
Abstract
Background
The use of pretargeting technology for cancer imaging and treatment has made significant progress in the last few years. This approach takes advantage of the fact that biotin binds strongly to proteins avidin and streptavidin. Thus, a non-toxic tumor cell specific antibody is conjugated with avidin/streptavidin, and is administered to patients. After the antibody binds to tumor cells (usually 24–48 h); a clearing agent is given to remove the residual circulating antibodies in blood. Lastly, a toxic biotin-radioisotope conjugate is administered. Due to the small size of the biotin-radioisotope molecule and tight binding between biotin and avidin/streptavidin, the biotin-radioisotope rapidly binds to tumor cells with high specificity. CC-1065 (1) is one of a few classes of extremely potent antitumor agents, and a biotinalyted CBI-bearing CC-1065 analogue is a promising candidate to be used in the pretargeting technology to treat cancer.
Results
A biotinalyted CBI-bearing CC-1065 analogue, 6, was synthesized. The IC50 of 6 was 0.7 nM against U937 cells. Compound 6 caused apototsis of U937 cells.
Conclusions
For the first time, a biotinalyted CBI-bearing CC-1065 analogue, 6, was synthesized. The biotinylated 6 can serve as a model compound to explore the usefulness of non-radioactive small molecule anticancer drugs in the pretargeting strategy for cancer imaging and therapy.
Similar content being viewed by others


Background
Anticancer drugs generally act on metabolically active or rapidly proliferating cells, and cannot distinguish between cancer and normal cells; thus, toxicities to normal cells limit the dose of drugs that can be given to patients. Therefore, much research has focused on development of more specific therapeutic strategies to reduce toxicity to normal cells. One of these strategies is monoclonal antibody-directed pretargeting technology. The use of pretargeting technology for cancer imaging and treatment has made significant progress in the last few years [1–6]. This approach takes advantage of the fact that biotin binds strongly to proteins avidin and streptavidin [7, 8]. Thus, a non-toxic tumor cell specific antibody is conjugated with avidin/streptavidin, and is administered to patients. After the antibody binds to tumor cells (usually 24–48 h); a clearing agent is given to remove the residual circulating antibodies in blood. Lastly, a toxic biotin-radioisotope conjugate is administered. Due to the small size of the biotin-radioisotope molecule and tight binding between biotin and avidin/streptavidin, the biotin-radioisotope rapidly binds to tumor cells with high specificity. This approach decouples the lengthy antibody to target process and the administration of a toxic moiety, but takes full advantage of both the high target specificity of the antibody and the favorable pharmacokinetics of a small toxic molecule. While progress using radioisotopes in this approach has been achieved, few reports on using a potent non-radioactive small molecule anticancer drug for this purpose have been seen. A potent non-radioactive small molecule drug for this approach is potentially better than a radioisotope because the latter is much more difficult to handle than a non-radioactive chemical agent.
CC-1065 (1) is one of a few classes of extremely potent antitumor agents [9–12]. It binds to double-stranded B-DNA within the minor groove with the sequence preference for 5'-d(A/GNTTA)-3' and 5'-d(AAAAA)-3', and alkylates the N3 position of the 3'-adenine with its left-hand CPI segment [13–15]. CC-1065 also inhibits gene transcription by interfering with binding of the TATA box binding protein to its target DNA [16]. Despite its high potency and broad spectrum of antitumor activity, CC-1065 cannot be used in humans because it causes delayed death in experimental animals [17]. To pursue compounds retaining the potent antitumor activity but devoid of the toxic side effects of the parent compound, many CC-1065 analogues have been synthesized and tested [18–26].
Because of the high potency, broad spectrum of antitumor activity, and novel mechanism of action, the CC-1065 class of compounds has the potential to be useful in the pretargeting approach. Herein, we report synthesis and preliminary cytotoxicity study of a CBI-biotin CC-1065 analogue designed as a model compound to explore its use in the pretargeting approach for cancer imaging and therapy.
Results and discussion
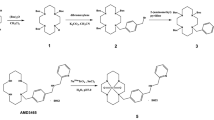
Target compound 6 was synthesized using two methods. The synthesis using Method A is illustrated in Scheme 1. Alternatively, 6 was also synthesized using Method B (Scheme 2).
Using Method A, a radioisotope-labeled N-hydroxysuccinimidobiotin can be conveniently added at the last step to synthesize radioisotope-labeled materials for metabolic and other biological studies. Because of the lengthy process and low overall yield in the synthesis of starting material 2, Method B is more economical for large-scale synthesis.
The cytotoxicity of 5, 6 and doxorubicin, used as a positive control, were tested against U937 leukemia cells, and the results were presented in Table 1. The biotinylated 6 (IC50: 0.7 nM) is approximately 7-fold less toxic than its precursor 5 (IC50: 0.09 nM). However, 6 is still much more potent than doxorubicin (IC50: 100 nM). As observed for other CC-1065 class of compounds [26, 28, 29], 6 caused DNA fragmentation and the cells died by apoptosis (data not shown).
Conclusions
The preliminary in vitro studies suggest that a biotin moiety can be incorporated into a CBI-bearing CC-1065 analogue to produce a potent biotinylated agent. When a biotin-nuclide conjugate is used, the conjugate does not need to be internalized to destroy the tumor cells. However, for a biotinylated CC-1065 analogue to work in this pretargeting strategy, the biotinylated drug must first bind to the avidin/streptavidin-antibody conjugate on the tumor cell surface. The drug must then gain entry to the cell to act by binding to DNA. Whether compound 6 can be internalized, after binding to the avidin/streptavidin-antibody conjugate on the tumor cell surface, is unclear at the present time. We will report experimental results addressing this question in due course. Nevertheless, we think that the biotinylated 6 can serve as a model compound to explore the usefulness of non-radioactive small molecule anticancer drugs in the pretargeting strategy for cancer imaging and therapy.
Materials and methods
Method A
3-[(5-Amino-1H-indol-2'-yl)carbonyl]-1-(chlorom-ethyl)-5-hydroxy-1, 2-dihydro-3H-benz [e]indole (5)
A solution of CBI [27], 2, (15 mg, 76 μmol) in 3 mL of ethyl acetate saturated with anhydrous hydrogen chloride was stirred at room temperature for 30 min in the dark. The suspension was concentrated in vacuo to give intermediate 3. Without further purification, the latter was dissolved in DMF (0.5 mL) and treated with 5-nitroindole-2-carboxylic acid (18 mg, 87 μmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDCI, 43 mg, 225 μmol). The reaction mixture was stirred at room temperature overnight and purified by thin layer chromatography eluting with 50% hexane in ethyl acetate to give 4. Without further purification, the latter was dissolved in a solution containing DMF (0.5 mL) and ethyl acetate (5 mL). Pd/C (10%, 10 mg) was added and the reaction mixture was hydrogenated for 1 h at ambient temperature under 1 atm pressure. The product was filtered, and the filter cake was washed with methanol (20 mL). The solvent was removed in vacuo, and ether was added. The solid was filtered, and washed to afford 5 (13 mg, 44% yield from 2). mp>300°C. 1H NMR (DMF-d7, ppm): 11.16 (brs, 1 H, NH), 10.46 (s, 1 H, OH), 8.24–6.81 (m, 9 H, Ar-H), 4.84–4.79 (dd, 1 H, J = 9.3, 11.1 Hz, N HH), 4.71–4.67 (m, 3 H, NCH H, NH2), 4.31–4.21 (m, 1 H, CH2ClC HCH2N), 4.18–4.12 (dd, 1 H, J = 1.0, 18.7 Hz, ClC HH), 3.93–3.88 (dd, 1 H, J = 7.9, 11.2 Hz, ClCH H). HRMS calcd for C22H18ClN3O2 391.1088 found 391.1068.
3-[5-(Biotin-amino)-1H-indol-2'-yl]carbonyl]-1-(chloromethyl)-5-hydroxy-1, 2-dihydro-3H-benz [e]-indole(6)
To a solution of 5 (2 mg, 5.1 μmol) in DMF (0.2 mL) was added N-hydroxysuccinimidobiotin (5.2 mg, 15.3 μmol), and the solution was stirred for 48 h at ambient temperature. The product was purified by thin layer chromatography eluting with ethyl acetate and methanol (4/1, v/v) to give 6 (1.5 mg, 48%) as a gray solid. 1H NMR (DMF-d7, ppm): 11.56 (brs, 1 H, NH), 10.55 (s, 1 H, OH), 9.91 (s, 1 H, NH), 8.25-7.21 (m, 9 H, Ar-H), 6.32 (s, 1 H, biotin NH), 6.24 (s, 1 H, biotin NH), 4.90-4.85 (t, 1 H, J = 11.0 Hz, N HH), 4.74-4.70 (dd, 1 H, J = 2.0, 11.0 Hz, NH H), 4.49-4.45 (m, 1 H, biotin H), 4.32-4.28 (m, 2 H, ClCH2C HCH2, biotin H), 4.14-4.10 (dd, 1 H, J= 3.6, 11.1 Hz, C HHCl), 3.96-3.91 (dd, 1 H, J = 7.8, 11.1 Hz, CH HCl), 3.20-3.27 (m, 1 H, biotin H), 2.45-2.41 (t, 2 H, J = 7.7 Hz, COCH2), 1.8-1.5 (m, 6 H, CO(CH2)3). HRMS calcd for C32H32ClN5O4S (M + Na+) 640.1761, found 640.1759.
Method B
5-(Biotin-amino)indole-2-carboxylic acid (9)
To a solution of 7 (60 mg, 294 μmol) in DMF (1 mL) was added N-hydroxysuccinimidobiotin (110 mg, 323 μmol) and the solution was stirred overnight at room temperature. The product was purified by flash chromatography eluting first with ethyl acetate followed by acetone. The solvent was removed in vacuo. Without further purification, methanol (5 mL) was added followed by 1 N NaOH (2 mL). The reaction mixture was stirred overnight at room temperature. The precipitate was filtered, and the filtrate was neutralized using 20% HCl. The precipitate was filtered, and the filter cake was washed with water. The product was dried in air to afford 49 mg (41% yield from 7) of gray solid. 1H NMR (DMSO-d6, ppm): 12.55 (brs, 1 H, COOH), 11.60 (s, 1 H, NH), 9.70 (s, 1 H, NH), 7.98-7.02 (m, 4 H, Ar-H), 6.39 (s, 1 H, biotin NH), 6.31 (s, 1 H, biotin NH), 4.33-4.29 (t, 1 H, J = 7.5 Hz, C4'-H), 4.17-4.13 (m, 1 H, C8'-H), 3.14-3.11 (m, 1 H, C5'-H), 2.85-2.81 (dd, 1 H, J = 5.2, 12.7 Hz, C7'-HH), 2.60-2.57 (d, 1 H, J = 12.1 Hz, C7'-H H), 2.32-2.28 (t, 1 H, J = 7.4 Hz, CH2CO), 1.70-1.30 (m, 6 H, (CH2)3CH2CO). HRMS calcd for C19H23N4O4S (M + H+) 403.1440 found 403.143 5.
To a solution of 3 (from 15 mg of CBI) in DMF (1 mL) was added 9 (30 mg, 73 μmol) and EDCI (44 mg, 230 μmol) sequentially, and the reaction mixture was stirred overnight at room temperature. The product was purified by thin layer chromatography eluting first with ethyl acetate followed by ethyl acetate and methanol (4/1, v/v). The solvent was removed in vacuo, and ether was added. The solid was filtered, and washed with ether to give 17 mg (37% yield) of 6, whose NMR spectrum is identical to the product made using Method A.

Structure of CC-1065

Synthesis of 6 (Method A)

Synthesis of 6 (Method B)
References
Hnatowich DJ, Virzi F, Rusckowski M: Investigations of avidin and biotin for imaging applications. J Nucl Med. 1987, 28: 1294-1302.
Paganelli G, Pervez S, Siccardi AG, Rowlinson G, Deleide G, Chiolerio F, Malcovati M, Scassellati GA, Epenetos AA: Intraperitoneal radiolocalization of tumors pre-targeted by biotinylated monoclonal antibodies. Int J Cancer. 1990, 45: 1184-1189.
Axworthy DB, Fritaberg AR, Hylarides MD, Mallet R, Theodore LJ, Gustavson LM, Su FM, Beamuier PL, Reno JM: Durable complete remissions of breast, lung, and colon tumor xenografts with a single dose of pretargeted Y-90 in a mouse model. J Nucl Med. 1995, 36: 217P-
Stoldt HS, Aftab F, Chinol M, Paganelli G, Luca F, Testori A, Geraghty JG: Pretargeting strategies for radio-immunoguided tumour localisation and therapy. Eur J Cancer. 1997, 33: 186-192. 10.1016/S0959-8049(96)00477-7.
Knox SJ, Goris ML, Tempero M, Weiden PL, Gentner L, Breitz H, Adams GP, Axworthy D, Gaffigan S, Bryan K, Fisher DR, Colcher D, Horak ID, Weiner LM: Phase II trial of Yttrium-90-DOTA-biotin pretargeted by NR-LU-10 antibody/streptavidin in patients with metastatic colon cancer. Clin Cancer Res. 2000, 6: 406-414.
Gautherot E, Rouvier E, Daniel L, Loucif E, Bouhou J, Manetti C, Martin M, Le Doussal JM, Barbet J: Pretargeted radioimmunotherapy of human colorectal xenografts with bispecific antibody and 1311-labeled bivalent hapten. J Nucl Med. 2000, 41: 480-487.
Green NM: Avidin. Adv Protein Chem. 1975, 29: 85-133.
Green NM: Avidin and streptavidin. Methods Enzymol. 1990, 184: 51-67.
Hanka LJ, Dietz A, Gerpheide SA, Kuentzel SL, Martin DG: CC-1065 (NSC-218223), A new antitumor antibiotic. Production, in vitro biological activity, microbiological assays, and taxonomy of the producing microorganisms. J Antibiot. 1978, 31: 1211-1217.
Martin DG, Chidester CG, Duchamp DJ, Mizsak SA: Structure of CC-1065 (NSC-218223), a new antitumor antibiotic. J Antibiot. 1980, 33: 902-903.
Martin DG, Biles C, Gerpheide SA, Hanka LJ, Kroeger WC, McGovern JP, Mizsk SA, Neil GL, Stewart JC, Visser J: CC-1065 (NSC-218223), a potent new antitumor agent, improved production and isolation, characterization and antitumor activity. J Antibiot. 1981, 34: 1119-1125.
Bhuyan BK, Newell KA, Crampton SL, Von Hoff DD: CC-1065 (NSC-218223), a most potent antitumor agent: Kinetics of inhibition of growth, DNA synthesis and cell survival. Cancer Res. 1982, 42: 3532-3537.
Swenson DH., Li LH, Hurley LH, Rokem JS, Petzold GL, Dayton BD, Wallace TL, Lin AH, Krueger WK: Mechanism of interaction of CC-1065 (NSC-218223) with DNA. Cancer Res. 1982, 42: 2821-2828.
Hurley LH, Reynolds VL, Swenson DH, Petzold GL, Scahill TA: Reaction of the antitumor antibiotics CC-1065 with DNA: Structure of a DNA adduct with DNA sequence specificity. Science. 1984, 226: 843-844.
Reynolds VL, Molineaux IJ, Kaplan DJ, Swenson DH, Hurley LH: Reaction of antitumor antibiotic CC-1065 with DNA. Location of the site of thermally induced strand breakage and analysis of DNA sequence specificity. Biochemistry. 1985, 27: 6228-6237.
Chiang SY, Welch J, Rauscher III FJ, Beerman TA: Effects of minor groove binding drugs on the interaction of TATA box binding protein and TFIIA with DNA. Biochemistry. 1994, 33: 7033-7040.
McGovren JP, Clarke GL, Pratt EA, DeKoning TF: Preliminary toxicity studies with the DNA-binding antibiotic CC-1065. J Antibiot. 1984, 37: 63-70.
Aristoff PA: CC-1065 Analogs: Sequence Specific DNA-alkylating Antitumor Agents. Adv Med Chem. 1993, 2: 67-110. (b) Boger DL, Boyce C. W, Garbaccio RM, Goldberg JA:CC-1065 and the Duocarmycins: Synthetic Studies.Chem Rev 1997, 97:787-820, and references cited
Wang Y, Wright SC, Larrick JW: DNA-binding indole derivatives, their prodrugs and immunoconjugates,. U. S. Patent US 5,843,937,. 1998
Boger DL, Santillán A, Searcey M, Jin Q: Critical role of the linking amide in CC-1065 and the duocarmycins: implications on the source of DNA alkylation catalysis. J Am Chem Soc. 1998, 120: 11554-11557. 10.1021/ja9818093.
Fukuda Y, Seto S, Furuta H, Ebisu H, Oomori Y, Terashima S: The novel cyclopropapyrroloindole (CPI) bis-alkylators bearing 3,3'-(1,4-phenylene)diacryloyl group as a linker. Bioorg Med Chem Lett. 1998, 8: 2003-2004. 10.1016/S0960-894X(98)00346-1.
Boger DL, Turnbull P: Synthesis and evaluation of a carbocyclic analogue of the CC-1065 and duocarmycin alkylation subunit: Role of the vinylogous amide and implications on DNA alkylation catalysis. J Org Chem. 1998, 63: 8004-8011. 10.1021/jo981698q.
Boger DL, Santillán A, Searcey M, Jin Q: Synthesis and evaluation of duocarmycin and CC-1065 analogues containing modifications in the subunit linking amide. J Org Chem. 1999, 64: 5241-5244. 10.1021/jo990452y.
Milbank JBJ, Tercel M, Atwell GJ, Wilson WR, Hogg A, Denny WA: Synthesis of 1-substituted 3-(chloromethyl)-6-aminoindoline (6-amino-seco-CI) DNA minor groove alkylating agents and structure-activity relationships for their cytotoxicity. J Med Chem. 1999, 42: 649-658. 10.1021/jm980545s.
Boger DL, Stauffer F, Hedrick MP: Substituent effects within the DNA binding subunit of CBI analogues of the duocarmycins and CC-1065. Bioorg Med Chem Lett. 2001, 11: 2021-2024. 10.1016/S0960-894X(01)00372-9.
Wang Y, Yuan H, Ye W, Wright SC, Wang H, Larrick JW: Synthesis and Preliminary Biological Evaluations of CC-1065 Analogs: Effects of Different Linkers and Terminal Amides on Biological Activity. J Med Chem. 2000, 43: 1541-1549. 10.1021/jm990514c.
Aristoff PA, Johnson PD: Synthesis of CBI-PDE-I-dimer, the benzannelated analogue of CC-1065. J Org Chem. 1992, 57: 6234-6239.
Wrasidlo W, Johnson DS, Boger DL: Induction of endonucleolytic DNA fragmentation and apoptosis by the duocarmycins. Bioorg Med Chem. 1994, 4: 631-636. 10.1016/S0960-894X(01)80168-2.
Wright SC, Schellenberger U, Wang H, Wang Y, Kinder DH: Chemotherapeutic drug activation of the AP24 protease in apoptosis: Requirement for caspase 3-like proteases. Biochem Biophys Res Commun. 1998, 245: 797-803. 10.1006/bbrc.1998.8508.
Acknowledgment
We thank Jolande Murray for help with the manuscript. This work was supported in part by a grant from the National Institutes of Health (CA79357-01 to Y.W).
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Wang, Y., Yuan, H., Wright, S.C. et al. Synthesis and cytotoxicity of a biotinylated CC-1065 analogue. BMC Chem Biol 2, 1 (2002). https://doi.org/10.1186/1472-6769-2-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6769-2-1