
Abstract
Background
Primary hepatocytes, one of the most widely used cell types for toxicological studies, have a very limited life span and must be freshly derived from mice or even humans. Attempts to use stable cell lines maintaining the enzymatic pattern of liver cells have been so far unsatisfactory. Stress proteins (heat shock proteins, HSPs) have been proposed as general markers of cellular injury and their use for environmental monitoring has been suggested. The aim of this work is to develop a bi-transgenic hepatocyte cell line in order to evaluate the ability of various organic and inorganic chemicals to induce the expression of the HSP70 driven reporter gene.
We previously described transgenic mice (Hsp70/hGH) secreting high levels of human Growth Hormone (hGH) following exposure to toxic compounds in vivo and in vitro in primary cultures derived from different organs. In addition, we also reported another transgenic model (AT/cytoMet) allowing the reproducible immortalization of untransformed hepatocytes retaining in vitro complex liver functions.
Results
The transgenic mouse line Hsp70/hGH was crossed with the AT/cytoMet transgenic strain permitting the reproducible immortalization of untransformed hepatocytes. From double transgenic animals we derived several stable hepatic cell lines (MMH-GH) which showed a highly-differentiated phenotype as judged from the retention of epithelial cell polarity and the profile of gene expression, including hepatocyte-enriched transcription factors and detoxifying enzymes. In these cell lines, stresses induced by exposure to inorganic [Sodium Arsenite (NaAsO2) and Cadmium Chloride (CdCl2)], and organic [Benzo(a)Pyrene (BaP), PentaChloroPhenol (PCP), TetraChloroHydroQuinone (TCHQ), 1-Chloro-2,4-DiNitro-Benzene (CDNB)] compounds, specifically induced hGH release in the culture medium.
Conclusions
MMH-GH, an innovative model to evaluate the toxic potential of chemical and physical xenobiotics, provides a simple biological system that may reduce the need for animal experimentation and/or continuously deriving fresh hepatocytes.
Similar content being viewed by others


Background
The identification of chemical and physical agents potentially hazardous for human health is a great challenge involving a great deal of time and money, since inexpensive high-throughput assays are not yet available. In addition, research involving living animals conflicts with the personal beliefs of a large strata of the population.
Transgenic technology was proposed to address the issue of genotoxicity and toxicology [1–5]. We previously described transgenic mice that bear the human growth hormone (hGH) as a reporter gene whose expression is driven by the regulatory sequences of the heat shock protein 70 gene (hsp70/hGH mice) [4]. In the blood of these mice, high levels of hGH were specifically detected after administration of known toxic, non-genotoxic inorganic compounds, such as arsenic and cadmium. In addition, we showed that hsp70/hGH transgenic-derived primary cells, including hepatocytes, specifically release hGH in the culture medium after exposure to toxic inorganic compounds.
Cultured mammalian hepatocytes retaining differentiated hepatic functions would be greatly useful in toxicology. However, primary hepatocytes have a very limited life span and must be freshly derived from mice or even humans. Various attempts to use transformed hepatocyte cell lines or to genetically engineer primary hepatocytes for toxicological studies have been unsatisfactory, since they rapidly modify their repertoire of tissue-specific gene expression including metabolising enzymes [6–9]. In this regard, a major breakthrough came from the discovery that the liver-specific transgenic expression of a truncated form of the hepatocyte growth factor receptor (cytoMET) under the control of the α1-antitrypsin (AT) promoter reproducibly permits immortalization of untransformed and highly-differentiated hepatocyte cell lines (MMH) [10, 11].
Based on these considerations, we crossed hps70/hGH with AT-cytoMET mice, obtaining double transgenic mice and from them stable hepatocyte cell lines (MMH-GH). The results shown in the present paper indicate that these MMH-GH cell lines, which specifically expressed the reporter hGH gene after exposure to inorganic and organic compounds with known toxicity, may represent a useful tool in toxicological studies.
Results
Several stable cell lines (MMH-GH1, MMH-GH2, MMH-GH5, MMH-GH9, MMH-GH10, MMH-GH11) were derived from liver explants of double transgenic 18 days post coitum embryos as described in the Methods section. As expected, all the 6 lines tested failed to grow in soft agar and to form tumors in nude mice (data not shown), in accordance with previous observations showing that cytoMET transgenic expression renders hepatocytes permissive for immortalization without cellular transformation [10].
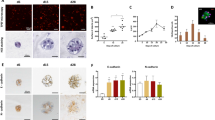
To gather further evidence on the differentiated hepatic phenotype of these lines, we documented epithelial polarity and liver specific gene expression. Immunofluorescence analysis showed in Figure 1 highlights the expression and the cellular localization of polarity markers such as ZO-1 and the epithelium-specific adhesion molecule E-cadherin in two different MMH-GH clones and as positive and negative controls, in a well polarized and previously described MMH line (MMHE14) and in a undifferentiated, non polarized and transformed MMH line (MMHE14/Myc) respectively. Both proteins are visible as a fine network on the membrane areas of cell-cell contact, indicating that the cells of both lines retain a simple epithelial polarity. Moreover, the staining for cytokeratins reveals an abundant network of these intermediate filaments, outlining the epithelial growth habit of the cells.

MMH-GHs maintain a highly-differentiated phenotype at morphological and molecular levels. Panel A shows the immunofluorescence analysis of E-Cadherin, ZO-1 and cytokeratins expression and localization in MMH-GH clones, in a well polarized MMHline (MMHE14) and in a transformed, undifferentiated MMH line(MMH/Myc). Phase-contrast images and microphotographs of E-Cadherin immunostaining represent the same fields. Panel B shows the RT-PCR analysis of RNA extracted from an MMH clone, from adult liver as control and from two different MMH-GH cell lines (MMH-GH5 and MMH-GH9). The amplified fragments are shown in the left side of the panel, while in the right side the Southern Blot analysis of the same amplified sequences performed with oligonucleotides used as internal probes is shown. Normalization of RT-PCR was obtained by amplification of beta-actin cDNA. The genes considered are: liver-enriched transcription factors (HNF1a, HNF1b, HNF4), drug and carcinogen-activating and -detoxifying enzymes (Glutamine Synthethase, GLNS; UDP-glucuronosyl-transferase, UDP-GT; Cytochrome P450 1A1, Cyp1A1; Epoxide hydrolase, Eph) and gluconeogenesis supporting enzymes (Lactate dehydrogenase, LDH; Glutamate-oxaloacetate-transaminase 1, Got1; Glutamate-oxaloacetate-transaminase 2, Got2). Oligonucleotides used as forward and reverse primers for PCR and as internal probes in Southern Blot analysis are listed in Additional file: 2, together with the NCBI accession numbers of the sequences from which they were derived.
In order to explore their application in toxicological studies we analyzed by RT-PCR some key enzymes involved in the detoxifying machinery, highlighting a coherent pattern of gene expression. As shown in Figure 1b, liver-enriched transcription factors (HNFs), phase I (drug and carcinogen-activating), phase II (detoxifying) and gluconeogenesis supporting enzymes are expressed in MMH-GH lines; similar results were obtained at early (3) and late (23) passages after the first thawing.
Six independent clones of MMH-GH cells were then tested for their ability to respond to in vitro toxic stimulation. Cells were treated with NaAsO2 and CdCl2 at concentrations ranging from 10-5 to 10-4 M for 5 or 24 h (data not shown). These conditions were chosen according to results previously obtained with hsp70/hGH transgenic primary hepatocytes [4]. The MMH-GH5 clone, showing a basal level of secreted hGH below the detection limit and a high response to treatments, was selected for further studies.
The hGH secretion and the cellular viability of MMH-GH5 and PHGH cells from hsp70/hGH mice in response to the toxic inorganic compounds NaAsO2 and CdCl2 were evaluated (Fig 2). While the response to NaAsO2 was comparable at each concentration tested in both cell lines, after exposure to CdCl2 we observed differing behaviour in the two models. PHGH showed an inverse correlation between cadmium concentration and hGH release while MMH-GH5 showed a linear response (panel A). This result is based on the different viability of the two cell populations (panel B). Notably, the MMH-GH5 cells showed a higher cell viability after exposure to high doses of toxic inorganic compounds, which results in a dose-response linearity of the hGH release.

MMH-GH5 and PHGH response to toxic inorganic compounds Panel A. Results represent Mean ± SE of hGH levels secreted by PHGH and MMH-GH5 cells after a 5 hour treatment with different concentrations of NaAsO2 (As1: 10-5 M, As2: 5 × 10-4 M, As3: 10-4 M) and CdCl2 (Cd1: 10-5 M, Cd2: 5 × 10-4 M, Cd3: 10-4 M). Panel B. Mean ± SE of viability of PHGH and MMH-GH5 at the indicated treatment conditions as determined by the Trypan blue dye exclusion method. These results were obtained from 6 independent experiments with treatments carried out in triplicate.
To test whether primary and immortalized hepatocytes were able to respond also to toxic organic compounds, we tested four known toxic agents: Benzo(a)pyrene (BaP), considered mainly a genotoxic agent, PentaChloroPhenol (PCP), TetraChloroHydroQuinone (TCHQ, the major in vivo metabolite of PCP) and 1-Chloro-2,4-DiNitro-Benzene (CDNB). PHGH and MMH-GH5 cells were exposed to two different concentrations of these compounds for 1 and 5 hours. hGH levels were measured in the medium either immediately after the treatment or after 24 hours of recovery with control medium.
As shown in Additional file: 1, PHGH cells, while capable of responding to TCHQ and CDNB, did not show elevated hGH levels in response to BaP and PCP, due to the cytotoxicity of these compounds, as indicated by the low percentage of residual viable cells. On the other hand, MMH-GH5, showing a higher viability, responded to all four compounds, thus providing a suitable cell-based assay also for these compounds. It is possible that this different behaviour is due to the fact that stable cell lines better adapt to culture conditions than primary cells and are more resistant to toxic insults.
Discussion
In spite of many assays developed in lower organisms, mammalian hepatocytes still represent a compulsory step in the evaluation of toxic compounds because they carry out modifications leading to the production of various metabolites, which are the ultimate causes of toxicity [12, 13]. However, their use so far has been restricted to two models, primary cultured hepatocytes and hepatoma cell lines both of which have several drawbacks. Primary hepatocytes, used as cultures or tissue slices must be continuously derived from living animals or even humans, their functional activities decline rapidly after a few days under conventional culture conditions and their response is dependent on many variables, while hepatoma cell lines lose features of the hepatocyte, such as drug- and toxic-activating enzymes [13, 14]. Engineered immortalized human cell lines have also been developed, with different results [6–9].
To overcome these limitations, we have developed new bi-transgenic immortalized hepatocyte cell lines, named MMH-GH. As detailed in previous reports, the transgenic expression of cytoMET, a truncated form of the c-MET oncogene, permits the immortalization of hepatocytes (MMH) that retain and perform in vitro complex liver functions [10]. In the work reported here, we report that these cells represent a novel tool that can be used in toxicology studies. As shown above, each inorganic and organic toxic compound tested so far caused specific hGH release, albeit with different kinetics in agreement with the results obtained by Ait-Aissa et al [15], who showed that hsp70 induction is different depending on the classes of compounds. Notably, for the correct interpretation of these tests, both hGH levels and cellular viability parameters must be taken into account.
Conclusions
We believe that MMH-GH cell lines provide a cheap, reproducible, rapid, reliable and ethically acceptable tool that has several innovative features. The major advantages are: 1 – the use of stable cell lines allows a drastic reduction of animals sacrificed for toxicology research; 2 – the cells are of the histotype most widely used for toxicological studies; 3 – the cells retain in culture the morphology, phenotype and functions mimicking hepatocytes in the living animal; 4 – this assay, based on clonal cell populations, has with respect to primary cultures greater potentiality for standardization; 5 – the presence of the reporter gene under the control of a stress-responsive promoter allows us to perform a quantitative, rapid and low-cost assay able to detect early cell damage.
The fact that our detection system is able to identify hsp70 promoter induction at low doses ([5]; Figure 2 and Additional file: 1) is an advantage over traditional hepatocyte culture systems which rely upon viability as the key end point. Standardization of an in vitro assay based on MMH-GH5 cells will allow a significant reduction in the number of animals sacrificed, a very sensitive issue shared by both public Institutions and pharmaceutical industries [16, 17]. Several clones can be derived from a small number of transgenic mice and can be used for a indefinite number of tests. Moreover, cellular clones with the best features may be selected, different species (rats or fish [18]) could be engineered, different promoters and different reporter genes could be used, as already suggested [5], to further exploit the potentiality of this approach. In addition, promoter elements for stress proteins of hsp genes with more restricted responses to stress could be applicable for further refining this system. Finally, by thoroughly investigating the metabolic pathways regulated by truncated cytoMet, human immortalized hepatocytes maintaining features of differentiated liver cells could be produced and used in toxicology studies.
Methods
Chemicals
NaAsO2 and CdCl2 were purchased from Merck (Darmstad, Germany); B(a)P, PCP, TCHQ, CDNB, were purchased from SIGMA (St Louis, MO). All the cell culture reagents were from SIGMA and all the plastic material from Falcon (San Jose, CA).
Isolation of primary hepatocytes
The day before the treatment, the parenchymal component of liver cells was isolated as previously described [4] and seeded in 24 well plates in William's E medium supplemented with 10% heat inactivated FCS and Pen/Strep. After 2 hours the medium was replaced to remove floating cells.
Derivation of hepatic stable cell lines and cell culture
The experimental protocol was approved by the Scientific Committee of the Istituto di Tecnologie Biomediche, CNR. The production and characterization of the two transgenic strains (hsp70/hGH and cytoMET) used in the present study have been previously described [4, 10, 11]. Homozygous mice from the two strains were crossed to generate double heterozygous animals. Animal care and sacrifice were performed according to the EU guidelines [19] and hepatocytes were obtained and cultured as previously described [4, 10]. Briefly, livers from 18 dpc bi-transgenic foetuses were dissected, mechanically dissociated and plated at high density on collagen I (Sigma) coated Nunc Petri dishes in RPMI 1640 containing 10% FCS, 50 ng/ml EGF, 30 ng/ml IGF II, 10 micrograms/ml insulin (Boehringer Mannheim) and antibiotics. The cultures were maintained without transfer for several weeks. Individual epithelial islands were slowly expanded in collagen I-coated dishes of increasing size, and frozen after around 25 cell generations. Passages are calculated from initial thawing.
The day before the treatment with chemicals, MMH-GH cells were seeded at 80% confluence in 12 well plates pre-coated with collagen in complete medium. The treatments were performed in the same medium without FCS.
Immunofluorescence
For indirect immunofluorescence staining, cells were grown on collagen I-coated glass cover slips, fixed by immersion in methanol/acetone (3:7, vol/vol) for 15 min at 20°C and then air dried. Coverslips were then incubated for 1 h at room temperature in humidified atmosphere with the primary antibody. After three washes with PBS, the coverslips were incubated with the secondary fluorochrome-conjugated antibody diluted in PBS plus 3% BSA, washed repeatedly with PBS, and mounted with 70% glycerol in PBS. The samples were analyzed under phase contrast and appropriate fluorescent light.
The antibodies were obtained and used as follows: mouse monoclonal anti-pan-Cytocheratin antibody from Sigma Chemical Co., diluted 1/100; rat monoclonal anti-zonula occludens (ZO)-1 antibody from Zymed diluted 1/100; mouse monoclonal anti-E-Cadherin antibody from Signal Transduction Laboratories (Lexington, KY), diluted 1/100; sheep anti-mouse Ig FITC linked whole antibody from Amersham International, used 1/100; rabbit polyclonal anti-rat IgG FITC conjugated antibody from Sigma Chemical Co., diluted 1/100.
The cell lines used as positive and negative controls of epithelial polarity were MMH line E14 described in [10] and the same cell line transduced with activated, transforming form of cMyc E14/Myc (unpublished) respectively.
RNA analysis by RT-PCR
Single-stranded cDNAs were obtained from reverse transcription of 1 microgram of total RNA extracted from MMH, MMH-GH5 and MMH-GH9 cells and wild type liver using oligo(dT)-primers and M-MLV reverse transcriptase (Promega, Madison, WI), in a final reaction volume of 25 microliters according to the manufacturer's conditions. The cDNAs were amplified by PCR according to the standard protocol using forward and reverse specific primers. The PCR products were electrophoresed on 1% agarose gels and then transferred to Quiabrene Nylon membrane (Quiagen, Germany) for hybridization. Blots were probed with internal 32P-labeled oligonucleotides. The amplified genes [20–23], the primers used for amplification and the internal probes are listed in Additional file: 2, together with their NCBI accession numbers.
In vitro treatments
All the stock solutions were dissolved at the final concentration in culture medium without FCS. NaAsO2, CdCl2 were dissolved in H2O at the concentration of 10-2 M as stock solution and then diluted to 10-5 (dose 1), 5 × 10-5 M (dose 2) and 10-4 M (dose 3).
B(a)P, PCP, TCHQ and CDNB were dissolved according to the manufacturer's instructions in different solvents at 5 mM concentration as stock solution and then diluted in culture medium. B(a)P was administered at 10 and 50 microM, CDNB at 5 and 50 microM, while PCP and TCHQ were administered at 50 and 100 microM concentrations. These concentrations were chosen on the basis of data previously reported by Ait-Aissa et al [15].
Each treatment was carried out for 1 and 5 hours, then the supernatant was removed, frozen at -20° and replaced with fresh medium without FCS for another 24 hours. Every treatment was carried out in duplicate and every experiment was repeated at least 5 times. In every experiment control treatments were included in which cells received only culture medium.
Viability
Cell viability was determined at the end of treatments by Trypan blue dye exclusion method, as previously described [4].
Determination of hGH release
The concentration of hGH released in the supernatant after the treatments was determined as previously described [4] with an ELISA test, (hGH ELISA detection kit, Roche). Each determination was carried out in duplicate. The results were expressed as pg of hGH/106 cells.
References
Gossen JA, Vijg J: Transgenic mice as model systems for studying gene mutations in vivo. Trends Genet. 1993, 9: 27-31. 10.1016/0168-9525(93)90069-T.
Tennant RW, Stasiewicz S, Mennear J, French JE, Spalding JW: Genetically altered mouse models for identifying carcinogens. IARC Sci Publ. 1999, 146: 123-150.
Amanuma K, Takeda H, Amanuma H, Aoki Y: Transgenic zebrafish for detecting mutations caused by compounds in aquatic environments. Nat Biotechnol. 2000, 18: 62-65. 10.1038/71938.
Sacco MG, Zecca L, Bagnasco L, Chiesa G, Parolini C, Bromley P, Cato EM, Roncucci R, Clerici LA, Vezzoni P: A transgenic mouse model for the detection of cellular stress induced by toxic inorganic compounds. Nat Biotechnol. 1997, 15: 1392-1397.
Cannon RE, Tennant RW: Shock toxicology with transgenics. Nat Biotechnol. 1997, 15: 1349-
Pfeifer AM, Cole KE, Smoot DT, Weston A, Groopman JD, Shields PG, Vignaud JM, Juillerat M, Lipsky MM, Trump BF, Lechner JF, Harris CC: Simian virus 40 large tumor antigen-immortalized normal human liver epithelial cells express hepatocyte characteristics and metabolize chemical carcinogens. Proc Natl Acad Sci U S A. 1993, 90: 5123-5127.
Roberts EA, Letarte M, Squire J, Yang S: Characterization of human hepatocyte lines derived from normal liver tissue. Hepatology. 1994, 19: 1390-1399. 10.1016/0270-9139(94)90233-X.
Kono Y, Yang S, Letarte M, Roberts EA: Establishment of a human hepatocyte line derived from primary culture in a collagen gel sandwich culture system. Exp Cell Res. 1995, 221: 478-485. 10.1006/excr.1995.1399.
Fukaya K, Asahi S, Nagamori S, Sakaguchi M, Gao C, Miyazaki M, Namba M: Establishment of a human hepatocyte line (OUMS-29) having CYP 1A1 and 1A2 activities from fetal liver tissue by transfection of SV40 LT. In Vitro Cell Dev Biol Anim. 2001, 37: 266-269. 10.1290/1071-2690(2001)037<0266:EOAHHL>2.0.CO;2.
Amicone L, Spagnoli FM, Spath G, Giordano S, Tommasini C, Bernardini S, De Luca V, Della Rocca C, Weiss MC, Comoglio PM, Tripodi M: Transgenic expression in the liver of truncated Met blocks apoptosis and permits immortalization of hepatocytes. EMBO J. 1997, 16: 495-503. 10.1093/emboj/16.3.495.
Spagnoli FM, Amicone L, Tripodi M, Weiss MC: Identification of a bipotential precursor cell in hepatic cell lines derived from transgenic mice expressing cyto-Met in the liver. J Cell Biol. 1998, 14: 1101-1112. 10.1083/jcb.143.4.1101.
Guengerich FP: In vitro techniques for studying drug metabolism. J Pharmacokinet Biopharm. 1996, 24: 521-533.
Davila JC, Rodriguez RJ, Melchert RB, Acosta D: Predictive value of in vitro model systems in toxicology. Annu Rev Pharmacol Toxicol. 1998, 38: 63-96. 10.1146/annurev.pharmtox.38.1.63.
Ferrini JB, Pichard L, Domergue J, Maurel P: Long-term primary cultures of adult human hepatocytes. Chem Biol Interact. 1997, 107: 31-45. 10.1016/S0009-2797(97)00072-0.
Ait-Aissa S, Porcher J, Arrigo A, Lambre C: Activation of the hsp70 promoter by environmental inorganic and organic chemicals: relationships with cytotoxicity and lipophilicity. Toxicology. 2000, 145: 147-157. 10.1016/S0300-483X(00)00145-1.
Goodman S: Race is on to find alternative to animal tests. Nature. 2002, 418: 116-10.1038/418116a.
Kling J: In vitro models for in vivo drug profiles. Nat Biotechnol. 1996, 14: 1655-1656.
Burkhart JG: A fish story. Nat Biotechnol. 2000, 18: 11-10.1038/71869.
European Convention for the protection of vertebrate animals used for experimental and other scientific purposes. Official Journal of the European Communities. 1999, [http://europa.eu.int/eur-lex/pri/en/oj/dat/1999/l_222/l_22219990824en00290037.pdf]
Frain M, Swart G, Monaci P, Nicosia A, Stampfli S, Frank R, Cortese R: The liver-specific transcription factor LF-B1 contains a highly diverged homeobox DNA binding domain. Cell. 1989, 59: 145-157. 10.1016/0092-8674(89)90877-5.
De Simone V, De Magistris L, Lazzaro D, Gerstner J, Monaci P, Nicosia A, Cortese R: LFB3, a heterodimer-forming homeoprotein of the LFB1 family, is expressed in specialized epithelia. EMBO J. 1991, 10: 1435-1443.
Sladek FM, Zhong WM, Lai E, Darnell JE: Liver enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990, 4: 2353-2365.
Gonzalez FJ: The study of xenobiotic-metabolizing enzymes and their role in toxicity in vivo using targeted gene disruption. Toxicol Lett Dec. 1998, 102–103: 161-166. 10.1016/S0378-4274(98)00302-6.
Acknowledgments
We thank Prof. R. Dulbecco and Prof. A. Albertini for their encouragement. The technical assistance of L. Susani and Claudio Cavallari is acknowledged. The financial supports of CIB (Consorzio Italiano Biotecnologie) to M.T., AIRC to M.G.S. and M.T., MIUR to M.G.S. and M.T., MIUR-FIRB (grant RBNE019J9W and RBNE01R4MJ-04) to P.V. and M.G.S. are gratefully acknowledged. This is manuscript no. 68 of the Genoma 2000/ITBA Project funded by CARIPLO.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
LA, DF and MT carried out the isolation and characterization of bi-transgenic MMH-GH cell lines. MGS, EMC and PV carried out the isolation of PHGH cells, all the in vitro treatments, the hGH ELISA assay and the analysis of the results. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Sacco, M.G., Amicone, L., Catò, E.M. et al. Cell-based assay for the detection of chemically induced cellular stress by immortalized untransformed transgenic hepatocytes. BMC Biotechnol 4, 5 (2004). https://doi.org/10.1186/1472-6750-4-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1472-6750-4-5