic organization of the MAL loci in CEN.PK113-7D. Loci marked with (#) were individually amplified, sequenced and assembled.
In contrast to S288C, strains from the CEN.PK lineage are able to grow on maltose as sole carbon source. Both the MAL3 locus and the BIO2/IMA1 locus have double sequence coverage in CEN.PK113-7D (Figure 1A). In S288C the contiguous gene pair BIO2/IMA1 is located upstream of the MAL1 locus on chromosome VII (Figure 1C). As already observed for PHO12 the presence of paralogs complicated the hybridization pattern. IMA1, encoding for an isomaltase, has four paralogs in S288C. Therefore, in addition to the hybridization band on CHRVII corresponding to IMA1, we observed three more hybridized chromosomes, CHRIX (IMA3), CHRX (IMA4, IMA5) and CHRXV (IMA2). In CEN.PK113-7D, the BIO2 and IMA1 hybridization patterns consistently revealed not one but two additional copies of the paired genes on CHRXI and CHRIII. In S288C, BIO2 and IMA1 are physically linked to the locus MAL1 (CHRVII). However, CEN.PK paired-end read mapping to the S288C genome established that the BIO2/IMA1 duplication was located on the right arm subtelomeric region of chromosome III (Figure 1B). Hybridization chromosomal profiles with the MAL32 probes confirmed the co-localization of BIO2/IMA1 and MAL loci on three chromosomes CHRVII, and CHRIII but also on CHRXI as extra copies of BIO2 and IMA1. It was therefore hypothesized that the chromosome III BIO2/IMA1 genes preceded the MAL2 locus as well, given the gene order also observed on the MAL1 locus. This hypothesis was validated by long-range PCR amplification of MAL1, MAL2, and MAL3 in CEN.PK113-7D. The PCR products were sequenced and assembled individually, which was not possible using whole-genome shotgun data because of the high sequence homology. The sequence assembly results confirmed that the additional copy located on chromosome III linked the additional MAL2 locus to BIO2/IMA1. We were not able to reconstruct the MAL4 locus located on CHRXI that seems to share high similarity with CHRIII MAL2.
The maltase MAL22 and maltose permease MAL21 genes are very similar to their MAL3 paralogs, but the regulator gene MAL23 is very different from both the MAL1 and the MAL3 locus. This MAL23 mutant allele also known as MAL2-8 C[47] is responsible for the partial de-repression of the MAL genes in CEN.PK strains in the absence of maltose [48]. Interestingly, the two additional MAL (2 and 4) loci found in CEN.PK113-7D presented features of the two common loci in S288C (MAL1 and MAL3). They probably originated from a two-step event with an initial recombination between MAL1 and 3 at MALx3 genes, placing BIO2 and IMA1 in front of MAL2 or 4, followed by a duplication of newly recombined MAL locus (Figure 1D).
The last region that was called duplicated contained two genes, LEU2 and NFS1. However, this duplication could not be confirmed by Southern blotting of karyotype, nor by direct resequencing of the LEU2 locus.
Single nucleotide variation
Single Nucleotide Variation (SNV) and insertions/deletion (indel) analysis was performed by alignment of the CEN.PK113-7D assembly and the reference sequence of S288C. This analysis identified a total of 21,889 SNVs (Additional file 6: Table S2), of which 13,235 were located in 1,843 open reading frames. 4,677 SNVs (35%) resulted in amino acid changes and affected a total number of 1,406 proteins.
An earlier study by Otero et al (2010), restricting their analysis to genes involved in metabolism, revealed that galactose uptake and ergosterol biosynthesis pathways were enriched for non-synonymous SNVs. In addition to confirming these results, our sequencing data enabled the identification of new variations (Additional file 7: Table S4).
An emblematic mutation in the CEN.PK strain family is located in the CYR1 gene, which encodes for adenylate cyclase, a key enzyme for cAMP production and cAMP-dependent protein kinase signalling. The CEN.PK113-7D CYR1 gene carries a non-synonymous mutation that results in an amino acid substitution (Lys1876Met) [49]. The metabolic repercussions of this mutation are very limited. It was accompanied by the absence of a cAMP peak and a reduced trehalase activation after sudden exposure to high glucose concentration and a delayed mobilization of storage carbohydrates. While we confirmed the occurrence of this SNV in CYR1, several other mutations were found in genes encoding for components of cAMP signalling pathway including PLC1, GPA2, GPB2, IRA2 (Figure 2, Additional file 8: Table S5). The IRA2 gene exhibits a very early frameshift that disrupts the coding sequence and most importantly the C-terminus of the protein that is essential for its activity [50]. The repercussion of these mutations might be suppressed by the presence of the lcr1 (cyr1met1876) mutation, especially for genes encoding for components of the cAMP signalling pathway located upstream to CYR1 (i.e. GPA2 and IRA2) [49].

cAMP signalling pathway in S. cerevisiae. Superscript text indicated number of missense (MS), sense (SE) and indel mutations in the genes.
In total (including the mutations found in the PKA pathway) 20 genes encoding for proteins involved in mitogen-activated protein kinase (MAPK) signalling pathway (Additional file 9: Figure S4) showed non-synonymous differences with the S288C sequence. The STE20 gene implicated in pheromone response, pseudohyphal/invasive growth, vacuole inheritance and down-regulation of sterol uptake carried eight such differences. The gene MSN2, involved in the regulation of over 200 genes in response to several stresses, carried one non-synonymous difference [51].
Small insertions and deletions
In total 2,859 small indels (< 90 bp) were called with an average length of 3 bp. Indels often reside within low-complexity regions, mostly in tandem repeats [52]. 420 indels were found inside a gene, together affecting 297 genes. In 132 genes with indels the start and stop codons usage was not affected (Additional file 10: Table S3).
The DAVID functional annotation tool [53] was applied on the genes containing indels to find enrichment of Gene Ontology (GO) terms using the GO Fat subset [54] (Figure 3). The biological process term 'regulation of transcription' (44 genes of 297 genes with indels) was significantly overrepresented (p = 9.3 × 10-3, Bonferroni corrected). The percentage of gene products involved in 'regulation of transcription' even increased to nearly a quarter of all genes with indels (44 of 220, 20%) when genes classified as uncharacterized or dubious in the Saccharomyces Genome Database (SGD) [55] were not considered. Eventually, in 72% (32 of the 44) of the cases, the indel did not cause a frameshift in the transcription factor genes (i.e. start and stop codon usage was conserved). For example, the CEN.PK113-7D SWI1 allele carries an in-frame 69 bp deletion, conversely CEN.PK113-7D SNF11 contains a 12 bp extension relative to the S288C SNF11 allele. Swi1 and Snf11 are both subunits of the Swi/Snf ATP-dependent chromatin remodelling complex and are part of the RNA polymerase II holoenzyme complex. Sequence alignment of the CEN.PK113-7D SWI1 and SNF11 genes with the sequences from different sequenced S. cerevisiae strains showed a high diversity in gene and protein length (Figure 4 and Additional file 11: Figure S5).

Enrichment analysis of non-synonymous SNVs ( n = 1406) and indels ( n = 297) performed on the Gene Ontology Fat subset and their Bonferroni corrected p -values are shown. All significantly enriched GO terms are shown (α = 0.05).

Tandem repeat variation in SNF11 amongst sequenced S. cerevisiae strains. Multiple sequence alignment of SNF11 was performed with ClustalW [56]. The cladogram was created using whole genome distances and the UPGMA algorithm. Edge lengths in the cladogram are meaningless. Genbank accessions are listed in Additional file 12: Table S7.
Interestingly, the indels in SWI1 and SNF11 occur in tandem repeat regions. Such tandem repeats have been shown to predominantly occur in promoter regions and in genes encoding for transcription factors and cell-surface proteins [52]. In S. cerevisiae recombination of intragenic repeats of cell surface proteins has been suggested to be a reversible mechanism of evolutionary adaptation to the environment by creating cell surface diversity [57]. Although 128 genes that encode for transmembrane and cell wall proteins carried at least one indel, this number was not deemed significant (p < 0.05, Bonferroni corrected). However, similarly to analysis of human genome sequences [52], category enrichment analysis of genes with indels identified overrepresentation of the functional categories 'regulation of transcription' and 'DNA binding' (Figure 3). These repetitive sequences induce contraction and expansion of the gene length by recombination and slippage of the DNA polymerase [52]. In humans, repeat expansion in coding sequences has been implicated in several degenerative diseases as Huntington disease and spinocerebellar ataxias. The genetic cause of Huntington disease is a trinucleotide expansion in exon 1 of the IT15 gene that encodes for huntingtin, a protein involved in several cellular functions including transcription [58].
The severe effects of tandem repeat length variation in humans raises the question of the effect of this phenomenon in S. cerevisiae. Intriguingly, the physiological implications of these polymorphisms in transcription factors have not yet been studied. Dedicated research in the future should address this to understand the role of such variation.
Absent and specific genes in CEN.PK113-7D relative to the reference S288C
A substantial number of S. cerevisiae genes are redundant as a result of genome duplication, thus allowing the loss of one copy of each duplicated gene. To systematically analyze the genes that were absent in CEN.PK113-7D sequence relative to S288C, a methodical search of S288C homologous genes with at least 95% identity in CEN.PK113-7D was performed. To prevent false negatives (i.e. genes called absent because they were not assembled), genes with a lower copy number (log2 ratio < -0.6) in CEN.PK113-7D than in S288C using the read mapping analysis were listed and compared to those S288C genes that did not present a homolog in CEN.PK113-7D. This analysis identified 83 genes absent in CEN.PK113-7D relative to S288C (Additional file 13: Table S6). Absence of 62 of these genes can be explained by the absence of several large fragments that, in strain S288C, are located in subtelomeric regions (Additional file 13: Table S6), such as on the left arm of CHRI (25 Kbp), on the left arm of CHRVIII (12 kb), on the left arm of CHRXII (17 kb) and on the right arm of CHRXIV (14 kb).
Only 21 deletions were found outside subtelomeric regions. Our assembly confirmed the size difference of the PMR2 locus in the CEN.PK113-7D strain [4]. In S288C the PMR2 locus on chromosome IV harbors five copies of ENA genes (ENA1-ENA5) that encode plasma membrane sodium pumping ATPases. Sequence analysis of the PMR2 locus in S. cerevisiae CEN.PK113-7D revealed the presence of a single and new ENA6 allele that showed substantial sequence differences, both at the nucleotide level and at the predicted amino acid sequence level, with previously described ENA genes (Figure 5). The presence of this single and atypical ENA gene correlated with hypersensitivity to sodium and, in particular, to lithium ions. This CEN.PK113-7D locus was also found in several industrial S. cerevisiae strains and natural isolates. The closest homolog was found in S. cerevisiae YJM269 (strain isolated from Blauer Portugieser grapes (GenBank: AEWN00000000.1)), which exhibited a complete coverage of the locus with an identity of 99% at the nucleotide level (Figure 5A).

Occurrence analysis of three regions present in the CEN.PK113-7D genome, but not in the S288C genome. A) Venn diagram that represents the occurrence of the three regions over the available S. cerevisiae sequenced strains in Genbank (Additional file 12: Table S7). B and C) Annotation of the regions and RNA-seq expression profiles. RNA-seq data from glucose- and nitrogen limited anaerobic chemostat cultures (red and blue, respectively) were plotted (one bar every 10th base) for the CEN.PK113-7D specific ENA locus (B) and the two specific contigs (C). Expression data, expressed as the number of times a base is covered by a read, are ranged from are [0-750] for contig379 and contig151 and [0-250] for contig596.
The assembled CEN.PK113-7D genome sequence contained several sequences that are absent in S288C. The CEN.PK specific genes were organized in two large blocks: contig151 and contig596 (Figure 5C). The first block contig151, showed similarity with sequence data of S. cerevisiae strains YJM269 (79%) and PW5 (isolated from Nigerian Raphia palm wine) http://www.genome.wustl.edu/genomes/view/saccharomyces_cerevisiae_pw5 (63%) (Figure 5A). Of the five adjacent genes on contig151, the three new genes could not be functionally annotated. However, their expression could be confirmed by RNA-seq on samples taken from anaerobic carbon- and nitrogen-limited chemostats, commonly used cultivation conditions to study yeast physiology. Interestingly, uncharacterized genes 430, 431 and 432 on contig151 exhibited differential expression and were significantly up-regulated under nitrogen-limitation relative to carbon-limitation [59, 60] (Figure 5C). Furthermore, Southern blotting of chromosomal separation gels established that contig151 could be placed on chromosome XVI (Additional file 14: Figure S3). These observations support the existence and functionality of these open reading frames in CEN.PK113-7D (Figure 5). Functional analysis of these new genes should be part of a larger, emerging challenge: the functional analysis of the growing pangenome of the species S. cerevisiae that arises from the advent of next-generation sequencing.
CEN.PK113-7D specific sequences lead to biotin prototrophy
The second sequence block present in CEN.PK113-7D but not in S288C is contig596 (Figure 5C). This contig contained three predicted open reading frames. The first of the three shared strong similarity (95% identity) with PHO12 that encodes an acid phosphatase. As mentioned earlier, PHO12 had three paralogs in the CEN.PK113-7D genome, which preclude the exact localization of this PHO12 family member found on contig596. The second and third open reading frames identified on this contig shared 72% identity with BIO1 (YJR154W) that encodes for a pimeloyl-CoA synthase and 95% identity with BIO6 that encodes for the following step in biotin biosynthesis, a 7-keto-8-amino pelargonic acid (KAPA) synthase (Figure 6A). Southern blotting of S288C and CEN.PK113-7D chromosomal separation gels indicated that these two open reading frames alongside the newly identified PHO12 family member were located on CHRI (Figure 6B).

Biotin biosynthesis characterization in CEN.PK113-7D and S288C. A) Biotin biosynthesis pathway in CEN.PK113-7D. B) Southern blotting of S288C and CEN.PK113-7D chromosomes using specific probes for BIO1 and BIO6 genes. C) Maximal specific growth rate of CEN.PK113-7D and S288C in shake flask cultivations with synthetic medium [62] in absence (white bar), and presence (grey bar) of biotin. Growth rates were calculated from biomass measurements expressed as optical density measured at 660 nm.
The CEN.PK113-7D BIO1 and BIO6 did exhibit similarity within the S. cerevisiae pangenome. The genes are conserved (over 99%) in several other S. cerevisiae strains, such as Sigma1278b (laboratory strain used in pseudohyphal studies, GenBank: ACVY00000000.1), YJM269 and UC5 (isolated from Sene sake in Kurashi, Japan, GenBank: AFDD00000000.1) (Figure 5A). Although, the systematic sequence of S288C as reported in SGD indicates that these genes were absent, Hall and Dietrich (2007) identified two pairs of pseudo genes on CHRI ([YAR069W-A (BIO6) and YHR214W-F (BIO8)]) and CHRVIII [YAR070W-A (BIO1) and YHR214W-G (BIO7)] in S288C. Southern blot chromosomal localization of BIO1 and BIO6 genes in S288C and CEN.PK113-7D established that these two genes were indeed exclusively present in CEN.PK113-7D and were located on CHRI. If the pseudogene pairs were indeed present in S288C as previously proposed [61], they displayed very low degree of similarity. In addition BIO1 and 6 transcripts were detected in glucose- and nitrogen-limited conditions (Figure 5C).
To verify whether these differences in genotype between S288C and CEN.PK113-7D were reflected in phenotype, we tested their growth in chemically defined medium with and without added biotin. After two consecutive shake-flask cultures in biotin-free medium to exhaust biotin reserves, while S288C strain failed to grow (μ < 0.0005 h-1 in 100 hours), the CEN.PK113-7D strain successfully proliferated in the absence of biotin, at a specific growth rate of 0.045 h-1 (compared to 0.38 h-1 in presence of biotin). Biotin is an indispensable water-soluble vitamin that is mainly used as a cofactor by biotin-dependent carboxylases. In the literature, most S. cerevisiae strains are considered to have lost the ability to grow in absence of biotin. To complement this auxotrophy, biotin is routinely included in chemical defined fermentation media. Biotin needs to be dissolved at high pH at which it is not stable. This step is critical in medium preparation and can cause batch-to-batch variation. Strains prototrophic for biotin might offer a solution to this, and CEN.PK113-7D is an excellent model to evaluate the use of biotin prototrophic S. cerevisiae strains for industrial application. Such biotin prototrophy would be economically interesting. A quick estimate based on the amount of biotin used in chemically defined medium [62] designed to grow around 3 g.l-1 biomass, an average catalog price for biotin of 100 €.g-1 and a fermentation volume of 150 m3 would reduce the medium cost by 1000€. This cost saving might be increased by 5 to 20-fold with media elaborated to grow higher biomass densities (e.g. fed-batch for protein production) [63].
CEN.PK113-7D shares genomic material with both laboratory and industrial strains
Our analysis so far focused on the genome comparison of CEN.PK113-7D and the main reference sequence of the laboratory strain S288C. Phylogenetic analysis that aimed at comparing CEN.PK113-7D genome to the other S. cerevisiae genomes available in GenBank (Additional file 12: Table S7), showed the CEN.PK113-7D genome was located in between the laboratory strains (S288C and Sigma1278b) and industrial strains, such as the bioethanol (JAY291), wine (EC1118, RM11-1a, AWRI1631, AWRI1796, Vin13, VL3, T73 and Lalvin QA23) and beer (CLIB382, FostersO and FostersB) strains (Figure 7). Pioneering works from Liti [2] and co-workers as weel as Schacherer and co workers [64] indeed performed similar analyses on a wide range of S. cerevisiae strains, however CEN.PK113-7D could never be positioned accurately. Liti and workers did not include CEN.PK in their work while Schacherer and co-workers did include it, but derived the phylogenetic distances between yeast strains from SNP distribution by array comparative genome hybridization experiments in which all strains were compared to the sequence of S288c excluding all sequences not found in S288c. Our approach based on full genome sequence of all involved strains depicts a clearer positioning of the CEN.PK strains in yeast phylogenetic tree. CEN.PK113-7D is not a typical laboratory strain, but has both features typically found in industrial S. cerevisiae strains and features that make it a suitable laboratory strain. Regions more closely related to either industrial strains or laboratory strains were distributed in a mosaic structure across the CEN.PK113-7D genome (Figure 8, Additional file 12: Table S7). The industrial background of CEN.PK113-7D may further be elucidated by sequencing its parental strains (ENY-WA-1A and MC996A), which have different phenotypes but are of unclear origin.


Mosaic CEN.PK113-7D genome. CEN.PK chromosomes colored by their identity to S. cerevisiae genomes, which were divided into the groups: laboratory (lab) (FL100:PRJNA60147, S288C:PRJNA128, Sigma1278b:PRJNA39317), industrial (e.g. wine, beer, bio-ethanol) (AWRI1631:PRJNA30553, AWRI796:PRJNA48559, CBS7960:PRJNA60391, CLIB382:PRJNA60145, EC1118:PRJEA37863, FostersB:PRJNA48569, FostersO:PRJNA48567, JAY291:PRJNA32809, Kyokai no. 7:PRJNA45827, Lalvin QA23:PRJNA48561, M22:PRJNA28815, PW5:PRJNA60181, RM11-1a:PRJNA13674, T73:PRJNA60195, UC5:PRJNA60197, Vin13:PRJNA48563, VL3:PRJNA48565, YJM269:PRJNA60389) and other (CLIB215:PRJNA60143, CLIB324:PRJNA60415, EC9-8:PRJNA73985, T7:PRJNA60387, Y10:PRJNA60201, YJM789:PRJNA13304, YPS163:PRJNA28813) (Additional file 12: Table S7). Each group was assigned one of the color channels of the RGB figure (red: lab, green: other and blue: industrial). The genome was divided in non-overlapping fragments of 1000 base pairs, represented by one pixel in the figure, which were aligned to the available S. cerevisiae genomes in GenBank (Additional file 12: Table S7). The identity of the best alignment in a group was set to be the value of the corresponding color channel. These values were scaled between 0 and 1; 0 meaning a maximal identity of 97% or lower, 1 meaning a maximal identity of 100%. For example, a white pixel color means 100% conservation of the fragment in all three groups. Blue and cyan mean conservation in industrial strains, but not in laboratory strains.
Conclusion
We have sequenced, assembled and annotated the genome of S. cerevisiae CEN.PK113-7D. Complementary to previous re-sequencing efforts [35], our de novo assembly provides additional insight in still unexplored differences between S. cerevisiae strains, such as large indels. Comparisons of the de novo assembled genome of CEN.PK113-7D to other S. cerevisiae genomes revealed many unstable tandem repeats in genes involved in transcriptional regulation. Repeat expansion and contraction in genes coding for regulatory proteins may be a mechanism of evolving transcription regulation, reconfiguring binding affinity and specificity of transcription factors. From a metabolic engineering perspective, varying tandem repeat lengths may be a promising approach to 'tune' transcription regulation networks.
De novo assembly of the genome allowed us to uncover genes in CEN.PK113-7D that are absent in S288C and relate to phenotypic characteristics specific to CEN.PK113-7D such as maltose metabolism and biotin biosynthesis. These findings led to the surprising discovery that CEN.PK113-7D is biotin prototroph, a phenotypic trait potentially interesting for industrial application. Phylogenetic analysis based on whole-genome sequence established that CEN.PK113-7D is not solely related to laboratory strains like S288C, but also to industrial strains. These genetic specificities are the basis of the robust physiological performance of this strain, which is used as a yeast model for industrial applications. Together, these results contribute to the elucidation of the genetic diversity within the S. cerevisiae pangenome. In addition, a high-quality annotated genome of this popular model strain is now available as a resource to the yeast research community (GenBank BioProject PRJNA52955; http://cenpk.bt.tudelft.nl).
Materials and methods
DNA purification and sequencing
The genomic DNA of S. cerevisiae CEN.PK 113-7D [21] was purified as previously described [66]. High-throughput sequencing DNA libraries for Illumina (Illumina, San Diego, CA) and 454 (454 Life Sciences, Brandford, CT) sequencing were prepared according the manufacturer's recommendations. The Illumina paired-end library was sequenced on Genome Analyzer IIx (Illumina) with a read length of 50 bp at ServiceXS (Leiden, The Netherlands). The 454 library was sequenced on a GS FLX + system (454 Life Sciences) with an average read length of 350 bp at GATC-Biotech (Konstanz, Germany). The sequence reads were deposited in the Sequence Read Archive (SRA study accession: SRP011487.1).
Assembly, scaffolding, annotation and RNAseq
The two sequencing methods used in this study yielded data with different properties in terms of error models and read length. Different assemblers, i.e. de Bruijn graph and overlap-layout-consensus assemblers, yield optimal results given these data types. We therefore chose a hybrid assembly strategy using the Velvet [67] assembler for the Illumina data and the Newbler assembler for the 454 data. The output of both assemblers was combined using the Maia algorithm [42]. Paired-end scaffolding was then performed using Velvet's scaffolder [67]. The resulting contigs were placed into chromosomal scaffolds based on homology with the S288C genome (Saccharomyces Genome Database, http://www.yeastgenome.org/, March 3rd, 2011) using MUMmer [68]. The chromosomal scaffolds were generated for visualization purposes. Variation analysis was performed on contigs rather than on scaffolds, as the bear no bias towards S288C.
ORF finding was performed using several tools combined in the Cyrille2 pipeline [69]. Ab initio gene prediction was performed using Augustus [70], SNAP [71] and GeneMark-S [72]. Comparative gene prediction was performed by mapping S288C ORFs, S288C proteins and UniProt fungal proteins with GenomeThreader [73].
The output of the above predictors was combined by Jigsaw [40] to predict the final ORFs for CEN.PK. These ORFs were aligned using BLAST [74] to the Saccharomyces Genome Database (SGD) to assign the name of the closest S288C homolog. The assembled data can be retrieved at GenBank [BioProject PRJNA52955] and at http://cenpk.bt.tudelft.nl).
RNAseq data
cDNAs from anaerobic carbon-limited, and anaerobic nitrogen-limited chemostat samples [60] were prepared as previously described [75]. The Illumina cDNA libraries were prepared according the manufacturer's recommendations. The libraries were sequenced on Genome Analyzer IIx with read length of 50 bp at Baseclear (Leiden, The Netherlands). The resulting reads were mapped onto the CEN.PK113-7D genome sequence using the Burrows-Wheeler Alignment tool BWA [45]. Visualization of the mapped reads was performed using the Integrated genomics viewer [76].
Gap and missing sequence analysis
We investigated the causes of fragmentation and potentially missing sequence between two CEN.PK113-7D contigs. Every CEN.PK113-7D contig was assigned a location in the S288C genome using alignment by MUMmer. Regions in S288C not covered by one of the CEN.PK113-7D contigs, i.e. the missing sequences or gaps, were analyzed. There are several possible causes for missing sequence: (1) the sequence is unique to S288C; (2) the regions were not well amplified in the PCR reaction in the sequencing instrument, because of too high or too low GC content; or (3) the reads are present but not assembled, possibly because of repeats. If the reads are present and the region is not repetitive, a contig break is unlikely. We therefore investigated the latter two causes of gaps: too low read coverage or repeats.
The Illumina reads were mapped to the S288C genome using BWA [45] and analyzed with Samtools [77]. First we counted the number of regions in the S288C genome between consecutively aligned CEN.PK113-7D contigs that contained at least one base not covered by any read. Such gaps we call missing sequence. Second, we determined if a repetitive sequence could be located in the gap. To this we aligned the S288C genome to itself using MUMmer. Regions were called repetitive if they had > 95% identity with sequence elsewhere in the genome. If a gap overlapped with one of these repetitive regions in S288C, it was labeled unassembled. The total size of missing sequence gaps and unassembled gaps was then tallied, providing insight in the causes of the fragmentation of the assembly.
Variation analysis
SNV indels and novel DNA
CEN.PK113-7D contigs were aligned to S288C with MUMmer with an identity threshold of at least 97%. SNVs and indels were called from alignments with a non-ambiguous mapping. Small indels were detected as gapped alignments, maximized to a length of 90 bp by MUMmer's default settings.
We define a region in CEN.PK113-7D to be unique if it shows less than 95% identity to any region in S288C. To list these unique regions, first all non-unique regions were found by whole genome alignment of CEN.PK113-7D and S288C, keeping only alignments with more than 95% identity. For each position in the S288C genome only the best aligned CEN.PK113-7D region was considered; if other regions aligned, this was considered to be a duplication in CEN.PK113-7D, hence unique. All resulting unaligned regions were then called unique to CEN.PK113-7D and analyzed for genes. Conservation of these unique regions in other yeast strains was investigated by aligning them to the NCBI Whole-Genome-Shotgun Sequences database using blastn [74]. Species which contained a similar sequence were selected by sorting the hits on score and subsequently selecting the top hits with query coverage > 60% and identity > 90%.
Enrichment of Gene Ontology (GO) terms in the GO Fat subset [54] in the sets of SNVs and indels was performed in the DAVID functional annotation tool. P-values were calculated using the EASE score, a modified version of the Fisher exact test [78]. Bonferroni correction was applied by DAVID to account for multiple testing.
Deleted and duplicated genes
Copy number variation of regions in CEN.PK113-7D compared to S288C was investigated by mapping both S288C and CEN.PK113-7D reads to the S288C reference genome. The log2-ratio of the CEN.PK113-7D depth of coverage over that of S288C was calculated using CNV-seq version 0.2-6 [46] and normalized to an average of 1 per chromosome. For this analysis, 36 bp reads for both strains were obtained from [35]. The default CNV-seq threshold of 0.6 was used as a cut-off for the log2-ratio.
S288C genes that could not be mapped to the CEN.PK113-7D assembly by GenomeThreader with an identity score of at least 95% were labeled not present. An additional validation was performed to prevent false positive deletion calls of genes that should be present in the genome but are not assembled; using copy number variation between CEN.PK113-7D and S288C. Genes with a significantly lower copy number value in CEN.PK113-7D were considered not to be present in the data. The intersection of the set of genes not assembled and the set of genes not present in the data resulted in a high confidence list of deleted genes.
Transposon analysis
Ty retrotransposons mostly cause contig breaks in the assembly process, because their sequences are repetitive in the genome. The presence of the S288C transposons in the CEN.PK113-7D genome can be investigated by using these contig breaks. When inspecting the alignment of CEN.PK113-7D and S288C on locations these Ty elements in the S288C genome, three situations can be observed, the transposon is not present, therefore a gapped alignment of one contig spans the transposon location (GA) with a gap of size similar to the Ty retrotransposon size (~6-kbp); the transposon is present in CEN.PK113-7D, but unassembled, therefore two contigs align to both sides of the transposon location (CB); or the transposon is present in CEN.PK and assembled, therefore a gapless alignment is observed (AS). The first situation yields a not present call for the transposon, the second and third situation yield a present call. The presence of transposons unique to CEN.PK113-7D was not investigated, since this requires long insert mate-pair libraries. An estimate of the number of Ty elements in the CEN.PK113-7D genome for each member of the Ty family (Ty1-Ty5) was obtained by calculating the log2-ratio of the depth of coverage for each Ty element in S288C.
Southern blotting
The chromosomes of the CEN.PK113-7D strain were prepared and separated by clamped homogeneous electrical field (CHEF) electrophoresis as previously described in [38]. Transfer of the DNA from the gel to a nylon membrane (Amersham Hybond™-N+, GE Healthcare Europe GmbH, Diegem, Belgium), was performed as previously described in [79]. Genomic DNA probe fragments were amplified from genomic DNA of S. cerevisiae CEN.PK113-7D using Phusion Hot-Start Polymerase (Finnzymes, Landsmeer, The Netherlands) and the oligonucleotides listed in Additional file 5: Table S8. The probes were labelled according to the manufacturer's instructions (Amersham Gene Images AlkPhos Direct Labelling and Detection System, Buckinghamshire, UK). Hybridization was done overnight at 55°C in hybridization buffer (50% formamide, 5× Saline-Sodium Citrate (SSC) buffer, 2% blocking reagent (Roche), 0.1% Na-lauroylsarcosyl, 0.02% Sodium Dodecyl Sulfate (SDS)). Membranes were washed twice with primary wash buffer (2 M urea, 0.1% SDS, 50 mM sodium phosphate, 50 M sodium chloride, 1 mM magnesium chloride, 0.2% blocking reagent) for 15 minutes at 55°C and twice with secondary wash buffer (50 mM Tris base, 0.1 M sodium chloride, 2 mM magnesium chloride) for 5 minutes at room temperature. Digoxigenin-labelled probes were detected by chemiluminescence using CPD-star (Roche, Paris, France).
PCR amplification of the MAL loci
The genomic MAL1, 2 and 3 loci were amplified from genomic DNA of S. cerevisiae CEN.PK113-7D using the Qiagen LongRange PCR kit (Qiagen, Hilden, Germany) according to manufacturer's instructions in a Biometra TGradient Thermocycler (Biometra, Gottingen, Germany). The following primer combinations were used for amplification: ZUO1 Fw/MALx2 Rv (MAL1 locus), YCR102W-A Fw/MALx2 Rv (MAL2 locus) and PHO89 Fw/MALx2 Rv (MAL3 locus) (Additional file 5: Table S8). Sequencing DNA libraries of each of MAL PCR locus for Illumina were prepared and the PCR products were sequenced on Genome Analyzer IIx with read length of 50 bp at Baseclear (Leiden, The Netherlands). The subsequent sequences were assembled using Newbler (454 Life Sciences, Brandford, CT).
Phylogenetic tree construction
The S. cerevisiae genomes were downloaded from GenBank (accession date: 21 Nov. 2011) (Additional file 12: Table S7). Similarities between all pairs of genomes were determined using MUMmer with the settings as recommended in [80]. Pairwise distances were calculated using the coverage distance function [81]. The phylogenetic tree was created by performing hierarchical clustering (UPGMA) with the SplitsTree4 package [65].
Strains and cultivation conditions
The prototrophic S. cerevisiae strains CEN.PK113-7D [21] and S288C (ATCC 204508) [82] were grown in liquid cultures. The shake-flask cultivations with biotin were performed in 500 ml flasks containing 100 ml of medium, which were incubated at 30°C on an orbital shaker set at 200-rpm. The composition of the synthetic medium (SM) was as follows: glucose (20 g.l-1), (NH4)2SO4 (5 g.l-1) KH2PO4 (3 g.l-1), MgSO4 (0.5 g.l-1), trace elements and vitamin solutions [62]. A separate biotin-free vitamin solution was used for growth in absence of biotin. The pH of the medium was adjusted to 5.0 and sterilized by autoclaving. Glucose was autoclaved separately. Vitamins were filter-sterilized and added to the medium. Growth of the various strains was monitored by OD measurements at 660 nm.
References
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al: Life with 6000 genes. Science. 1996, 274: 546-567. 10.1126/science.274.5287.546.
Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, et al: Population genomics of domestic and wild yeasts. Nature. 2009, 458: 337-341. 10.1038/nature07743.
Lukjancenko O, Wassenaar TM, Ussery DW: Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol. 2010, 60: 708-720. 10.1007/s00248-010-9717-3.
Daran-Lapujade P, Daran JM, Luttik MA, Almering MJ, Pronk JT, Kotter P: An atypical PMR2 locus is responsible for hypersensitivity to sodium and lithium cations in the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D. FEMS Yeast Res. 2009, 9: 789-792. 10.1111/j.1567-1364.2009.00530.x.
Novo M, Bigey F, Beyne E, Galeote V, Gavory F, Mallet S, Cambon B, Legras JL, Wincker P, Casaregola S, Dequin S: Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proc Natl Acad Sci USA. 2009, 106: 16333-16338. 10.1073/pnas.0904673106.
Fendt SM, Oliveira AP, Christen S, Picotti P, Dechant RC, Sauer U: Unraveling condition-dependent networks of transcription factors that control metabolic pathway activity in yeast. Mol Syst Biol. 2010, 6: 432-
Wenger JW, Piotrowski J, Nagarajan S, Chiotti K, Sherlock G, Rosenzweig F: Hunger artists: yeast adapted to carbon limitation show trade-offs under carbon sufficiency. PLoS Genet. 2011, 7: e1002202-10.1371/journal.pgen.1002202.
Wisselink HW, Toirkens MJ: del Rosario Franco Berriel M, Winkler AA, van Dijken JP, Pronk JT, van Maris AJ: Engineering of Saccharomyces cerevisiae for efficient anaerobic alcoholic fermentation of L-arabinose. Appl Environ Microbiol. 2007, 73: 4881-4891. 10.1128/AEM.00177-07.
Conrad TM, Lewis NE, Palsson BO: Microbial laboratory evolution in the era of genome-scale science. Mol Syst Biol. 2011, 7: 509-
Hong KK, Vongsangnak W, Vemuri GN, Nielsen J: Unravelling evolutionary strategies of yeast for improving galactose utilization through integrated systems level analysis. Proc Natl Acad Sci USA. 2011, 108: 12179-12184. 10.1073/pnas.1103219108.
Oud B, van Maris AJ, Daran JM, Pronk JT: Genome-wide analytical approaches for reverse metabolic engineering of industrially relevant phenotypes in yeast. FEMS Yeast Res. 2011, 12: 183-196.
Wei W, McCusker JH, Hyman RW, Jones T, Ning Y, Cao Z, Gu Z, Bruno D, Miranda M, Nguyen M, et al: Genome sequencing and comparative analysis of Saccharomyces cerevisiae strain YJM789. Proc Natl Acad Sci USA. 2007, 104: 12825-12830. 10.1073/pnas.0701291104.
Doniger SW, Kim HS, Swain D, Corcuera D, Williams M, Yang SP, Fay JC: A catalog of neutral and deleterious polymorphism in yeast. PLoS Genet. 2008, 4: e1000183-10.1371/journal.pgen.1000183.
Borneman AR, Forgan AH, Pretorius IS, Chambers PJ: Comparative genome analysis of a Saccharomyces cerevisiae wine strain. FEMS Yeast Res. 2008, 8: 1185-1195. 10.1111/j.1567-1364.2008.00434.x.
Argueso JL, Carazzolle MF, Mieczkowski PA, Duarte FM, Netto OV, Missawa SK, Galzerani F, Costa GG, Vidal RO, Noronha MF, et al: Genome structure of a Saccharomyces cerevisiae strain widely used in bioethanol production. Genome Res. 2009, 19: 2258-2270. 10.1101/gr.091777.109.
Borneman AR, Desany BA, Riches D, Affourtit JP, Forgan AH, Pretorius IS, Egholm M, Chambers PJ: Whole-genome comparison reveals novel genetic elements that characterize the genome of industrial strains of Saccharomyces cerevisiae. PLoS Genet. 2011, 7: e1001287-10.1371/journal.pgen.1001287.
Dowell RD, Ryan O, Jansen A, Cheung D, Agarwala S, Danford T, Bernstein DA, Rolfe PA, Heisler LE, Chin B, et al: Genotype to phenotype: a complex problem. Science. 2010, 328: 469-10.1126/science.1189015.
Akao T, Yashiro I, Hosoyama A, Kitagaki H, Horikawa H, Watanabe D, Akada R, Ando Y, Harashima S, Inoue T, et al: Whole-genome sequencing of sake yeast Saccharomyces cerevisiae Kyokai no. 7. DNA Res. 2011, 18: 423-434. 10.1093/dnares/dsr029.
Saccharomyces cerevisiae RM11-1a Database. [http://www.broadinstitute.org/annotation/genome/saccharomyces_cerevisiae/]
Saccharomyces cerevisiae Strain Project. [http://genome.wustl.edu/genomes/saccharomyces_cerevisiae_strain_project_genomes]
Entian KD, Kötter P: Yeast genetic strain and plasmid collections. Method Microbiol. 2007, 36: 629-666.
van Dijken JP, Bauer J, Brambilla L, Duboc P, Francois JM, Gancedo C, Giuseppin ML, Heijnen JJ, Hoare M, Lange HC, et al: An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strains. Enzyme Microb Tech. 2000, 26: 706-714. 10.1016/S0141-0229(00)00162-9.
Canelas AB, Harrison N, Fazio A, Zhang J, Pitkanen JP, van den Brink J, Bakker BM, Bogner L, Bouwman J, Castrillo JI, et al: Integrated multilaboratory systems biology reveals differences in protein metabolism between two reference yeast strains. Nat Commun. 2010, 1: 145-10.1038/ncomms1150.
Kresnowati MT, van Winden WA, Almering MJ, ten Pierick A, Ras C, Knijnenburg TA, Daran-Lapujade P, Pronk JT, Heijnen JJ, Daran JM: When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Mol Syst Biol. 2006, 2: 49-
Kuyper M, Hartog MM, Toirkens MJ, Almering MJ, Winkler AA, van Dijken JP, Pronk JT: Metabolic engineering of a xylose-isomerase-expressing Saccharomyces cerevisiae strain for rapid anaerobic xylose fermentation. FEMS Yeast Res. 2005, 5: 399-409. 10.1016/j.femsyr.2004.09.010.
Becker J, Boles E: A modified Saccharomyces cerevisiae strain that consumes L-Arabinose and produces ethanol. Appl Environ Microbiol. 2003, 69: 4144-4150. 10.1128/AEM.69.7.4144-4150.2003.
Wisselink HW, Toirkens MJ, Wu Q, Pronk JT, van Maris AJ: Novel evolutionary engineering approach for accelerated utilization of glucose, xylose, and arabinose mixtures by engineered Saccharomyces cerevisiae strains. Appl Environ Microbiol. 2009, 75: 907-914. 10.1128/AEM.02268-08.
Karhumaa K, Wiedemann B, Hahn-Hagerdal B, Boles E, Gorwa-Grauslund MF: Co-utilization of L-arabinose and D-xylose by laboratory and industrial Saccharomyces cerevisiae strains. Microb Cell Fact. 2006, 5: 18-10.1186/1475-2859-5-18.
Pagliardini J, Hubmann G, Bideaux C, Alfenore S, Nevoigt E, Guillouet SE: Quantitative evaluation of yeast's requirement for glycerol formation in very high ethanol performance fed-batch process. Microb Cell Fact. 2010, 9: 36-10.1186/1475-2859-9-36.
Guadalupe Medina V, Almering MJ, van Maris AJ, Pronk JT: Elimination of glycerol production in anaerobic cultures of a Saccharomyces cerevisiae strain engineered to use acetic acid as an electron acceptor. Appl Environ Microbiol. 2010, 76: 190-195. 10.1128/AEM.01772-09.
Schehl B, Muller C, Senn T, Heinisch JJ: A laboratory yeast strain suitable for spirit production. Yeast. 2004, 21: 1375-1389. 10.1002/yea.1189.
van Maris AJ, Geertman JM, Vermeulen A, Groothuizen MK, Winkler AA, Piper MD, van Dijken JP, Pronk JT: Directed evolution of pyruvate decarboxylase-negative Saccharomyces cerevisiae, yielding a C2-independent, glucose-tolerant, and pyruvate-hyperproducing yeast. Appl Environ Microbiol. 2004, 70: 159-166. 10.1128/AEM.70.1.159-166.2004.
van Maris AJ, Winkler AA, Porro D, van Dijken JP, Pronk JT: Homofermentative lactate production cannot sustain anaerobic growth of engineered Saccharomyces cerevisiae: possible consequence of energy-dependent lactate export. Appl Environ Microbiol. 2004, 70: 2898-2905. 10.1128/AEM.70.5.2898-2905.2004.
Zelle RM, de Hulster E, Kloezen W, Pronk JT, van Maris AJ: Key process conditions for production of C(4) dicarboxylic acids in bioreactor batch cultures of an engineered Saccharomyces cerevisiae strain. Appl Environ Microbiol. 2010, 76: 744-750. 10.1128/AEM.02396-09.
Otero JM, Vongsangnak W, Asadollahi MA, Olivares-Hernandes R, Maury J, Farinelli L, Barlocher L, Osteras M, Schalk M, Clark A, Nielsen J: Whole genome sequencing of Saccharomyces cerevisiae: from genotype to phenotype for improved metabolic engineering applications. BMC Genomics. 2010, 11: 723-10.1186/1471-2164-11-723.
Madsen KM, Udatha GD, Semba S, Otero JM, Koetter P, Nielsen J, Ebizuka Y, Kushiro T, Panagiotou G: Linking genotype and phenotype of Saccharomyces cerevisiae strains reveals metabolic engineering targets and leads to triterpene hyper-producers. PLoS One. 2011, 6: e14763-10.1371/journal.pone.0014763.
Wattanachaisaereekul S, Lantz AE, Nielsen ML, Nielsen J: Production of the polyketide 6-MSA in yeast engineered for increased malonyl-CoA supply. Metab Eng. 2008, 10: 246-254. 10.1016/j.ymben.2008.04.005.
Daran-Lapujade P, Daran JM, Kotter P, Petit T, Piper MD, Pronk JT: Comparative genotyping of the Saccharomyces cerevisiae laboratory strains S288C and CEN.PK113-7D using oligonucleotide microarrays. FEMS Yeast Res. 2003, 4: 259-269. 10.1016/S1567-1356(03)00156-9.
Schacherer J, Ruderfer DM, Gresham D, Dolinski K, Botstein D, Kruglyak L: Genome-wide analysis of nucleotide-level variation in commonly used Saccharomyces cerevisiae strains. PLoS One. 2007, 2: e322-10.1371/journal.pone.0000322.
Allen JE, Salzberg SL: JIGSAW: integration of multiple sources of evidence for gene prediction. Bioinformatics. 2005, 21: 3596-3603. 10.1093/bioinformatics/bti609.
Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES: Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003, 423: 241-254. 10.1038/nature01644.
Nijkamp J, Winterbach W, van den Broek M, Daran JM, Reinders M, de Ridder D: Integrating genome assemblies with MAIA. Bioinformatics. 2010, 26: i433-i439. 10.1093/bioinformatics/btq366.
Wolfe KH, Shields DC: Molecular evidence for an ancient duplication of the entire yeast genome. Nature. 1997, 387: 708-713. 10.1038/42711.
Kim JM, Vanguri S, Boeke JD, Gabriel A, Voytas DF: Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 1998, 8: 464-478.
Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25: 1754-1760. 10.1093/bioinformatics/btp324.
Xie C, Tammi MT: CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009, 10: 80-10.1186/1471-2105-10-80.
Kopetzki E, Zellner E, Schumacher G, Zimmerman FK: Nucleotide sequence of the Saccharomyces cerevisiae positive regulatory mutant gene MAL2-8cp. Nucleic Acids Res. 1989, 17: 5390-10.1093/nar/17.13.5390.
Daran-Lapujade P, Jansen ML, Daran JM, van Gulik W, de Winde JH, Pronk JT: Role of transcriptional regulation in controlling fluxes in central carbon metabolism of Saccharomyces cerevisiae. A chemostat culture study. J Biol Chem. 2004, 279: 9125-9138. 10.1074/jbc.M309578200.
Vanhalewyn M, Dumortier F, Debast G, Colombo S, Ma P, Winderickx J, Van Dijck P, Thevelein JM: A mutation in Saccharomyces cerevisiae adenylate cyclase, Cyr1K1876M, specifically affects glucose- and acidification-induced cAMP signalling and not the basal cAMP level. Mol Microbiol. 1999, 33: 363-376. 10.1046/j.1365-2958.1999.01479.x.
Tanaka K, Nakafuku M, Satoh T, Marshall MS, Gibbs JB, Matsumoto K, Kaziro Y, Toh-e A: S. cerevisiae genes IRA1 and IRA2 encode proteins that may be functionally equivalent to mammalian ras GTPase activating protein. Cell. 1990, 60: 803-807. 10.1016/0092-8674(90)90094-U.
Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA: Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell. 2001, 12: 323-337.
Gemayel R, Vinces MD, Legendre M, Verstrepen KJ: Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet. 2010, 44: 445-477. 10.1146/annurev-genet-072610-155046.
da Huang W, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009, 4: 44-57.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25: 25-29.
Christie KR, Hong EL, Cherry JM: Functional annotations for the Saccharomyces cerevisiae genome: the knowns and the known unknowns. Trends Microbiol. 2009, 17: 286-294. 10.1016/j.tim.2009.04.005.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al: Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23: 2947-2948. 10.1093/bioinformatics/btm404.
Verstrepen KJ, Jansen A, Lewitter F, Fink GR: Intragenic tandem repeats generate functional variability. Nat Genet. 2005, 37: 986-990. 10.1038/ng1618.
Gatchel JR, Zoghbi HY: Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005, 6: 743-755.
Daran-Lapujade P, Daran JM, van Maris AJ, de Winde JH, Pronk JT: Chemostat-based micro-array analysis in baker's yeast. Adv Microb Physiol. 2009, 54: 257-311.
Tai SL, Boer VM, Daran-Lapujade P, Walsh MC, de Winde JH, Daran JM, Pronk JT: Two-dimensional transcriptome analysis in chemostat cultures. Combinatorial effects of oxygen availability and macronutrient limitation in Saccharomyces cerevisiae. J Biol Chem. 2005, 280: 437-447.
Hall C, Dietrich FS: The reacquisition of biotin prototrophy in Saccharomyces cerevisiae involved horizontal gene transfer, gene duplication and gene clustering. Genetics. 2007, 177: 2293-2307. 10.1534/genetics.107.074963.
Verduyn C, Postma E, Scheffers WA, Van Dijken JP: Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast. 1992, 8: 501-517. 10.1002/yea.320080703.
Gasser B, Dragosits M, Mattanovich D: Engineering of biotin-prototrophy in Pichia pastoris for robust production processes. Metab Eng. 2010, 12: 573-580. 10.1016/j.ymben.2010.07.002.
Schacherer J, Shapiro JA, Ruderfer DM, Kruglyak L: Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature. 2009, 458: 342-345. 10.1038/nature07670.
Huson DH, Bryant D: Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006, 23: 254-267.
de Kok S, Nijkamp JF, Oud B, Roque FC, de Ridder D, Daran JM, Pronk JT, van Maris AJ: Laboratory evolution of new lactate transporter genes in a jen1Delta mutant of Saccharomyces cerevisiae and their identification as ADY2 alleles by whole-genome resequencing and transcriptome analysis. FEMS Yeast Res. 2012
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18: 821-829. 10.1101/gr.074492.107.
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL: Versatile and open software for comparing large genomes. Genome Biol. 2004, 5: R12-10.1186/gb-2004-5-2-r12.
Fiers MW, van der Burgt A, Datema E, de Groot JC, van Ham RC: High-throughput bioinformatics with the Cyrille2 pipeline system. BMC Bioinformatics. 2008, 9: 96-10.1186/1471-2105-9-96.
Stanke M, Tzvetkova A, Morgenstern B: AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 2006, 7 (Suppl 1): S11 11-18.
Korf I: Gene finding in novel genomes. BMC Bioinformatics. 2004, 5: 59-10.1186/1471-2105-5-59.
Besemer J, Lomsadze A, Borodovsky M: GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29: 2607-2618.
Kurtz S, Gremme G, Brendel V, Sparks ME: Engineering a software tool for gene structure prediction in higher organisms. Inf Softw Technol. 2005, 47: 965-978. 10.1016/j.infsof.2005.09.005.
Zhang Z, Schwartz S, Wagner L, Miller W: A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000, 7: 203-214. 10.1089/10665270050081478.
Boer VM, Daran JM, Almering MJ, de Winde JH, Pronk JT: Contribution of the Saccharomyces cerevisiae transcriptional regulator Leu3p to physiology and gene expression in nitrogen- and carbon-limited chemostat cultures. FEMS Yeast Res. 2005, 5: 885-897. 10.1016/j.femsyr.2005.04.003.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP: Integrative genomics viewer. Nat Biotechnol. 2011, 29: 24-26. 10.1038/nbt.1754.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009, 25: 2078-2079. 10.1093/bioinformatics/btp352.
Hosack DA, Dennis G, Sherman BT, Lane HC, Lempicki RA: Identifying biological themes within lists of genes with EASE. Genome Biol. 2003, 4: R70-10.1186/gb-2003-4-10-r70.
Sambrook J, Russell DW: The condensed protocols from Molecular cloning: a laboratory manual. 2006, Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press
Auch AF, Klenk HP, Goker M: Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci. 2010, 2: 142-148. 10.4056/sigs.541628.
Henz SR, Huson DH, Auch AF, Nieselt-Struwe K, Schuster SC: Whole-genome prokaryotic phylogeny. Bioinformatics. 2005, 21: 2329-2335. 10.1093/bioinformatics/bth324.
Mortimer RK, Johnston JR: Genealogy of principal strains of the yeast genetic stock center. Genetics. 1986, 113: 35-43.
Acknowledgements
We acknowledge the financial support from Netherlands Genomics Initiative (NGI). The PhD project of JFN is funded by the Kluyver Centre for Genomics of Industrial Fermentation. We thank our colleagues of the Industrial Microbiology Section for their support.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JFN, DdR, JTP and JMD designed the experimental work. JFN, MvdB, MR and DdR carried-out the genome assembly. ED, RCvH, JFN, MvdB and DdR performed the genome annotation. SdK, LB, MAL, PDL performed molecular biology work. WV, JN, PK, WHMH, PKl, CJP, DP, PKö, JTP, JMD shared raw sequencing data. JFN, DdR, MR, JTP and JMD prepared the manuscript. All authors read and approved the final manuscript.
Dick de Ridder and Jean-Marc Daran contributed equally to this work.
Electronic supplementary material
12934_2012_667_MOESM2_ESM.DOC
Additional file 2: Table S1. Repetitive transposon sequences were hard to assemble from whole genome shotgun data. Evidence of transposons was obtained in two ways. First, depth-of-coverage of CEN.PK113-7D and S288C reads on Ty retrotransposons sequences in the S288C genome was analysed. The number of retrotransposons was estimated from these ratios. Second, evidence for transposons in the assembly was obtained by counting the presence of contig breaks (CB) on transposon loci in S288C and the presence of assembled (AS) transposons (Figure S1). An assembled transposon locus with a gapped alignment (GA) around the transposon sequence in S288C indicates the transposon is absent from the CEN.PK genome. (DOC 30 KB)
12934_2012_667_MOESM3_ESM.PDF
Additional file 3: Figure S1. Analysis of transposon composition by alignment of the CEN.PK113-7D and S288c genomes. When an S288c transposon is not present in CEN.PK113-7D it results in a gapped alignment (GA) of about 6 Kbp. Transposons that are present can cause contig breaks (CB) in the assembly. Only YCLWTy5-1 was fully assembled (AS). (PDF 12 KB)
12934_2012_667_MOESM7_ESM.XLSX
Additional file 7: Table S4. Mutations in the galactose uptake and ergosterol biosynthesis pathways compared to the SNVs found previously in CEN.PK113-7D [35]. (XLSX 14 KB)
12934_2012_667_MOESM12_ESM.DOC
Additional file 12: Table S7. S. cerevisiae with an assembled genome deposited in GenBank. The classification assigned in the 'group' column was used to generate Figure 8. (DOC 47 KB)
12934_2012_667_MOESM13_ESM.XLSX
Additional file 13: Table S6. List of deleted genes, which is defined as not having a homologous hit in the CEN.PK113-7D genome for at least 95% and having a CEN.PK113-7D/S288c log2 ratio of less then -0.6. The PMR2 locus has a blue background color. (XLSX 16 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Nijkamp, J.F., van den Broek, M., Datema, E. et al. De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb Cell Fact 11, 36 (2012). https://doi.org/10.1186/1475-2859-11-36
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2859-11-36