
Abstract
Background
Breast cancer is one of the most dreading types of cancer among women. Herbal medicine has becoming a potential source of treatment for breast cancer. Herbal plant Dillenia suffruticosa (Griff) Martelli under the family Dilleniaceae has been traditionally used to treat cancerous growth. In this study, the anticancer effect of ethyl acetate extract of D. suffruticosa (EADs) was examined on human breast adenocarcinoma cell line MCF-7 and the molecular pathway involved was elucidated.
Methods
EADs was obtained from the root of D. suffruticosa by using sequential solvent extraction. Cytotoxicity was determined by using MTT assay, mode of cell death by cell cycle analysis and apoptosis induction by Annexin-FITC/PI assay. Morphology changes in cells were observed under inverted light microscope. Involvement of selected genes in the oxidative stress-mediated signaling pathway was explored using multiplex gene expression analysis.
Results
The treatment of EADs caused cytotoxicity to MCF-7 cells in a dose- and time-dependent manner at 24, 48 and 72 hours with IC50 of 76 ± 2.3, 58 ± 0.7 and 39 ± 3.6 μg/mL, respectively. The IC50 of tamoxifen-treated MCF-7 cells was 8 ± 0.5 μg/mL. Induction of apoptosis by EADs was dose- and time- dependent. EADs induced non-phase specific cell cycle arrest at different concentration and time point. The multiplex mRNA expression study indicated that EADs-induced apoptosis was accompanied by upregulation of the expression of SOD1, SOD2, NF-κB, p53, p38 MAPK, and catalase, but downregulation of Akt1.
Conclusion
It is suggested that EADs induced apoptosis in MCF-7 cells by modulating numerous genes which are involved in oxidative stress pathway. Therefore, EADs has the potential to act as an effective intervention against breast cancer cells.
Similar content being viewed by others
Background
Breast cancer is the most typical cancer diagnosed among women thus far[1]. The mortality rate of breast cancer declines over the year and the latest five years data affirm that breast cancer incidence rate is stable[2]. Death rate in female patients reduces by 15.3% since year 1991 due to amelioration in early detection and treatment[3]. Nevertheless, breast cancer cases are still accounting for 23% of total new cancer cases globally[4]. With advance in molecular knowledge, more novel anti-cancer agents with great selectivity and specificity ought to be developed to overcome limitation of current treatments.
Deregulated cell proliferation and inhibition of cell death evoke uncontrolled development of cancer[5]. Cancer cells exhibit resistance to apoptosis or cell death in order to survive and metastasize[6]. The common feature of cancer progression, inclusive of breast cancer, is genetic alteration in apoptotic pathways such as alteration of pro- and anti-apoptotic genes that provides an insight to a target of treatment[7]. The treatment strategy aims to destroy cancer cells by activating their apoptotic signaling pathways, ideally to induce selective apoptosis cell death on cancer cells, and exert no harmful effects on normal cells[8, 9].
Apoptosis form of cell death is chosen to eliminate cancer cells instead of other alternative mechanism because it is a series of regulated cell events that perform cellular suicide without triggering inflammatory response, and neither harmful to neighboring cells. It differs from necrosis that causes membrane rupture thus elicits inflammatory response[10]. Most importantly, apoptotic form of cell death is occasionally altered in cancer cells. The understanding of apoptosis unfolds a gate to tumor-specific apoptosis therapy[11].
Reactive oxygen species (ROS) such as hydroxyl radical (OH-), superoxide anions (O2 -), hydrogen peroxide (H2O2) and peroxyl radicals (ROO-) are common products of aerobic metabolism that can be useful or harmful to biological system[12]. Low concentration of ROS may facilitate signal transduction, enzyme activation and other cellular functions, but high concentration of ROS generates damage to DNA, protein and lipid which can lead to cells transformation such as cancer[13, 14]. To offset the ROS detrimental effect, cells are complement with antioxidant defense system transformation that includes superoxide dismutases, catalase, glutathione and others as protective mechanism[15]. In fact, ROS and antioxidants exist in balance under normal circumstances. When the equilibrium between ROS and anti-oxidants is disrupted, collective generation of ROS is described as oxidative stress[16]. Evidence showed that ROS not only function as regulator of subcellular events but are also able to induce cell death through apoptotic pathway[17]. Recently, many anticancer agents such as 5-fluorouracil, tamoxifen and paclitaxel exploit this channel to eliminate cancer cells by continually exert cellular ROS to a threshold that can kill cancerous cells effectively[14, 18–20].
Plant has long history as a source of anticancer agents, and gives prominent impact on modern drug development process[21]. Over the last 20 years, exceeding 25% of drugs are plant derived while another 25% are originated from chemically modified natural products[22]. Plant-based anticancer drugs such as etoposide (topoisomerase II inhibitor) from epipodophyllotoxins, topotecan and irinotecan (topoisomerase I inhibitor) from camptothecins, vincristine and vinblastine (tubulin-binding agent) from vinca alkaloids, induce apoptosis in chemotherapeutic therapy against various types of cancer[23, 24]. The main target of action for those anti-cancer drugs may be distinct, but eventually they lead to identical cell death pathway, which is apoptosis[25, 26]. Therefore, in order to discover more plant-based anticancer agent, various plant extracts were investigated for their apoptosis-inducing ability. Interestingly, although plants are frequently reported to possess antioxidant activity, some of them are found to exert distinguish apoptosis inducing ability through the induction of oxidative stress[27–30].
Dillenia suffruticosa (Griff) Martelli (D. suffruticosa), which belongs to family Dilleniaceae, is a plant native in Peninsular Malaysia, Kalimantan, Sumatra and Singapore[31, 32]. The evergreen shrub can be found in secondary forest and swampy ground. The fruit of the plant has the ability to treat cancerous growth[33]. Other traditional use of the plant is to relieve rheumatism[34]. Methanolic extract of D. suffruticosa showed a broad spectrum of antimicrobial activity against Bacillus cereus, Bacillius subtilis, Candida albicans, and Pseudomonas aeruginosa[35]. Water extract of D. suffruticosa also exhibited inhibitory action against replication of dengue virus type 2[36]. Armania et al.[37] reported that extract of D. suffruticosa showed high antioxidant and cytotoxic activities towards various cell lines including Hela, MCF-7, MDA-MB-231, A549 and HT-29 cell lines. In this study, root extract was selected for elaborated study. As the previous study demonstrated that root extract of the plant exhibited the most potent cytotoxic activity, in comparison to fruit, leaf, and flower parts of the plant.
The aim of this study was to investigate the anticancer effect of ethyl acetate of D. suffruticosa (EADs) in breast cancer cells, MCF-7, and to explore the apoptotic signaling pathway underlying it.
Methods
Chemicals and reagents
Hexane, dichloromethane, ethyl acetate and dimethyl sulfoxide (DMSO) were purchased from FS Chemicals (Francfort, Germany) (analytical grade). RPMI 1640 was purchased from Nacalai Tesque (Kyoto, Japan). Fetal bovine serum, trypsin, streptomycin and penicillin were obtained from PAA Laboratories GmBH (Pasching, Austria). 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT), propidium iodide and RNAse A were purchased from Sigma (St. Loius, USA). Tissue culture flasks, 6-well plates and 96-well plates were obtained from TPP (Trasadingan, Switzerland). Annexin-V FITC Kit was obtained from eBioscience Inc. (San Diego, USA). Real Genomics Total RNA extraction kit (RBC Biosciences, Taiwan) and GenomeLab GeXP Start Kit (Beckman Coulter, USA) were also procured.
Cell culture
The human adenocarcinoma breast cancer cell line, MCF-7, and mouse fibroblast cell line, 3T3 were obtained from the American Type and Culture Collection (Rockville, USA). Cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin, and maintained in humidified incubator at 37°C in atmosphere of 5% CO2.
Preparation of EADs
The root powder of D. suffruticosa was supplied by Primer Herber Sdn. Bhd. (Malaysia). The plant with voucher specimen number SK1937/11 was deposited in the herbarium of Institute of Bioscience, Universiti Putra Malaysia. Briefly, 100 g of the powder was soaked in 300 mL of hexane at a ratio of 1:3 (w/v) with occasional shaking using a rotary shaker for three times at 3:1:1 day interval. The mixture solvent was collected and filtered using Whatman No. 1 filter paper. The residue was dried in an oven at 40°C and subsequently used for successive extraction of dichloromethane followed by ethyl acetate using the same methods. Lastly, filtered ethyl acetate extract was evaporated using a vacuum rotary evaporator (Buchi, Switzerland)[37]. The yield was weighed and kept at -20°C until required. For subsequent experiment, the stock of EADs in DMSO (30 mg/mL) was used. The final concentration of DMSO was 0.33% in all the extracts prepared. DMSO at 0.33% is non-toxic to the cell line mentioned above[38].
Cytotoxicity of EADs
Cytotoxicity of EADs on MCF-7 cells was determined by the MTT (3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide) assay[39]. Briefly, 1x105 of cells were seeded in each well of a 96-well plate. After 24 hours incubation, cells were treated with EADs (3.13 to 100 μg/mL). Untreated control cells were also included. After incubation with EADs for 24, 48 and 72 hours, 20 μL of 5 mg/mL of MTT was added into each well and incubated for 3 hours. Active mitochondria in live cells reduced MTT to crystalline purple blue formazan. The number of living cells was proportionate to the amount of crystalline purple blue formazan produced. After incubation, media in each well was discarded and 100 μL of DMSO was added to solubilize the purple blue formazan. The absorbance was measured with an ELISA plate reader (Biotek, USA) at wavelength of 570 nm, and 630 nm as background. A graph of percentage of cell viability versus concentration of EADs was plotted and the IC50 (concentration that inhibits 50% of cell growth compared to control) was determined.
Cell morphology study of apoptosis by inverted light microscope
Briefly, 3 × 105 of MCF-7 cells were seeded in each well of a 6-well plate. After 24 hours of incubation, cells were treated with EADs at concentration of 25 and 50 μg/mL. Control untreated cells were also included. Morphological changes of cells untreated and treated with EADs were examined under an inverted light microscope (Olympus, Tokyo, Japan) after 24, 48 and 72 hours. The cells were captured at the same spot at different time interval.
Cell cycle analysis
Briefly, 3 × 105 of MCF-7 cells were seeded in each well of a 6-well plate and treated with EADs at 25 and 50 μg/mL. Control untreated cells were also included. After incubation for 24, 48 and 72 hours, cells were trypsinized and washed with PBS. After centrifugation, cell suspension was resuspended repeatedly into single cells prior fixation with 70% ethanol. Fixed cells were kept at -20°C for at least 2 hours. Later, fixed cells were washed with PBS twice and the supernatant was discarded. Cell pellets were resuspended with 425 μL of PBS in a round bottom tube. Next, 50 μL of RNAse and 25 μL of propidium iodide were added into the cell suspension and incubated for 15 minutes on ice in the dark. FACS Calibur (BD Biosciences, USA) and Cell Quest Pro software (BD Biosciences, USA) was used to determine the cell cycle distribution. A total of 10,000 of cells were acquired each time using FACS Calibur flowcytometer. Flowcytometric data were analyzed using Modfit software and displayed in histogram cell count (у-axis) against DNA content (x-axis).
Annexin V/PI apoptosis assay
Briefly, 3 × 105 of MCF-7 cells were seeded in each well of a 6-well plate and treated with EADs at 25 and 50 μg/mL. Control untreated cells were also included. After incubation for 24, 48 and 72 hours, cells were trypsinized, washed twice with PBS and the supernatant was discarded. The cell pellets were mixed with 185 μL of 1X binding buffer. Next, 5 μL of Annexin-V FITC and 10 μL of propidium iodide (PI) were added into the suspension and incubated at room temperature for 10 minutes in the dark. Subsequently, 300 μL of 1X binding buffer was added prior to measurement using FACS calibur flowcytometer and Cell Quest Pro software (BD Biosciences, USA). Samples were kept on ice. This assay was carried out following manufacturer’s kit from BenderMedsystem (Vienna, Austria). The fluorescence colour was detected through 530 and 585 nm band pass filter. A total of 10,000 cells were acquired. Flowcytometric data were analyzed using FlowJo 7.6 software and displayed in dot plot of Annexin V/FITC (y-axis) against PI (x-axis).
Multiplex mRNA expression analysis using GeXP analysis system
RNA isolation
Briefly, 3 × 105 of MCF-7 cells were seeded in each well of a 6-well plate and treated with EADs at 25 and 50 μg/mL. Untreated control cells were also included. Untreated and EADs treated cells were trypsinized and washed twice with PBS. RNA extraction was performed using the Real Genomics Total RNA extraction kit (RBC Biosciences, Taiwan).
Reverse transcription and polymerase chain reaction
Samples were prepared according to the GenomeLab GeXP Start Kit (Beckman Coulter, USA). Briefly, 2 μL of customized reverse primers of the desired genes were mixed with 11 μL of RNA free water, 4 μL of reverse transcription buffer, 1 μL of reverse transcriptase and 1 μL of 50 ng/μL of sample. The reverse transcription reaction was run for 1 minute at 48°C, 60 minutes at 42°C and 5 minutes at 95°C. Subsequently, the cDNA produced was amplified by PCR reaction. Next, 4 μL of 5X PCR buffer, 4 μL of magnesium chloride, 2 μL of customized forward primers mixture (Table 1), 0.7 μL of Taq polymerase and 9.3 μL of cDNA were mixed and run at specified time and temperature.
GeXP multiplex analysis
The GenomeLab GeXP genetic analysis system (Beckman Coulter, USA) was used to examine the expression level of genes involved in apoptosis pathway. The forward and reverse primers were supplied by First Base Ltd. (Selangor, Malaysia). The genes and their primer sequences were listed in Table 1. Briefly, 1 μL of PCR product was mixed with 38.5 μL of sample loading solution and 0.5 μL of DNA size standard, and added into sample plate to start the sample run using GeXP Genetic Analysis System (Beckman Coulter, USA). The amplified fragments were separated according to their respective size by capillary gel electrophoresis in the GeXP system. Results were analyzed using the Fragment Analysis module of the GeXP system software and eXpress Profiller software. The normalization was performed using beta actin.
Statistical analysis
Data were represented as mean ± SD of at least three independent experiments. Data were analyzed using IBM SPSS version 20. Statistical test one way ANOVA and Tukey post hoc test were conducted for pairwise comparisons. P value less than 0.05 was considered statistically significant.
Results
Cytotoxic properties of EADs on MCF-7 cells
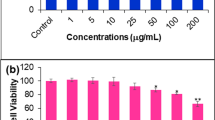
As shown in Figure 1, significant cytotoxic effect of EADs in MCF-7 cells was noted at 25, 50 and 100 μg/mL compared to the control at various time points (P < 0.05). The cytotoxic effect was time-and dose-dependent. Treatment with EADs at 25 and 50 μg/mL reduced the cell viability from 86.3% to 29.6% and 85.5% to 18.3%, respectively, from 48 to 72 hours (P < 0.05). IC50 values of EADs were 76 ± 2.3, 58 ± 0.7 and 39 ± 3.6 μg/mL, respectively, at 24, 48 and 72 hours. Based on the cytotoxic effect, 25 and 50 μg/mL of EADs, and incubation time of 24 and 48 hours were selected for further analysis.

Effect of EADs on the viability of MCF-7 cells as determined by MTT assay. Cytotoxic effect of EADs was investigated on (a) breast adenocarcinoma cell line, MCF-7 and (b) non-neoplastic mouse fibroblast cell line, 3 T3. The result indicated that EADs was relatively non-selective towards cancerous cell line. Antiproliferative effect of EADs was compared to reference drug, tamoxifen on MCF-7 cell line (Figure 1c). Data are represented as mean percentage of viable cells in bars ± SD of at least three replicates in three independent tests. An asterisk * indicates statistically significant different from untreated control (P < 0.05).
Morphological changes of MCF-7 cells following treatment with EADs
The cell number reduced at 50 μg/mL of EADs at 72 hours. Cell detachment, cell rounding, cytoplasmic condensation and cell shrinkage were observed at 48 and 72 hours in MCF-7 cells treated with 50 μg/mL of EADs (Figure 2). At 25 μg/mL of EADs, cell shrinkage and cytoplasmic condensation were noted but the cell number increased over time.

Morphological changes of MCF-7 cells treated with EADs observed under an inverted light microscope. The cells exhibited morphological changes and characteristics of apoptosis such as cell shrinkage and rounding (black arrow), and detachment from the substatum (white arrow). Decrease in cell population was noted with the increase in the concentration of the extract. (a) 100X magnification (b) 400X magnification
EADs induced cell cycle arrest in MCF-7 cells
The cell cycle phase distribution of MCF-7 cells treated with EADs at 24 and 48 hours is depicted in Figure 3. The cell cycle arrest by EADs was time- and concentration-dependent. At 24 and 48 hours, an increase in cell population in G1 at 25 μg/mL of EADs was noted (P < 0.05). On the other hand, 50 μg/mL of EADs at 24 hours elevated the number of cells in S and G2/M compared to control, accompanied by a decline in G1 phase cell population (P < 0.05). Meanwhile, at 48 hours, G2/M phase cell population was ascended compared to the control following treatment with 50 μg/mL of EADs (P < 0.05). Increase in the population of cells at sub-G1 phase was observed following treatment with EADs (P < 0.05).

Cell cycle analysis of MCF-7 breast cancer cells treated with EADs at 24 and 48 hours. Effects of EADs on the cell cycle distribution in MCF-7 cells were analysed using flowcytometry analysis. Bar charts representing the percentage of cell populations in MCF-7 cells treated with EADS for (a) 24 hours and (b) 48 hours. DNA histogram (Figure 3c) displayed cell cycle phase distribution of control and EADs-treated cells at 24 and 48 hours. The data are presented as mean ± standard deviation of three replicates in three independent tests. An asterisk * indicates statistically significant different from untreated control (P < 0.05).
EADs induced apoptosis in MCF-7 cells
Induction of apoptosis by EADs was quantitatively determined by Annexin V-FITC and propidium iodide fluorescence staining. The percentage of early apoptotic cells increased in a dose and time dependent manner (Figure 4). At 24 hours, the early apoptotic cells increased from 16.7% at 25 μg/mL to 42.7% at 50 μg/mL of EADs compared to 8.3% in the control. The number of late apoptotic cells increased from 9.9% to 18.3% in 50 μg/ml of EADS (P < 0.05). At 48 hours, the number of early apoptotic cells increased to 36.1% at 25 μg/mL of EADs, and elevated to 45.5% at 50 μg/mL of EADs.

Induction of apoptosis in MCF-7 cells by EADs determined using Annexin V-PI flowcytometry technique. The data representing three independent tests which displayed similar results. The lower left quadrant represents intact viable cells (Annexin-FITC and PI negative). The lower right quadrant represents early apoptotic cells (Annexin-FITC positive and PI negative). The upper right region represents late apoptotic cells or secondary necrotic cells (Annexin-FITC and PI positive). The data are presented as dot plots of Annevin V/FITC against PI of at least three independent tests.
EADs altered the expression of oxidative stress pathway related genes
The MCF-7 cells treated with 25 and 50 μg/mL of EADs significantly upregulated the expression level of SOD1, SOD2, p53, p38 MAPK, catalase and NF-κB genes but downregulated Akt1 (P < 0.05) compared to the control (Figure 5).

Expression level of genes involved in oxidative stress-induced apoptotic pathway following treatment with EADs using GeXP analysis system. The data are presented as mean ± standard deviation of three replicates in at least three independent tests. An asterisk * indicates statistically significant different from untreated control (P < 0.05).
Discussion
Cancer cells evolve to avoid apoptosis-inducing signaling pathway in order to survive[40]. Thus, induction of apoptosis in cancer cells can be a promising treatment method in cancer therapy. Natural-derived products, regardless of crude extracts or isolated active compounds, had drawn growing attention as agent in cancer therapy, due to their ability to modulate apoptosis[41–43].
In this study, EADs has been shown to be cytotoxic and inhibit the proliferation of MCF-7 cells in a time- and dose-dependent manner. The cytotoxic property may be due to the presence of phytochemicals such as saponins, triterpenes, tannins and polyphenolic compounds in the extract[37]. Man et al.[44] reported that saponins exhibited anti-tumorigenic effects via multiple anticancer pathways because of the great diversity of their structures. For instance, a steroidal saponin known as Dioscin was described to inhibit tumor through induction of oxidative stress[45]. In addition, triterpenes from Antrodia camphorata were found to exhibit cytotoxic effect towards HT-29 human colon cancer cells[46]. Polyphenolic compounds exist in plants are associated with anticancer activity by interaction with key enzymes in cellular signaling pathways, cell cycle, apoptosis and metastasis[47, 48].
The treatment of EADs elicited non-phase specific cell cycle arrest in MCF-7 cells. For instance, at 25 μg/mL of EADs, cell cycle arrest at G1 at 48 hours was noted. On the other hand, at 50 μg/mL of EADs, the cell cycle arrest was at S and G2M at 24 hours, but G2M cell cycle arrest at 48 hours. The cell cycle phase non-specific nature of EADs denotes that it kills tumor cells in either resting or dividing state. The non-specific phase drugs are among the most effective drugs against slow-growing tumors[49]. Ozawa et al.[50] demonstrated that the action of a non-phase specific antitumor agent is basically dependent on the concentration and time. This finding is in accordance with the cytotoxicity of EADs.
MCF-7 cells treated with EADs exhibited certain apoptotic features such as cell rounding, cell shrinkage and cytoplasmic condensation, and also the presence of sub-G1 phase population. The induction of apoptosis was then further confirmed by flowcytometric Annexin V-FITC/PI. One of the hallmarks of apoptosis is the externalization of phospholipid phosphatidylserine (PS) by translocation from the inner to outer layer of plasma membrane for recognition of phagocytes during early stage of apoptosis[51]. Hence, phosphatidylserine can serve as specific target for the detection of early apoptotic cells. Annexin V-FITC which has high binding affinity for phosphatidylserine is appropriate conjugate for identification of early stage apoptosis[52, 53]. Simultaneously, propidium iodide is included for dye exclusion to differentiate between apoptotic and necrotic cells[54].
Interestingly, other prominent characteristics of apoptosis such as membrane blebbing, DNA fragmentation and formation of apoptotic bodies were absent. MCF-7 cells are previously reported lack in caspase-3, an important component in the cascade of apoptosis, due to deletion of a 47 base pair in the exon 3 of the caspase gene[55]. The caspase-3 deficient MCF-7 cells do not display some typical morphological characteristics of apoptosis such as chromatin condensation, DNA fragmentation and membrane blebbing[56]. During normal condition in apoptosis, caspase-3 is activated and responsible for morphological and biochemical changes related to the apoptosis execution[57]. It is speculated that other caspases such as caspase-6 or caspase-7 or caspase-independent pathway are involved in apoptosis induced by EADs[58].
Many anticancer agents induced apoptotic cell death by introducing oxidative stress to a threshold that compromises cell viability, disturbing the equilibrium between ROS and antioxidants within cancer cells[59]. From the GeXP analysis data, the expression of SOD1, SOD2, and catalase genes was upregulated. It is postulated that the antioxidant defense system in MCF-7 cells is triggered in response to increase cellular oxidative stress generated by EADs. Superoxide dismutases (SODs) act as the sole enzyme that dismutates superoxide radicals (O2 -) into hydrogen peroxide (H2O2) and oxygen (O2). SOD1 is mainly located in the cytosol and intermembrane space of mitochondria while SOD2 is present in the matrix of mitochondria[16]. The product of SODs action, H2O2, is metabolized by an antioxidant known as catalase into water and oxygen[60]. In this case, SOD1, SOD2 and catalase were upregulated in order to scavenge the elevating level of ROS induced by EADs. Nonetheless, in spite of the protective mechanism of the antioxidants, MCF-7 cells still underwent apoptosis. Hence, it is believed that the ROS level induced by EADs was high and has surpassed the antioxidant capacity, leading to apoptosis in MCF-7 cells[59, 61].
In the present study, Akt1 expression was downregulated in a concentration-dependent manner suggesting the involvement of Akt pathway in EADs-induced apoptosis. Previous studies have shown that apoptosis in MCF-7 cells was related to the inhibition of Akt signaling pathway after treatment with Wogonin or retinoic-acid[62, 63]. Generally, Akt is a serine-threonine kinase that facilitates the control of balance between survival and apoptosis. Oxidative stress has been associated with the regulation of Akt pathway[64, 65]. Studies have reported that in response to oxidative stress, Akt can be downregulated and it is important in apoptosis process[66]. Akt signalling pathway deregulation in cancer cells has been one of the targets in the search of potential cancer treatment[67].
In this study, upregulation of p53 level was noted, which is possibly related to oxidative stress. Other than antioxidant defense, cells counterbalance the effect of oxidative stress by activation of p53-dependent pathways. p53 is a nuclear transcription factor that can be activated in response to oxidative stress to promote apoptosis by regulation of multitude of downstream effectors. Once it is activated, cell cycle is arrested for DNA repair process to restore normal cell function[68]. We propose that EADs-induced oxidative stress will increase the p53 level and subsequently lead to the non-phase specific cell cycle arrest in MCF-7 cells. However, if cells are not able to overcome the oxidative stress damage and DNA damage cannot be repaired, p53 becomes a mediator to induce apoptosis[68, 69].
EADs treatment in MCF-7 cells was also found to upregulate NF-κB expression. It is believed that increased activity of NF-κB in EADs-induced apoptosis is again due to oxidative stress. The nuclear transcription factor NF-κB regulates genes involved in a number of biological processes such as inflammation, cell survival, cell differentiation and cell growth[70]. NF-κB enhances the pro-inflammatory and anti-apoptotic genes expression, and acts as a protective barrier for cells against oxidative stress. However, NF-κB has also been associated with apoptosis and brings to activation of certain apoptosis-related genes[58]. Furthermore, many studies supported the pro-apoptotic effect exerted by NF-κB in response to oxidative stress[71]. It has been concluded that pro-apoptotic or anti-apoptotic effect of NF-κB depends on stimuli received, signaling pathway interactions, transcriptional regulation and function of genes it modulates[72]. ROS activate NF-κB via the dissociation of IκB from NF-κB through phosphorylation, thereby enable NF-κB to enter nucleus and activate transcription by binding to DNA[73].
Another intracellular signaling molecule that is involved in regulation of oxidative stress is p38 MAPK which belongs to MAPK superfamily. In accordance, a marked increase in gene expression of p38 MAPK in MCF-7 cells treated with EADs was observed. The p38 MAPK strongly responds to stress-inducing signals such as oxidative stress and cause apoptosis as a result of cellular injuries[16]. The p38 MAPK plays a role in regulation of cellular biological functions like inflammation, proliferation, differentiation, survival.[74, 75]. This pathway has been known as tumor suppressor because it is often activated by cellular stress and control signals that inhibit proliferation or enhance apoptosis[76]. Investigation of ROS activated p38 MAPK has been carried out widely. In addition, p38 MAPK and Akt pathways were found to be interconnected in numerous cases. For example, a study reported that suppression of Akt signaling pathway excites p38 MAPK related apoptosis and vice versa[77]. Our result also indicated that oxidative stress stimulates downregulation of Akt and facilitates upregulation of p38 MAPK, hence give rise to apoptosis in EADs treated MCF-7 breast cancer cells.
Nonetheless, several studies documented that Akt pathway is involved in the activation of NF-κB pathway under the treatment of TNF-α and growth factor[78]. However, it has also been demonstrated that NF-κB pathway may function independently from Akt pathway. NF-κB binding and transcription activity can still be activated despite of the inhibition of the Akt pathway[79]. Hence, it is postulated that although some crosstalks exist between Akt pathway and NF-κB pathway in response to EADs-induced oxidative stress in MCF-7 cells, but it appears that these two pathways can act independently. As discussed above, in response to oxidative stress, p53 is able to exert pro-apoptotic effect. Furthermore, it has been shown that p38 MAPK can phosphorylate p53, and play part in regulation of p53 expression under stress situation by stabilizing the p53 protein.
Conclusions
In summary, EADs was found to exhibit cytotoxicity towards MCF-7 cell line possibly via introduction of oxidative stress that activates Akt, NF-κB, p53, and p38 MAPK signaling pathway. It shows the potential of EADs to be developed into an anticancer agent. Nevertheless, the shift of attention towards bioactive compound that are responsible for the anti-breast cancer activity of EADs, and understanding of its mechanism of action are utmost essential to discover the potential of the extract in breast cancer intervention.
Abbreviations
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide
- ANOVA:
-
Analysis of variance
- DNA:
-
Deoxyribonucleic acid
- DMSO:
-
Dimethyl sulfoxide
- ELISA:
-
Enzyme-linked immunosorbent assay
- EADs:
-
Ethyl acetate extract of D. suffruticosa
- p38 MAPK:
-
P38 mitogen-activated protein kinases
- NF-κB:
-
Nuclear factor-kappaB
- TNF-α:
-
Tumor necrosis factor alpha
- PBS:
-
Phosphate buffered saline
- Akt:
-
Protein 53 (p53).protein kinase B
- ROS:
-
Reactive oxygen species.
References
United States Cancer Statistics: 1999–2007 Incidence and Mortality Web-based Report.http://www.cdc.gov/uscs,
ACS: Cancer Facts and Figures 2012. 2012, Cancers with Increasing Incidence Trends,http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-031941.pdf,
Siegel R, Naishadham D, Jemal A: Cancer statistics. CA Cancer J Clin. 2012, 62 (1): 10--29. 10.3322/caac.20138.http://dx.doi.org/10.3322/caac.20138,
Forman D, Ward E, Ferlay M, Jemal A, Bray F: Global cancer statistics. CA Cancer J Clin. 2011, 61 (2): 69-90. 10.3322/caac.20107.http://dx.doi.org/10.3322/caac.20107,
Kaufmann SH, Gores GJ: Apoptosis in cancer: cause and cure. BioEssays. 2000, 22 (11): 1007-1017. 10.1002/1521-1878(200011)22:11<1007::AID-BIES7>3.0.CO;2-4.http://dx.doi.org/10.1002/1521-1878(200011)22:11<1007::AID-BIES7>3.0.CO;2-4,
McGill G, Fisher DE: Apoptosis in tumorigenesis and cancer therapy. Frontiers in Biosci: a J and virtual Lib. 1997, 2: d353-d379.http://europepmc.org/abstract/MED/9230063,
Sledge GW, Miller KD: Exploiting the hallmarks of cancer: the future conquest of breast cancer. Eur J Cancer. 2003, 39 (12): 1668-1675. 10.1016/S0959-8049(03)00273-9.http://dx.doi.org/10.1016/S0959-8049(03)00273-9,
Kasibhatla S, Tseng B: Why target apoptosis in cancer treatment?. Mol Cancer Ther. 2003, 2 (6): 573-580.http://mct.aacrjournals.org/content/2/6/573.abstract,
Wong RSY: Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011, 30: 87-10.1186/1756-9966-30-87.http://www.jeccr.com/content/30/1/87,
Kroemer G, Dallaporta B, Resche-Rigon M: The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998, 60: 619-642. 10.1146/annurev.physiol.60.1.619.http://dx.doi.org/10.1146/annurev.physiol.60.1.619,
Lowe SW, Lin AW: Apoptosis in cancer. Carcinogenesis. 2000, 21 (3): 485-495. 10.1093/carcin/21.3.485.http://dx.doi.org/10.1093/carcin/21.3.485,
Simic MG, Bergtold DS, Karam LR: Generation of oxy radicals in biosystems. Mutation Res/Fundamental and Mol Mech of Mutagenesis. 1989, 214 (1): 3-12. 10.1016/0027-5107(89)90192-9.http://dx.doi.org/10.1016/0027-5107(89)90192-9,
Kryston TB, Georgiev AB, Pissis P, Georgakilas AG: Role of oxidative stress and DNA damage in human carcinogenesis. Mutation Res/Fundamental and Mol Mech of Mutagenesis. 2011, 711 (1–2): 193-201.http://dx.doi.org/10.1016/j.mrfmmm.2010.12.016,
Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H, Lleonart ME: Oxidative stress and cancer: an overview. Ageing Res Rev. 2013, 12 (1): 376-390. 10.1016/j.arr.2012.10.004.http://dx.doi.org/10.1016/j.arr.2012.10.004,
Dreher D, Junod AF: Role of oxygen free radicals in cancer development. Eur J Cancer. 1996, 32 (1): 30-38. 10.1016/0959-8049(95)00531-5.http://dx.doi.org/10.1016/0959-8049(95)00531-5,
Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M: Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006, 160 (1): 1-40. 10.1016/j.cbi.2005.12.009.http://dx.doi.org/10.1016/j.cbi.2005.12.009,
Wen J, You K-R, Lee S-Y, Song C-H, Kim D-G: Oxidative stress-mediated apoptosis: the anticancer effect of the sesquiterpene lactone parthenolide. J Biol Chem. 2002, 277 (41): 38954-38964. 10.1074/jbc.M203842200.http://dx.doi.org/10.1074/jbc.M203842200,
Afzal S, Jensen S, Sørensen J, Henriksen T, Weimann A, Poulsen H: Oxidative damage to guanine nucleosides following combination chemotherapy with 5-fluorouracil and oxaliplatin. Cancer Chemother Pharmacol. 2012, 69 (2): 301-307. 10.1007/s00280-011-1700-2.http://dx.doi.org/10.1007/s00280-011-1700-2,
Alexandre J, Hu Y, Lu W, Pelicano H, Huang P: Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Res. 2007, 67 (8): 3512-3517. 10.1158/0008-5472.CAN-06-3914.http://dx.doi.org/10.1158/0008-5472.can-06-3914,
Nazarewicz RR, Zenebe WJ, Parihar A, Larson SK, Alidema E, Choi J, Ghafourifar P: Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res. 2007, 67 (3): 1282-1290. 10.1158/0008-5472.CAN-06-3099.http://dx.doi.org/10.1158/0008-5472.can-06-3099,
Graham JG, Quinn ML, Fabricant DS, Farnsworth NR: Plants used against cancer – an extension of the work of Jonathan Hartwell. J Ethnopharmacol. 2000, 73: 347-377. 10.1016/S0378-8741(00)00341-X.http://www.sciencedirect.com/science/article/pii/S037887410000341X,
Farnsworth NR: The Role of Etnopharmacology in Drug Development. 1990, England: John Wiley Chichester,http://www.ncbi.nlm.nih.gov/pubmed/2086037,
Liu JH: Activation of apoptosis pathways by different classes of anticancer drugs. 2001, Hubei: University of Ulm,http://vts.uni-ulm.de/docs/2001/798/vts_798.pdf,
Hannun YA: Apoptosis and the dilemma of cancer chemotherapy. Blood. 1997, 89 (6): 1845-1853.http://bloodjournal.hematologylibrary.org/content/89/6/1845.short,
Debatin K-M: Activation of apoptosis pathways by anticancer treatment. Toxicol Lett. 2000, 112–113 (0): 41-48.http://dx.doi.org/10.1016/S0378-4274(99)00252-0,
Hickman J: Apoptosis induced by anticancer drugs. Cancer Metastasis Rev. 1992, 11 (2): 121-139. 10.1007/BF00048059.http://dx.doi.org/10.1007/BF00048059,
Yeh CC, Tseng CN, Yang JI, Huang HW, Fang Y, Tang JY, Chang FR, Chang HW: Antiproliferation and induction of apoptosis in Ca9-22 oral cancer cells by ethanolic extract of gracilaria tenuistipitata. Molecules. 2012, 17 (9): 10916-10927.http://www.mdpi.com/1420-3049/17/9/10916,
Yoo C-B, Han K-T, Cho K-S, Ha J, Park H-J, Nam J-H, Kil U-H, Lee K-T: Eugenol isolated from the essential oil of Eugenia caryophyllata induces a reactive oxygen species-mediated apoptosis in HL-60 human promyelocytic leukemia cells. Cancer Lett. 2005, 225 (1): 41-52. 10.1016/j.canlet.2004.11.018.http://dx.doi.org/10.1016/j.canlet.2004.11.018,
Altinok B, Sunguroglu A, Karadag A, Gurleyik E, Ozkan T, Aydos OS, Avci A: Antiproliferative, apoptotic and antioxidant activities of wheatgrass (Triticum aestivum L.) extract on CML (K562) cell line. Turkish J of Med Sci. 2011, 41 (4): 657-663.http://journals.tubitak.gov.tr/medical/issues/sag-11-41-4/sag-41-4-13-0912-425.pdf,
Xiao D, Zeng Y, Prakash L, Badmaev V, Majeed M, Singh SV: Reactive oxygen species-dependent apoptosis by gugulipid extract of ayurvedic medicine plant commiphora mukul in human prostate cancer cells is regulated by c-Jun N-terminal kinase. The Am Soc for Pharmacol and Exp Ther. 2010, 79: 499-507.http://dx.doi.org/10.1124/mol.110.068551,
Chong KY, Tan HTW, Corlett RT: A checklist of the total vascular plant flora of Singapore: native, naturalised and cultivated species. Raffles Museum of Biodiversity Research, National University of Singapore. 2009, Singapore: Raffles Museum of Biodiversity Research, National University of Singapore, 273-http://rmbr.nus.edu.sg/raffles_museum_pub/flora_of_singapore_tc.pdf,
U.S. Department Agriculture ARS: National Genetic Resources Program. 2011, USA: Germplasm Resources Information Network (GRIN). Online searchable database,http://www.ars-grin.gov/cgi-bin/npgs/html/index.pl,
Ahmad FB, Holdsworth DK: Traditional medicinal plants of Sabah, Malaysia part III. The Rungus people of Kudat. Int J Pharmacogn. 1995, 33: 262-264. 10.3109/13880209509065377.http://dx.doi.org/10.3109/13880209509065377,
Hanum F, Hamzah N: The use of medicinal plant species by the Temuan tribe of Ayer Hitam Forest, Selangor, Peninsular Malaysia. Pertanika J of Tropical Agri. 1999, 22 (2): 85-94.http://psasir.upm.edu.my/3802/1/The_Use_of_Medicinal_Plant_Species_by_the_Temuan_Tribe_of_Ayer_Hitam.pdf,
Wiart C, Mogana S, Khalifah S, Mahan M, Ismail S, Buckle M, Narayana AK, Sulaiman M: Antimicrobial screening of plants used for traditional medicine in the state of Perak, Peninsular Malaysia. Fitoterapia. 2004, 75 (1): 68-73. 10.1016/j.fitote.2003.07.013.http://www.sciencedirect.com/science/article/pii/S0367326X03002089,
Muliawan SY: Effect of Dillenia suffruticosa extract on dengue virus type 2 replication. Universa Med. 2008, 27 (1): 1-5.http://www.univmed.org/wp-content/uploads/2011/02/dr.sylvia.pdf,
Armania N, Yazan L, Musa SN, Ismail IS, Foo JB, Chan KW, Noreen H, Hisyam AH, Zulfahmi S, Ismail M: Dillenia suffruticosa exhibited antioxidant and cytotoxic activity through induction of apoptosis and G2/M cell cycle arrest. J Ethnopharmacol. 2013, 146 (2): 525-535. 10.1016/j.jep.2013.01.017.http://dx.doi.org/10.1016/j.jep.2013.01.017,
Wang CZ, Li XL, Wang QF, Mehendale SR, Yuan CS: Selective fraction of Scutellaria baicalensis and its chemopreventive effects on MCF-7 human breast cancer cells. Phytomedicine. 2010, 17 (1): 63-68. 10.1016/j.phymed.2009.07.003.http://dx.doi.org/10.1016/j.phymed.2009.07.003,
Mosmann T: Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983, 65 (1–2): 55-63.http://dx.doi.org/10.1016/0022-1759(83)90303-4,
Hanahan D, Weinberg Robert A: Hallmarks of cancer: the next generation. Cell. 2011, 144 (5): 646-674. 10.1016/j.cell.2011.02.013.http://dx.doi.org/10.1016/j.cell.2011.02.013,
Fulda S: Modulation of apoptosis by natural products for cancer therapy. Planta Med. 2010, 76 (EFirst): 1075-1079.http://dx.doi.org/10.1055/s-0030-1249961,
Bailly C: Ready for a comeback of natural products in oncology. Biochem Pharmacol. 2009, 77 (9): 1447-1457. 10.1016/j.bcp.2008.12.013.http://dx.doi.org/10.1016/j.bcp.2008.12.013,
Taraphdar AK, Roy M, Bhattacharya RK: Natural products as inducers of apoptosis: implication for cancer therapy and prevention. Curr Sci. 2001, 80 (11): 1387-1396.http://www.iisc.ernet.in/currsci/jun102001/1387.pdf,
Man S, Gao W, Zhang Y, Huang L, Liu C: Chemical study and medical application of saponins as anti-cancer agents. Fitoterapia. 2010, 81 (7): 703-714. 10.1016/j.fitote.2010.06.004.http://dx.doi.org/10.1016/j.fitote.2010.06.004,
Zhiyu W, Yue C, Neng W, Mei WD, Wei LY, Feng H, Gang SJ, De Po Y, Yuan GX, Jian-Ping C: Dioscin induces cancer cell apoptosis through elevated oxidative stress mediated by downregulation of peroxiredoxins. Cancer Biol & Therapy. 2012, 13 (3): 138-147. 10.4161/cbt.13.3.18693.http://www.landesbioscience.com/journals/cbt/article/18693/,
Yeh CT, Rao YK, Yao CJ, Yeh CF, Li CH, Chuang SE, Luong JHT, Lai GM, Tzeng YM: Cytotoxic triterpenes from Antrodia camphorata and their mode of action in HT-29 human colon cancer cells. Cancer Lett. 2009, 285 (1): 73-79. 10.1016/j.canlet.2009.05.002.http://dx.doi.org/10.1016/j.canlet.2009.05.002,
Lamoral-Theys D, Pottier L, Dufrasne F, Neve J, Dubois J, Kornienko A, Kiss R, Ingrassia L, Lamoral-Theys D, Pottier L, Dufrasne F, Neve J, Dubois J, Kornienko A, Kiss R, Ingrassia L: Natural polyphenols that display anticancer properties through inhibition of kinase activity. Curr Med Chem. 2010, 17 (814): 812-825.http://dx.doi.org/10.2174/092986710790712183,
Kuttan G, Kumar KB, Guruvayoorappan C, Kuttan R: Antitumor, anti-invasion, and antimetastatic effects of curcumin. Adv Exp Med Biol. 2007, 595: 173-184. 10.1007/978-0-387-46401-5_6.http://link.springer.com/chapter/10.1007%2F978-0-387-46401-5_6,
Barton-Burke M, Wilkes GM: Cancer Therapies. 2006, United States of America: Jones and Barlett’s books, 10
Ozawa S, Sugiyama Y, Mitsuhashi Y, Kobayashi T, Inaba M: Cell killing action of cell cycle phase-non-specific antitumor agents is dependent on concentration-time product. Cancer Chemother Pharmacol. 1988, 21 (3): 185-190.http://dx.doi.org/10.1007/bf00262767,
Fadok VA, Voelker DR, Campbell PA, Cohen JJ DLB, Henson PM: Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992, 148: 2207-http://www.jimmunol.org/content/148/7/2207.long,
Vermes I, Haanen C, Steffens-Nakken H, Reutellingsperger C: A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods. 1995, 184 (1): 39-51. 10.1016/0022-1759(95)00072-I.http://dx.doi.org/10.1016/0022-1759(95)00072-I,
Zhang G, Gurtu V, Kain SR, Yan G: Early detection of apoptosis using a fluorescent conjugate of annexin V. BioTechniques. 1997, 23: 525-531.http://www.biotechniques.com/multimedia/archive/00011/97233pf01_11715a.pdf,
Schutte B, Nuydens R, Geerts H, Ramaekers F: Annexin V binding assay as a tool to measure apoptosis in differentiated neuronal cells. J Neurosci Methods. 1998, 86 (1): 63-69. 10.1016/S0165-0270(98)00147-2.http://dx.doi.org/10.1016/S0165-0270(98)00147-2,
Jänicke RU, Sprengart ML, Wati MR, Porter AG: Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998, 273 (16): 9357-9360. 10.1074/jbc.273.16.9357.http://dx.doi.org/10.1074/jbc.273.16.9357,
Jänicke R: MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res Treat. 2009, 117 (1): 219-221. 10.1007/s10549-008-0217-9.http://dx.doi.org/10.1007/s10549-008-0217-9,
Mooney LM, Al-Sakkaf KA, Brown BL, Dobson PR: Apoptotic mechanisms in T47D and MCF-7 human breast cancer cells. Br J Cancer. 2002, 87 (8): 909-917. 10.1038/sj.bjc.6600541.http://europepmc.org/abstract/MED/12373608,
Kasibhatla S, Genestier L, Green DR: Regulation of Fas-ligand expression during activation-induced cell death in T lymphocytes via nuclear factor κB. J Biol Chem. 1999, 274 (2): 987-992. 10.1074/jbc.274.2.987.http://dx.doi.org/10.1074/jbc.274.2.987,
Kong Q, Beel JA, Lillehei KO: A threshold concept for cancer therapy. Med Hypotheses. 2000, 55 (1): 29-35. 10.1054/mehy.1999.0982.http://dx.doi.org/10.1054/mehy.1999.0982,
Kinnula VL, Crapo JD: Superoxide dismutases in malignant cells and human tumors. Free Radic Biol Med. 2004, 36 (6): 718-744. 10.1016/j.freeradbiomed.2003.12.010.http://dx.doi.org/10.1016/j.freeradbiomed.2003.12.010,
Salganik RI: The benefits and hazards of antioxidants: controlling apoptosis and other protective mechanisms in cancer patients and the human population. J Am Coll Nutr. 2001, 20 (5): 464S-472S.http://www.ncbi.nlm.nih.gov/pubmed/11603657,
Miller WH, Ratna S, Rincón SVD, Rousseau : Retinoic acid-induced growth arrest of MCF-7 cells involves the selective regulation of the IRS-1/PI 3-kinase/AKT pathway. Oncogene. 2002, 22 (3353): 3360-http://dx.doi.org/10.1038/sj.onc.1206485,
Diao Y, Huang YQ, Zhang GD, Huang KF: Wogonin induces apoptosis and down-regulates survivin in human breast cancer MCF-7 cells by modulating PI3K–AKT pathway. Int Immunopharmacol. 2012, 12 (2): 334-341. 10.1016/j.intimp.2011.12.004.http://dx.doi.org/10.1016/j.intimp.2011.12.004,
Testa JR, Bellacosa A: AKT plays a central role in tumorigenesis. Proc Natl Acad Sci. 2001, 98 (20): 10983-10985. 10.1073/pnas.211430998.http://dx.doi.org/10.1073/pnas.211430998,
Martindale JL, Holbrook NJ: Cellular response to oxidative stress: signaling for suicide and survival. J of Cell Physiol. 2002, 192 (1): 15-http://dx.doi.org/10.1002/jcp.10119,
Chang YJ, Huang YP, Li ZL, Chen CH: GRP78 Knockdown enhances apoptosis via the down-regulation of oxidative stress and Akt pathway after epirubicin treatment in colon cancer DLD-1 cells. PLoS ONE. 2012, 7 (4): e35123-10.1371/journal.pone.0035123.http://dx.doi.org/10.1371/journal.pone.0035123s,
Toker A, Yoeli-Lerner M: Akt signaling and cancer: surviving but not moving on. Cancer Res. 2006, 66 (8): 3963-3966. 10.1158/0008-5472.CAN-06-0743.http://dx.doi.org/10.1158/0008-5472.can-06-0743,
Bates S, Vousden KH: Mechanisms of p53-mediated apoptosis. Cell Mol Life Sci. 1999, 55: 28-37. 10.1007/s000180050267.http://www.ncbi.nlm.nih.gov/pubmed/10065149sss,
Robbins D, Zhao Y: Oxidative stress induced byMnSOD-p53 interaction: Pro- or anti-tumorigenic?. J of Signal Transduct. 2012, 2012: 1-13.http://dx.doi.org/10.1155/2012/101465,
Sethi G, Sung B, Aggarwal BB: Nuclear factor-κB activation: from bench to bedside. Exp Biol and Med. 2008, 233 (1): 21-31. 10.3181/0707-MR-196.http://dx.doi.org/10.3181/0707-mr-196,
Aoki M, Nata T, Morishita R, Matsushita H, Nakagami H, Yamamoto K, Yamazaki K, Nakabayashi M, Ogihara T, Kaneda Y: Endothelial apoptosis induced by oxidative stress through activation of NF-κB: antiapoptotic effect of antioxidant agents on endothelial cells. Hypertension. 2001, 38 (1): 48-55. 10.1161/01.HYP.38.1.48.http://dx.doi.org/10.1161/01.hyp.38.1.48,
Martin AG: NFkB anti-apoptotic or pro-apoptotic, maybe both. Cell Cycle. 2010, 9 (16): 3131-3132. 10.4161/cc.9.16.12780.http://www.mendeley.com/download/public/14520753/4840739732/054ce21125d09cd5018cfb8b4e375f6646499037/dl.pdf,
Janssen-Heininger YMW, Poynter ME, Baeuerle PA: Recent advances torwards understanding redox mechanisms in the activation of nuclear factor κb. Free Radic Biol Med. 2000, 28 (9): 1317-1327. 10.1016/S0891-5849(00)00218-5.http://dx.doi.org/10.1016/S0891-5849(00)00218-5,
Zarubin T, Han J: Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15: 11-18. 10.1038/sj.cr.7290257.http://dx.doi.org/doi:10.1038/sj.cr.7290257,
Park K-R, Nam D, Yun H-M, Lee S-G, Jang H-J, Sethi G, Cho SK, Ahn KS: β-Caryophyllene oxide inhibits growth and induces apoptosis through the suppression of PI3K/AKT/mTOR/S6K1 pathways and ROS-mediated MAPKs activation. Cancer Lett. 2011, 312 (2): 178-188. 10.1016/j.canlet.2011.08.001.http://dx.doi.org/10.1016/j.canlet.2011.08.001,
Massaoka MH, Matsuo AL, Figueiredo CR, Farias CF, Girola N: Jacaranone induces apoptosis in melanoma cells via ROS-mediated downregulation of Akt and p38 MAPK activation and displays antitumor activity in vivo. PLoS ONE. 2012, 7 (6): e38698-10.1371/journal.pone.0038698.http://dx.doi.org/10.1371/journal.pone.0038698,
Gratton JP, Morales-Ruiz M, Kureishi Y, Fulton D, Walsh K, Sessa WC: Akt down-regulation of p38 signaling provides a novel mechanism of vascular endothelial growth factor-mediated cytoprotection in endothelial cells. J Biol Chem. 2001, 276 (32): 30359-30365. 10.1074/jbc.M009698200.http://dx.doi.org/10.1074/jbc.M009698200,
Romashkova JA, Makarov SS: NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999, 401 (86): 90-http://dx.doi.org/10.1038/43474,
Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM: Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996, 274 (5288): 787-789. 10.1126/science.274.5288.787.http://dx.doi.org/10.1126/science.274.5288.787,
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1472-6882/14/55/prepub
Acknowledgements
The experiment was funded by the Fundamental Research Grant Malaysia (Vote number: 5523924). Special thanks are delivered to staff members of the Laboratory of Molecular Biomedicine, Laboratory of Vaccine and Immunotherapeutics, and Laboratory of Immunology from Universiti Putra Malaysia for their support and assistance in completing this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YST carried out the study and prepared the manuscript. YST and JBF collected and interpreted the data. NA and JBF contributed to the preparation of plant extract. MUI and NI contributed to GeXP analysis. LSY, RA, YKC and MI contributed to the design and conception of the study and interpretation of data. LSY critically revised manuscript and codirected with MI who supervised and provided reagents and facilities. All authors have read and approved the manuscript for publication.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tor, Y.S., Yazan, L.S., Foo, J.B. et al. Induction of apoptosis through oxidative stress-related pathways in MCF-7, human breast cancer cells, by ethyl acetate extract of Dillenia suffruticosa . BMC Complement Altern Med 14, 55 (2014). https://doi.org/10.1186/1472-6882-14-55
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6882-14-55