
Abstract
Background
Doxorubicin (DOX) is an effective antineoplastic drug; however, clinical use of DOX is limited by its dose-dependent cardiotoxicity. It is well known that reactive oxygen species (ROS) play a vital role in the pathological process of DOX-induced cardiotoxicity. For this study, we evaluated the protective effects of guggulsterone (GS), a steroid obtained from myrrh, to determine its preliminary mechanisms in defending against DOX-induced cytotoxicity in H9C2 cells.
Methods
In this study, we used a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay, lactate dehydrogenase (LDH) release measurements, and Hoechst 33258 staining to evaluate the protective effect of GS against DOX-induced cytotoxicity in H9C2 cells. In addition, we observed the immunofluorescence of intracellular ROS and measured lipid peroxidation, caspase-3 activity, and apoptosis-related proteins by using Western blotting.
Results
The MTT assay and LDH release showed that treatment using GS (1–30 μM) did not cause cytotoxicity. Furthermore, GS inhibited DOX (1 μM)-induced cytotoxicity in a concentration-dependent manner. Hoechst 33258 staining showed that GS significantly reduced DOX-induced apoptosis and cell death. Using GS at a dose of 10–30 μM significantly reduced intracellular ROS and the formation of MDA in the supernatant of DOX-treated H9C2 cells and suppressed caspase-3 activity to reference levels. In immunoblot analysis, pretreatment using GS significantly reversed DOX-induced decrease of PARP, caspase-3 and bcl-2, and increase of bax, cytochrome C release, cleaved-PARP and cleaved-caspase-3. In addition, the properties of DOX-induced cancer cell (DLD-1 cells) death did not interfere when combined GS and DOX.
Conclusion
These data provide considerable evidence that GS could serve as a novel cardioprotective agent against DOX-induced cardiotoxicity.
Similar content being viewed by others

Background
Doxorubicin (DOX), a member of the anthracycline class of chemotherapeutic drugs, is a potent antineoplastic drug that has exhibited a wide spectrum of antitumor activity including in leukemia, lymphomas, soft tissue sarcomas, and breast cancer for several decades. Despite the efficacy of DOX, its use has been limited by the dose-dependent cardiotoxicity associated with acute and chronic treatments in anticancer therapy[1–3]. Both acute and chronic DOX-induced cardiotoxicity may cause cardiac dysfunction or cardiomyopathy, and may ultimately lead to rigorous heart failure and death[3, 4]. Previous studies have suggested that one mechanism responsible for DOX cardiotoxicity is the formation of reactive oxygen species (ROS)[5, 6], which can harm membrane lipids and other cellular components, leading to cardiomyocyte apoptosis and death[7]. Certain antioxidants have been examined to reduce ROS formation, but many have demonstrated only a limited cardioprotective effect or have other side effects[8, 9].
Guggulsterone (GS) is a steroid compound that has been isolated from myrrh[10, 11]; the structure of which is shown in Figure1[12]. In 1972, two isomer derivatives, Z-GS and E-GS, were identified in Commiphora mukul (containing 4.50% (Z)-isomer and 1.42% (E)-isomer)[13] as antagonist ligands for the bile acid receptor farnesoid X receptor and as active ingredients responsible for hypolipidemic activity[14, 15]. During a 10-year period, myrrh was examined to treat various conditions, such as rheumatism, atherosclerosis, hypercholesterolemia, and obesity[16]. Both GS isomers were demonstrated to suppress LPS-induced inflammation by inhibiting IκB-α degradation and NF-κB activation[17, 18]. GS was mediated (possibly by activating protein-1) to upregulate the expression of the bile salt export pump[16]. In addition, GS was reported to protect cardiac and neuronal damages in animal models[19]. However, no research has yet been conducted on the use of GS in protecting against DOX-induced cardiotoxicity. This study is the first to evaluate the protective effects of GS to determine the preliminary mechanisms of defending against DOX-induced cytotoxicity in H9C2 cells.

Structure of Z -guggulsterone [ trans -4,17(20)-pregnadiene-3,16-dione][12].
Methods
Chemicals
Dulbecco’s Modified Eagle’s Medium, an RPMI 1640 medium, fetal bovine serum, penicillin/streptomycin, and medium supplements were purchased from Life Technologies (Gibco, Grand Island, NY). Monoclonal antibodies and a peroxidase-conjugated secondary antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). DOX, GS, and other agents were obtained from Sigma Chemical (St. Louis, MO).
Cell culture
All rat cardiac H9C2 myocardial cells, spontaneously immortalized ventricular rat embryo myoblasts, and DLD-1 cells (human colon adenocarcinoma) were purchased from the Food Industry Research and Development Institute, Taiwan (BCRC). The H9C2 cells were cultured in DMEM supplemented with 10% fetal bovine serum at 37°C in 5% CO2, and DLD-1 cells were cultured in the RPMI 1640 medium. The media were changed every 2–3 d.
Cell viability
Sub-confluent cells were trypsinized and seeded onto 96-well plates at a density of 1.5 × 105 cells/ml and incubated for 24 h before treatment. Thereafter, the cells were exposed to DOX 1 μM for 24 h and then incubated in a fresh medium with Z-GS at various concentrations for an additional 24 h. The effects of GS on DOX-induced cytotoxicity were assessed using the MTT assay, as previously described[20]. The unwashed dye was eluted and quantified spectrophotometrically at 550 nm using a microplate reader. Cell viability was determined as the percentage of surviving cells compared with that of the DOX-treated control.
Lactate dehydrogenase (LDH) release assay for cytotoxicity
GS-induced cytotoxicity leading to plasma membrane damage was measured using the LDH Cytotoxicity Detection Kit (Boehringer Mannheim, Mannheim, Germany). The LDH release assay has been widely used in cytotoxicity studies[21]. The detailed assay was performed as previously described[22].
Intracellular ROS
Intracellular ROS induced by DOX was measured using 2’7’-dichlorodihydrofluorescein diacetate (DCFH-DA) as a fluorescent probe[23]. H9C2 cells were loaded with DCFH-DA (20 μ) for 10 min, followed by 2 washes with HBSS. Dichlorodihydrofluorescein (DCF) fluorescence was detected using a fluorescence spectrophotometer with an excitation of 485 nm and an emission of 520 nm, and the fluorescence image was visualized using a fluorescence microscope.
Lipid peroxidation
Lipid peroxidation was assayed using the thiobarbituric acid (TBA) reaction. The amount of thiobarbituric acid reactive substance (TBARS) was determined using a standard curve of 1,1,3,3-tetramethoxypropane. The detailed assay procedure was performed as previously described[24].
Preparation of total cell lysates and nuclear and cytosolic extracts
H9C2 cells (5 × 105 cells/well) or DLD-1 cells in 6-well plates were incubated with or without concentrations of GS and DOX (1 μM) for 24 h. The total cell lysates were obtained using a lysis buffer (250 mM Tris–HCl (pH 6.8), 1% Triton-100, 0.1% SDS, 1 mM Na3VO4, 1 mM EDTA, 5 mM sodium fluoride, 1 mM PMSF, and 1 mg/ml leupeptin), and cell debris was removed using a centrifuge at 10 000 × g for 10 min at 4°C and stored at −80°C until required. The protein content of the cell lysates was determined using the Bradford assay[25].
Western blot analysis
Equal amounts of cell lysates (30 μg) were electroblotted onto a nitrocellulose membrane (Millipore, MA), following separation using 8%-12% SDS-polyacrylamide gel electrophoresis. The blot was probed using a primary antibody against poly (ADP-ribose) polymerase (PARP), caspase-3, bcl-2, bax, cytochrome C, and β-actin (Santa Cruz Biochemicals, Santa Cruz, CA). The intensity of each band was quantified using density analysis software (MetaMorph Imaging System, Meta Imaging Series 4.5), and the density ratio represented the relative intensity of each band against controls in each experiment.
Hoechst 33258 staining
Cells were rinsed twice in 4°C PBS and fixed in 4% formaldehyde at 4°C for 10 min. After washing, the cells were incubated using Hoechst 33258 (5 μg/ml) staining at room temperature for 10 min in the dark. The cells were then observed and imaged using a laser scanning confocal microscope (Bio-Rad MRC-1000, American Laboratory, USA) with an excitation of 350 nm and an emission of 460 nm[26].
Caspase-3 activity assay
Caspase-3 activity was determined using the ApoAlert Caspase Colorimetric Assay kit (Clontech Laboratories, Inc. USA), according to manufacturer’s protocol.
Data and statistical analysis
The results of all the experiments were expressed as the mean ± standard error (SE) obtained from the number of replicate treatments. Data were analyzed using analysis of variance (ANOVA), followed by Dunn’s post hoc test for comparison, and P values of < 0.05 were considered statistically significant.
Results
Effects of guggulsterone (GS) on cell viability and cytotoxicity
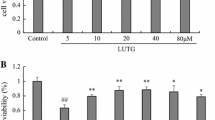
The cell survival percentages of H9C2 cells receiving GS treatment were measured using an MTT assay and an LDH release assay. As shown in Figure2A and2B, the cells were not significantly injured by GS treatment at up to 30 μM. These results suggest that GS concentrations were nontoxic to H9C2 cells below 30 μM. Thus, the cells were treated with GS in concentrations ranging from 1 to 30 μM for all follow-up experiments.

Effects of guggulsterone (GS) on the H9C2 cell line. (A) Cells were treated with various concentrations of GS for 24 h. Cell viability was determined using an MTT assay, as described in the Materials and Methods section. (B) GS cytotoxicity was analyzed using LDH release. The results are expressed as mean ± S.E. from 3 independent experiments. Asterisks indicate a significant difference for the control (GS 0 μM) (**P < 0.01).
GS protected H9C2 cells from DOX-induced cell death
To determine GS concentrations prevented in DOX-induced cytotoxicity, we examined the preventive abilities of GS in H9C2 cells at different concentrations. The results of the MTT assay and LDH release assay showed that exposure to DOX at a concentration of 1 μM for 24 h caused 25.9% cell death (Figure3A); however, no significant reduction of viable cells was found in 10–30 μM GS-treated cells after 24 h treatment (Figure3A and3B). These findings show that GS at 10–30 μM can reduce DOX-induced cytotoxicity in H9C2 cells. Microscopic examinations of the cell cultures showed a reversal of DOX-induced cell death in cell morphology when treated with differing concentrations of GS (10, 20, and 30 μM), as shown in Figure4.

GS prevention on DOX-induced H9C2 cell death. Cells were incubated with DOX (1 μM) in the presence and absence of GS for 24 h. The percentages of viable cells were determined using an MTT assay, as described in the Materials and Methods section. The results are expressed as mean±S.E. from 3 independent experiments. *P < 0.05 and **P < 0.01 indicate a significant difference from DOX treatment. #P < 0.01 indicates a significant difference from the control (GS 0 μM and DOX 0 μM).

Morphology of DOX-induced cell death in H9C2 cells in the presence or absence of GS for 24 h. Cells were viewed using a video camera (Nikon) attached to a microscope, and the image was projected onto a monitor. Images were obtained with a 200× objective.
GS relieved oxidative stress induced by DOX in H9C2 cells
As shown in Figure5, cells exposed to DOX were significantly brighter than the control cells with numerous highlights, where ROS was concentrated in dihydroethidium (DHE) staining. By contrast, the red fluorescence intensity of the cells pretreated with GS was considerably darker than or nearly as dark as the control cells. Moreover, the ROS level of the cells treated with GS alone was nearly the same as that of the control cells. Nevertheless, we measured the oxidation of DCFH and lipid peroxidation, which are widely used markers of intracellular ROS formation, in H9C2 cells treated for 24 h with 1 μM DOX in the presence or absence of GS, as shown in Figure6A and6B.

GS relieved the oxidative stress induced by DOX in H9C2 cells. Staining of intracellular ROS by dihydroethidium (DHE) staining in H9C2 cells (original magnification × 200).

Prevention of GS on DOX-induced oxidative stress. Subconfluent H9C2 cells were left untreated or exposed to DOX and co-treated with GS for 24 h and oxidative stress was evaluated by measuring DCFH oxidation (A) and the amount of TBARS formation (malondialdehyde, MAD) (B). The results are expressed as mean±S.E. from 3 independent experiments. **P < 0.01 indicates a significant difference from DOX treatment. ##P < 0.01 indicates a significant difference from the control (GS 0 μM and DOX 0 μM).
GS protected against apoptosis of H9C2 cells treated with DOX
We investigated the effects of GS on DOX-induced apoptosis in H9C2 cells by using Western blotting. PARP, caspase-3, and bcl-2 are major types of regulatory proteins associated with apoptosis. As shown in Figure7A, PARP and bcl-2, two apoptotic suppressors, were markedly decreased and the cleaved PARP had increased after treatment with DOX (1 μM) for 24 h. By contrast, caspase-3 (both the precursor and cleaved form), a pro-apoptotic protein, was markedly altered after DOX treatment. However, these effects of DOX-induced PARP, caspase-3, and bcl-2 were reversed in a concentration-dependent manner by pretreating with GS for 2 h. In addition, bax and cytochrome C release decreased significantly following GS treatment. As shown in Figure7B and7C, the density ratio of PARP (both the precursor and cleaved form), caspase-3 (both the precursor and cleaved form), bcl-2, bax, and cytochrome C is represented in quantitative determination. The protective effects of GS on H9C2 cells were detected using the Hoechst 33258 stained and caspase-3 activity assays. As shown in Figure8, normal cells were observed as round-shaped nuclei with homogeneous fluorescence intensity. DOX induced rapid nuclear changes of H9C2 with heterogeneous intensity and chromatin condensation. In the control and GS groups, most nuclei exhibited regular contours and were round and large. The GS + DOX group showed slight DNA condensation but only a few fragmentations of chromatin. This suggests that apoptosis was the prevalent form of cell death in H9C2 cells exposed to 1 μM of DOX. In addition, we further measured the activity of caspase-3, a major effector protein of apoptosis. As shown in Figure9, caspase-3 activity was increased by 1 μM of DOX, and GS can inhibit DOX-induced caspase-3 activity at 10–30 μM after 24 h of treatment.

Effects of GS on DOX-induced apoptosis-related protein in the cytoplasm of H9C2 cells. (A) Cells (5 × 105 cells/ well) were pretreated with the indicated concentrations of GS for 2 h before incubation with DOX (1 μM) for 24 h. The proteins (30 μg) of total cell lysates were analyzed at the expression levels of anti-PARP, caspase-3, bcl-2, bax, and cytosolic lysates that were analyzed for anti-cytochrome C by Western blotting at 10% SDS-PAGE. β-Actin is the internal standard to confirm equal loading. (B) Quantification of PARP, bcl-2, and caspase-3 expression, and cytochrome C release. (C) Quantification of the cleaved form of PARP and caspase-3, as well as the bax expression. Data are expressed as mean ± S.E. from 3 independent experiments. *P < 0.05, **P < 0.01, and PARP (both the precursor and cleaved form) expression compared to the DOX treatment alone; #P < 0.05 and ##P < 0.01, and caspase-3 (both the precursor and cleaved form) compared to the DOX treatment alone; ςP < 0.05 and ςςP < 0.01, and bcl-2 and bax compared to the DOX treatment alone. ξP < 0.05 and ξξP < 0.01, and cytochrome C compared to the DOX treatment alone.

The protective effects of GS on H9C2 cells against DOX-induced apoptosis by Hoechst 33258 staining. Fluorescence images of Hoechst 33258 stained H9C2 cells after a 2 h of exposure to DOX in the absence or presence of GS. Images were obtained using a 200× objective.

Effects of GS on DOX-induced apoptosis using caspase-3 activity assay in H9C2 cells. Subconfluent H9C2 cells were treated with DOX and co-treated with different concentrations of GS for 24 h, and caspase-3 activity was measured using colorimetry. The results are expressed as mean±S.E. from 3 independent experiments. **P < 0.01 indicates a significant difference from DOX treatment. ##P < 0.01 indicates a significant difference from the control (GS 0 μM and DOX 0 μM).
GS did not interfere with DOX-induced cell death in DLD-1 cells
To determine whether GS influences DOX in cancer therapy, GS and DOX were combined to treat DLD-1 cells (human colon adenocarcinoma). As shown in Figure10A, the morphology of DLD-1 cells indicated that they were injured after treatment with 1 μM of DOX; however, GS (10, 20 and 30μM) did not reverse this response. As shown in Figure10B, GS (1–50 μM) did not cause cytotoxicity in the DLD-1 cells. DOX (1 μM) can induce 42.6% cell death; however, the cell viability did not significantly increase after co-treatment of GS (1–50 μM) and DOX. In addition, we assessed PARP, caspase-3, and bax by Western blotting. As shown in Figure10C, DOX induced PARP and caspase-3 protein degradation, increased cleaved-PARP and cleaved-caspase-3 expression, and induced bax expression. However, these effects were not influenced by co-treatment with GS.

Effects of combining GS and DOX in cancer cells. (A) Morphology of DOX-induced cell death. (B) Cell viability of the MTT assay. (C) Western blotting of PARP, caspase-3 and bax protein expression in DLD-1 cells in the presence or absence of GS for 24 h. Images were obtained with a 200× objective of the microscope. The assay methods were performed in the same manner as that described for the H9C2 cells.
Discussion
DOX, as an anthracycline antibiotic, has been used in cancer therapy for nearly three decades. However, the clinical use of DOX is restricted considerably by an increased risk of cardiotoxicity associated with DOX-induced cardiomyocyte apoptosis[27–29]. Adult cardiomyocytes of the heart are well known to finally differentiated muscle cells that do not progress proliferation[30]. In addition, DOX seemed to affect specific enzymes (NADH and FADH2), transporters (Ca2+-ATPase and Na+, and K+-ATPase), and metabolic pathways (AMP-activated protein kinase) to varying extents in the cardiac muscle. The accumulation of these defects may ultimately result in irreversible cardiac failure[31]. This study elucidated the protection of GS from DOX-induced apoptosis and cell death by conducting the following experiments: MTT assay, LDH release, DHE-staining, fluorescence intensity of DCFH, lipid peroxidation, Hoechst 33258 staining, caspase-3 activity measurement and immunoblot analysis. The production of ROS as a derivative of the DOX metabolism has been suggested to be the main mechanism of DOX-induced cardiotoxicity[32–34]. However, superoxide radicals are involved in other ROS and generate hydroxyl radical and hydrogen peroxide[35]. According to current reports, generating these ROS causes mitochondrial damage, which may lead to cardiomyocyte apoptosis, necrosis, or death. In addition, these DOX-induced mitochondrial injuries in cardiomyocytes were prevented by some natural substances, such as curcumin, naringenin-7-O-glucoside, and plantainoside D[20, 36, 37]. In our study, we used DCFH-DA as an intracellular method of ROS detection[38]. The results demonstrated that GS can significantly reduce the intracellular ROS produced by DOX in H9C2 cells. We also measured lipid peroxidation (MDA formation) in H9C2 cells. The reduction of MDA content suggested that GS can attenuate the oxidative stress induced by DOX in H9C2 cells.
According to the identified apoptosis of cell signaling, PARP is a nuclear enzyme activated by strand breaks in DNA and implicated in DNA repair, apoptosis, organ dysfunction, or necrosis[39, 40]. In addition, it is known that members of the bcl-2 protein family are known to be major regulators of cytochrome C release and downstream caspases activation. Thus, bcl-2 plays a vital role in regulating cardiomyocyte apoptosis[29, 41]. Using Western blot analysis, we found that GS treatment attenuated DOX-induced apoptotic proteins (cleaved-PARP, cleaved-caspase-3, and bax). In contrast, treatment with GS also enhanced anti-apoptotic protein (bcl-2) expression in cardiomyocytes. Finally, another type of cancer cell line, DLD-1, was used to exclude the possible interference of GS with DOX in cancer therapy. The data showed that the efficacy of DOX did not diminish co-treatment of GS and DOX.
Conclusion
Numerous studies have shown that myocardial impairment caused by DOX may be due to cardiomyocyte apoptosis, and DOX may also cause injury to endothelial cells[42]. This study shows that GS is a novel potent protector against DOX-induced cardiotoxicity, providing protection using its antioxidative activity.
References
Singal PK, Iliskovic N: Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998, 339: 900-905. 10.1056/NEJM199809243391307.
Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L: Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004, 56: 185-229. 10.1124/pr.56.2.6.
Yeh E, Tong A, Lenihan D, Yusuf S, Swafford J, Champion C, Durand J, Gibbs H, Zafarmand A, Ewer M: Cardiovascular complications of cancer therapy: diagnosis, pathogenesis, and management. Circulation. 2004, 109: 3122-3131. 10.1161/01.CIR.0000133187.74800.B9.
Wallace KB: Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol. 2003, 93: 105-115. 10.1034/j.1600-0773.2003.930301.x.
Takemura G, Fujiwara H: Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. 2007, 49: 330-352. 10.1016/j.pcad.2006.10.002.
Neilan T, Blake S, Ichinose F, Raher M, Buys E, Jassal D, Furutani E, Perez-Sanz T, Graveline A, Janssens S: Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation. 2007, 116: 506-514. 10.1161/CIRCULATIONAHA.106.652339.
Menna P, Minotti G, Salvatorelli E: In vitro modeling of the structure-activity determinants of anthracycline cardiotoxicity. Cell Biol Toxicol. 2007, 23: 49-62. 10.1007/s10565-006-0143-8.
Elbl L, Hrstkova H, Tomaskova I, Michalek J: Late anthracycline cardiotoxicity protection by dexrazoxane (ICRF-187) in pediatric patients: echocardiographic follow-up. Support Care Cancer. 2006, 14: 128-136. 10.1007/s00520-005-0858-8.
Wouters KA, Kremer LC, Miller TL, Herman EH, Lipshultz SE: Protecting against anthracycline-induced myocardial damage: a review of the most promising strategies. Br J Haematol. 2005, 131: 561-578. 10.1111/j.1365-2141.2005.05759.x.
Urizar NL, Moore DD: Gugulipid: a natural cholesterol-lowering agent. Annu Rev Nutr. 2003, 23: 303-313. 10.1146/annurev.nutr.23.011702.073102.
Shen T, Lou HX: Bioactive constituents of myrrh and frankincense, two simultaneously prescribed gum resins in Chinese traditional medicine. Chem Biodivers. 2008, 5: 540-553. 10.1002/cbdv.200890051.
Bhatta RS, Kumar D, Chhonker YS, Jain GK: Simultaneous estimation of E- and Z-isomers of guggulsterone in rabbit plasma using liquid chromatography tandem mass spectrometry and its application to pharmacokinetic study. Biomed Chromatogr. 2011, 25: 1054-1060. 10.1002/bmc.1574.
Patil VD, Nayak UR, Dev S: Chemistry of Ayurvedic crude drugs-I : Guggulu (resin from Commiphora mukul)-1: Steroidal constituents. Tetrahedron. 1972, 28: 2341-2352. 10.1016/S0040-4020(01)93577-X.
Urizar NL, Liverman AB, Dodds DT, Silva FV, Ordentlich P: A natural product that lowers cholesterol as an antagonist ligand for FXR. Science. 2002, 296: 1703-1706. 10.1126/science.1072891.
Wu J, Xia C, Meier J, Li S, Hu X, Lala DS: The hypolipidemic natural product guggulsterone acts as an antagonist of the bile acid receptor. Mol Endocrinol. 2002, 16: 1590-1597. 10.1210/me.16.7.1590.
Deng R: Therapeutic effects of guggul and its constituent guggulsterone: Cardiovascular benefits. Cardiovasc Drug Rev. 2007, 25: 375-390.
Meselhy MR: Inhibition of LPS-induced NO production by the oleogum resin of Commiphora wightii and its constituents. Phytochemistry. 2003, 62: 213-218. 10.1016/S0031-9422(02)00388-6.
Shishodia S, Aggarwal BB: Guggulsterone inhibits NF-κB and IκBα kinase activation, suppresses expression of anti-apoptotic gene products, and enhances apoptosis. J Biol Chem. 2004, 279: 47148-47158. 10.1074/jbc.M408093200.
Kaul S, Kapoor N: Reversal of changes of lipid peroxide, xanthine oxidase and superoxide dismutase by cardio-protective drugs in isoproterenol induced myocardial necrosis in rats. Indian J Exp Biol. 1989, 27: 625-627.
Han X, Ren D, Fan P, Shen T, Lou H: Protective effects of naringenin-7-O-glucoside on doxorubicininduced apoptosis in H9C2 cells. Eur J Pharmacol. 2008, 581: 47-53. 10.1016/j.ejphar.2007.11.048.
Zuckerbraun HL, Babich H, May R, Sinensky MC: Triclosan: cytotoxicity, mode of action, and induction of apoptosis in human gingival cells in vitro. Eur J Oral Sci. 1998, 106: 628-636. 10.1046/j.0909-8836.1998.eos106204.x.
Wang CC, Choy CS, Liu YH, Cheah KP, Li JS, Wang JTJ, Yu WY, Lin CW, Cheng HW, Hu CM: Protective effect of dried safflower petal aqueous extracts and its main constituent, carthamus yellow, against lipopolysaccharide (LPS)-induced inflammation in RAW264.7 macrophage. J Sci Food Agric. 2011, 91: 218-225. 10.1002/jsfa.4172.
Choy CS, Cheah KP, Chiou HY, Li JS, Liu YH, Yong SF, Chiu WT, Liao JW, Hu CM: Induction of hepatotoxicity by sanguinarine is associated with oxidation of protein thiols and disturbance of mitochondrial respiration. J Appl Toxicol. 2008, 28: 945-956.
Hu CM, Li JS, Cheah KP, Lin CW, Yu WY, Chang ML, Yeh GC, Chen SH, Cheng HW, Choy CS: Effect of Sanguis draconis (a dragon's blood resin) on streptozotocin- and cytokine-induced β-cell damage, in vitro and in vivo. Diabetes Res Clin Pract. 2011, 94: 417-425. 10.1016/j.diabres.2011.08.014.
Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976, 72: 248-254. 10.1016/0003-2697(76)90527-3.
Fukuda M, Tsuchihashi Y, Takamatsu T, Nakanishi K, Fujita S: Fluorescence fading and stabilization in cytofluorometry. Histochemistry. 1980, 65: 269-276. 10.1007/BF00493176.
Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC: Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiomyopathy. Circulation. 2005, 111: 1601-1610. 10.1161/01.CIR.0000160359.49478.C2.
Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN: A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003, 111: 1497-1504.
Xin H, Liu XH, Zhu YZ: Herba leonurine attenuates doxorubicin-induced apoptosis in H9c2 cardiac muscle cells. Eur J Pharmacol. 2009, 612: 75-79. 10.1016/j.ejphar.2009.03.067.
Vidarsson H, Hyllner J, Sartipy P: Differentiation of human embryonic stem cells to cardiomyocytes for in vitro and in vivo applications. Stem Cell Rev. 2010, 6: 108-120. 10.1007/s12015-010-9113-x.
Tokarska-Schlattner M, Zaugg M, Zuppinger C, Wallimann T, Schlattner U: New insights into doxorubicin-induced cardiotoxicity: the critical role of cellular energetics. J Mol Cell Cardiol. 2006, 41: 389-405. 10.1016/j.yjmcc.2006.06.009.
Liu Z, Song XD, Xin Y, Wang XJ, Yu H, Bai YY, Liu JH, Zhang CN, Hui RT: Protective effect of chrysoeriol against doxorubicin-induced cardiotoxicity in vitro. Chin Med J (Engl). 2009, 122: 2652-2656.
Li K, Sung RY, Huang WZ, Yang M, Pong NH, Lee SM, Chan WY, Zhao H, To MY, Fok TE: Thrombopoietin protects against in vitro and in vivo cardiotoxicity induced by doxorubicin. Circulation. 2006, 113: 2211-2220. 10.1161/CIRCULATIONAHA.105.560250.
Zhang YW, Shi J, Li YJ, Wei L: Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch Immunol Ther Exp. 2009, 57: 435-445. 10.1007/s00005-009-0051-8.
Bast A, Haenen GR, Bruynzeel AM, Van der Vijgh WJ: Protection by flavonoids against anthracycline cardiotoxicity: from chemistry to clinical trials. Cardiovasc Toxicol. 2007, 7: 154-159. 10.1007/s12012-007-0018-0.
Hosseinzadeh L, Behravan J, Mosaffa F, Bahrami G, Bahrami A, Karimi G: Curcumin potentiates doxorubicin-induced apoptosis in H9c2 cardiac muscle cells through generation of reactive oxygen species. Food Chem Toxicol. 2011, 49: 1102-1109. 10.1016/j.fct.2011.01.021.
Kim DS, Woo ER, Chae SW, Ha KC, Lee GH, Hong ST, Kwon DY, Kim MS, Jung YK, Kim HM: Plantainoside D protects adriamycin-induced apoptosis in H9c2 cardiac muscle cells via the inhibition of ROS generation and NF-κB activation. Life Sci. 2007, 80: 314-323. 10.1016/j.lfs.2006.09.019.
Wang H, Joseph JA: Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med. 1999, 27: 612-616. 10.1016/S0891-5849(99)00107-0.
Eltze T, Boer R, Wagner T, Weinbrenner S, McDonald MC, Thiemermann C, Bürkle A, Klein T: Imidazoquinolinone, imidazopyridine, and isoquinolindione derivatives as novel and potent inhibitors of the poly(ADP-ribose) polymerase (PARP): a comparison with standard PARP inhibitors. Mol Pharmacol. 2008, 74: 1587-1598. 10.1124/mol.108.048751.
Aggeli IK, Beis I, Gaitanaki C: Oxidative stress and calpain inhibition induce alpha B-crystallin phosphorylation via p38-MAPK and calcium signalling pathways in H9c2 cells. Cell Signal. 2008, 20: 1292-1302. 10.1016/j.cellsig.2008.02.019.
Sugioka R, Shimizu S, Funatsu T, Tamagawa H, Sawa Y, Kawakami T, Tsujimoto Y: BH4-domain peptide from Bcl-xL exerts anti-apoptotic activity in vivo. Oncogene. 2003, 22: 8432-8440. 10.1038/sj.onc.1207180.
Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, Kotamraju S: Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol Cell Biochem. 2002, 234/235: 119-124. 10.1023/A:1015976430790.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1472-6882/12/138/prepub
Acknowledgments
This work was supported by the Chi Mei Medical Center, Taiwan, R.O.C. (99CM-TMU-11), and the National Science Council, Taiwan, R.O.C. (NSC99-2320-B-038-004-MY2).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WCW and YHU contributed equally to this work. WCW, YHU and CMH participated in the design and coordination of the study, carried out the analyses and wrote the manuscript. MLC, KPC and JSL helped to operate the confocal microscope and helped to draft the manuscript. WYY, KCL and CSC helped to analyze data for statistical analysis. CMH participated in the data interpretation and manuscript preparation. All authors read and approved the final manuscript.
Wen-Ching Wang, Yih-Huei Uen contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, WC., Uen, YH., Chang, ML. et al. Protective effect of guggulsterone against cardiomyocyte injury induced by doxorubicin in vitro. BMC Complement Altern Med 12, 138 (2012). https://doi.org/10.1186/1472-6882-12-138
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6882-12-138