
Abstract
Background
Long-acting β2-adrenergic agonists (LABAs) are recommended in combination with inhaled corticosteroids (ICSs) for asthma management. Abediterol is a novel, selective, potent, once-daily LABA in development for treatment of asthma and chronic obstructive pulmonary disease. This study aimed to determine abediterol doses with similar peak bronchodilatory effect to salbutamol 400 μg, and duration of action compatible with once-daily dosing in patients with persistent, stable asthma.
Methods
This was a Phase II, randomized, double-blind, double-dummy, crossover, placebo-controlled, dose-ranging study (ClinicalTrials.gov NCT01425801) in 62 patients with mild-to-moderate asthma who were also receiving an ICS. Patients received single doses of abediterol 0.313, 0.625, 1.25, or 2.5 μg, salbutamol 400 μg, or placebo in the morning. Spirometry was performed up to 36 h post-dose; safety and tolerability were assessed throughout the study. The primary endpoint was change from baseline in peak forced expiratory volume in 1 s (FEV1). Additional endpoints included trough FEV1, normalized area under the FEV1 curve (FEV1 AUC) up to 24 h post-dose, and peak and trough forced vital capacity (FVC).
Results
Abediterol produced dose-dependent improvements in peak FEV1 from baseline compared with placebo, from 0.274 (95% CI 0.221, 0.327) to 0.405 L (95% CI 0.353, 0.458) for abediterol 0.313 to 2.5 μg, respectively (p < 0.0001 all doses). Abediterol 0.625, 1.25, and 2.5 μg had similar magnitude of peak FEV1 effect to salbutamol. Dose-dependent changes from baseline in trough FEV1 versus placebo were 0.219 (95% CI 0.136, 0.302) to 0.400 L (95% CI 0.317, 0.483) for abediterol 0.313 to 2.5 μg, respectively (p < 0.0001). All abediterol doses achieved significant improvements versus placebo in FEV1 AUC 0–6, 0–12, and 0–24 h, and peak and trough FVC (p < 0.05). Less than 10% of patients experienced treatment-related adverse events for each dose of abediterol; most were mild to moderate in intensity and the most common were headache and nasopharyngitis. There were no clinically relevant changes in heart rate.
Conclusions
Abediterol 0.625–2.5 μg provided dose-dependent, clinically and statistically significant bronchodilation versus placebo in patients with asthma, with a peak effect similar to salbutamol and duration of action compatible with once-daily dosing. All doses of abediterol were well tolerated.
Similar content being viewed by others


Background
The use of β2-adrenergic agonist bronchodilators in combination with inhaled corticosteroids (ICSs) to manage the symptoms of asthma is recommended by Global Initiative for Asthma (GINA) guidelines [1]. Currently licensed bronchodilators include salbutamol, a short-acting β2-adrenergic agonist (SABA) administered as required [2], salmeterol and formoterol, two long-acting β2-adrenergic agonists (LABAs) administered twice daily in combination with an ICS [3, 4], and the recently licensed vilanterol, a LABA administered once daily in fixed-dose combination with the ICS fluticasone furoate [5]. There is ongoing interest to develop once-daily medications to further simplify treatment regimens and improve patient compliance [6].
Abediterol is a novel, once-daily LABA in clinical development as a fixed-dose combination with an ICS for the treatment of asthma and chronic obstructive pulmonary disease (COPD) [7]. Abediterol displays high affinity for β2-adrenoceptors, plus a higher functional selectivity for β2-adrenoceptors over β1-adrenoceptors than formoterol, indacaterol, vilanterol and olodaterol in cellular models [7–9]. The potency of abediterol is also higher than salmeterol and indacaterol in isolated human bronchi and, in addition to these two LABAs, vilanterol and olodaterol in animal models [7, 10]. Abediterol has a rapid onset and prolonged duration of action [7, 10].
A Phase II clinical trial in patients with asthma provided results that were consistent with the preclinical data; abediterol resulted in significant improvements in lung function at 5 min post-dose that were greater than improvements seen with salmeterol, and significant bronchodilation was sustained up to 24 h post-dose [11]. These characteristics make abediterol an ideal candidate for once-daily dosing. Moreover, abediterol has a favorable cardiovascular safety and tolerability profile: in anesthetized dogs, abediterol had a lesser effect on heart rate compared with salmeterol, formoterol, and indacaterol at concentrations that produced comparable bronchodilation [7, 12]. In early phase clinical trials, doses of abediterol ≤10 μg were safe and well tolerated and did not demonstrate any clinically relevant effects on heart rate in healthy subjects [13], patients with asthma [14], or patients with COPD [15].
Drug regulatory authorities have concerns that excessively high LABA doses may cause unacceptable side effects [16]. The trough forced expiratory volume in 1 s (FEV1), measured 24 h after dosing, is an important endpoint to assess once-daily LABA efficacy [16]. The maximum effect achieved soon after dosing, otherwise known as the ‘peak’ effect, is also of interest for efficacy and safety reasons. A very high peak effect may indicate high levels of acute drug exposure that could predispose patients to β-agonist-mediated side effects, such as cardiovascular adverse events, when receiving chronic treatment [17]. Moreover, the ideal pharmacologic characteristic of a once-daily LABA is a low ‘peak to trough ratio’, ensuring relatively stable lung function throughout the day [18].
The peak effect of salbutamol is well characterized, and causes acute bronchodilation without safety concerns. The bronchodilator response following single-dose administration of salbutamol (4 x 100 μg) is commonly used as one of the diagnostic tests for asthma, as recommended by a joint European Respiratory Society (ERS)/American Thoracic Society (ATS) taskforce [19]. This effect is, therefore, a reasonable benchmark for a novel LABA to achieve and this degree of acute bronchodilation is considered clinically meaningful. A major regulatory agency recommended that the peak effects of abediterol should be compared to those of salbutamol to guide dose selection for further clinical trials in order to avoid a dose that induces a much greater degree of bronchodilation than is necessary for clinical effectiveness, which could increase the potential for safety concerns during chronic treatment.
We report a placebo-controlled, dose-ranging, crossover study comparing the acute bronchodilator effects of abediterol to salbutamol in patients with persistent stable asthma. The primary aim of the study was to investigate the peak bronchodilatory effect of abediterol versus placebo and to confirm it was in the same range as that of salbutamol. The full profile of lung function over 24 h, including the trough FEV1, was also measured. The goal of this study was to enable the selection of abediterol doses for future long-term clinical trials, based on a peak effect similar to that of salbutamol 400 μg metered dose inhaler (MDI), coupled with a trough effect compatible with a 24-h duration of action. This trial design was a specific recommendation by a major regulatory agency.
Methods
Study design
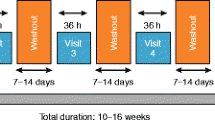
This was a Phase IIa, randomized, double-blind, double-dummy, 6-way crossover, single-dose, multicenter, dose-ranging study conducted between August 2011 and January 2012. The study was conducted in two sites in the UK and seven in Germany. The study protocol was approved by an independent ethics committee in each country (see Additional file 1) and complied with the Declaration of Helsinki and the International Conference on Harmonisation and Good Clinical Practice guidelines. Patients provided written, informed consent.
Following a screening visit and a 12–16 day run-in period to assess clinical stability, participants were randomized to 1 of 6 treatment sequences according to a balanced Williams design for crossover studies (1:1:1:1:1:1). Block randomization was performed by the Sponsor, using a computer-generated schedule to assign a treatment sequence to each patient randomization number. The block size and randomization schedule were not communicated to investigators. Patients received abediterol 0.313, 0.625, 1.25, or 2.5 μg, salbutamol 400 μg (administered as four inhalations with 100 μg per inhalation), or placebo (at 09:00 h ±1 h) at each visit, with a washout period of 7–14 days. A follow-up telephone call to assess safety was made approximately 2 weeks following the final treatment visit or premature discontinuation.
Abediterol and placebo to abediterol were administered via a multidose dry powder inhaler (Genuair®). Salbutamol and placebo to salbutamol were administered via a pressurized MDI (Ventolin™ Evohaler™). In order to preserve study blinding, independent, trained personnel prepared the medication, delivered the device to patients, and collected the device following inhalation.
Patients
Eligible participants were men or women aged 18–70 years with a clinical diagnosis of persistent asthma (according to GINA guidelines) for ≥6 months prior to screening. Eligible patients had an FEV1 >60% and ≤85% of the predicted normal value, with an FEV1 reversibility ≥12%, and an absolute increase in FEV1 of ≥200 mL over their baseline value following inhalation of salbutamol 400 μg at screening. Comparable FEV1 measurements were required at screening and pre-dose at each visit in order to ensure there were no carry-over effects from previous treatment periods (limit of variability ±20%). At enrollment, all patients were using only an ICS to control their asthma, and had been on a stable dose and regimen for at least 4 weeks prior to screening, up to the equivalent of 1600 μg/day of beclometasone dipropionate, and had not previously been exposed to abediterol.
Exclusion criteria included: a smoking history within the past 6 months or >10 pack-years; presence or history of relevant pulmonary disease or any other clinically relevant disease or abnormality as judged by the investigator; difficult-to-treat asthma; any respiratory tract infection ≤6 weeks prior to screening; hospitalization or emergency room treatment for asthma ≤3 months prior to screening; treatment with β2-antagonists; and positive laboratory tests for illicit drugs.
The use of anticholinergics, SABAs (other than salbutamol), LABAs, continuous oral or parenteral corticosteroids, methyl-xanthines, cromolyn sodium, nedocromil, leukotriene modifiers, anti-immunoglobulin E, β1-blocking agents, and other investigational drugs was prohibited during the study. Patients using prohibited medications underwent up to 6 weeks’ washout before the screening visit. Salbutamol pressurized MDI (100 μg/puff) was the only reliever medication permitted and was used on an as-needed basis and avoided 6 h prior to and during a treatment visit unless absolutely necessary.
Efficacy assessments
Pulmonary function tests were conducted at screening, pre-dose, and at 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, 14, 23, 24, and 36 h post-dose at each treatment visit using standardized spirometers at each investigational site (eResearchTechnology, Philadelphia, PA, USA). Prior to the first spirometry measure, the technician demonstrated the procedure and allowed the patient two practice attempts. At each study time point, the maneuvers were repeated until three technically adequate measurements were made according to ERS/ATS guidelines for acceptability and repeatability [20], with a maximum of eight attempts allowed.
Safety and tolerability assessments
Treatment-emergent adverse events (TEAEs), including serious adverse events, were recorded throughout the study and follow-up period. A physical examination was performed at screening and at 36 h post-dose following the last treatment period (Visit 6), and blood pressure and 12-lead electrocardiogram measures were made at screening, pre-dose, and at 0.75, 2, 6, 24 h post-dose for each treatment period, and 36 h post-dose at Visit 6. Clinical laboratory tests (blood chemistry, hematology, urinalysis, and a serum pregnancy test) were performed at screening and at 36 h post-dose at Visit 6. Blood glucose and serum potassium levels were assessed at screening, pre-dose, and at 4, 24 h post-dose for each treatment period, and 36 h post-dose at Visit 6.
Statistics
The primary efficacy endpoint was change from baseline in peak FEV1 (defined as the highest FEV1 value observed within 4 h of the administration of the investigational medicinal product [IMP]) on Day 1. Secondary endpoints included change from baseline in peak forced vital capacity (FVC) on Day 1, trough FEV1 (defined as the highest average of the maneuvers performed at 23–24 h post IMP administration) and FVC on Day 2, change from baseline in FEV1 and FVC at each time point up to 36 h post-dose, and change from baseline in normalized FEV1 area under the curve (AUC; defined as the area between baseline FEV1 and the FEV1 curve, from 0 to × hours divided by × hours) at 0–6, 0–12, and 0–24 h post-dose. In accordance with the GINA guidelines, a change in FEV1 of 200 mL was considered clinically relevant [1].
A population of 48 patients was necessary to achieve 80% power to detect a 150 mL change from baseline in peak FEV1 for any dose of abediterol compared with placebo. Assuming a 15% withdrawal rate, the protocol required at least 54 patients to be randomized.
All reported efficacy assessments were analyzed in the intention-to-treat population, defined as all randomized patients who took at least one dose of the IMP (placebo, abediterol, or salbutamol) and had at least one baseline and one post-dose FEV1 value for at least one treatment visit. Safety analyses are reported in the safety population, which consisted of all randomized patients who took at least one dose of the IMP. All statistical analyses were performed using Statistical Analysis System (SAS Institute Inc., Cary, NC, USA) version 9.1.3 software.
Efficacy variables were analyzed using an analysis of covariance model for crossover designs, with fixed-effect factors of sequence, treatment, and period, patient within sequence as a random effect, and baseline FEV1 or FVC as a covariate, as appropriate. Between-treatments comparisons were performed by means of contrasts on the treatment factor. The difference between treatments was estimated by the difference between the least squares means. Statistical comparisons between active treatments and placebo were two-sided hypothesis tests with a significance level of 0.05.
Results
Study population
A total of 115 patients were screened, with 62 patients randomized (40/62 [64.5%] male, mean age 40.9 ± 11.2 years). Patient demographics are presented in Table 1. A diagram of patient flow is presented in Figure 1. All 62 patients were included in the intention-to-treat and safety populations. One protocol violation occurred when a patient was inadvertently given the treatment scheduled for Visit 6 at Visit 5, and vice versa. Data for this patient were analyzed according to the treatments received. Four patients withdrew or were discontinued from the study: two due to inability to comply with the study schedule, one who did not fulfill stability criteria, and one for unknown reasons (patient did not return for the premature discontinuation visit).

Diagram of patient flow.
Efficacy
Peak FEV 1
Mean baseline FEV1 values were similar across all treatment periods, ranging from 2.63 L to 2.69 L. The full profile of lung function after each treatment is shown in Figure 2. The onset of action of abediterol was rapid, with significant bronchodilation compared with placebo observed at the first study time point (15 min) post-dose (p < 0.0001 for all doses of abediterol versus placebo, Figure 2a). All doses of abediterol produced significantly greater improvements in peak FEV1 from baseline compared with placebo (p < 0.0001 for all doses, Table 2), and the magnitude of effect of abediterol 0.625, 1.25, and 2.5 μg was not significantly different from salbutamol 400 μg (Figure 3, Table 3). Least squares means differences in change from baseline in peak FEV1 versus placebo were 0.274, 0.322, 0.371, and 0.405 L for abediterol 0.313, 0.625, 1.25, and 2.5 μg, respectively, and 0.353 L for salbutamol 400 μg (Table 3). Median time to peak FEV1 was 3 h post-dose for abediterol (all doses), 2 h post-dose for placebo, and 1 h post-dose for salbutamol 400 μg.

Change from baseline in FEV 1 over time. Data are presented as least squares means with 95% confidence intervals: a) p < 0.0001 versus placebo for salbutamol and all doses of abediterol at all time points; b) p < 0.0001 for all doses of abediterol 0–24 h post-dose and abediterol 1.25 and 2.5 μg 36 h post-dose versus placebo, p = 0.002 and p = 0.01 for abediterol 0.313 and 0.625 μg, respectively, at 36 h post-dose versus placebo. FEV1, forced expiratory volume in 1 s.

Change from baseline in peak FEV 1 at Day 1. Data are presented as least squares means ± standard error: p < 0.0001 versus placebo for all doses of abediterol and salbutamol 400 μg; **p < 0.01, ***p < 0.001, ****p < 0.0001 (analysis of covariance). FEV1, forced expiratory volume in 1 s.
Trough FEV 1
Statistically significant increases in FEV1 were sustained at 24 h post-dose compared with placebo (p < 0.0001 for all doses of abediterol 0–24 h post-dose, Figure 2b). Abediterol significantly improved trough FEV1 from baseline compared with placebo (p < 0.0001 for all doses; Figure 4). Least squares means differences in change from baseline in trough FEV1 versus placebo were 0.219 L (95% CI 0.136, 0.302), 0.259 L (95% CI 0.176, 0.342), 0.332 L (95% CI 0.249, 0.415), and 0.400 L (95% CI 0.317, 0.483) for abediterol 0.313, 0.625, 1.25, and 2.5 μg, respectively (Table 2). The effects of salbutamol on FEV1 were not sustained after 6 h post-dose, as expected for a SABA (Figure 2b).

Change from baseline in trough FEV 1 at Day 2. Data are presented as least squares means ± standard error: p < 0.0001 versus placebo for all doses of abediterol; **p < 0.01, ***p < 0.001, ****p < 0.0001 (analysis of covariance). FEV1, forced expiratory volume in 1 s.
FEV 1 AUC
Change from baseline in normalized FEV1 AUC0–6, FEV1 AUC0–12, and FEV1 AUC0–24 was significantly higher for all doses of abediterol versus placebo (p < 0.0001 for all, Table 2), with dose-related effects observed. The change from baseline in normalized FEV1 AUC0–6 was significantly greater for abediterol 0.625–2.5 μg versus salbutamol 400 μg (p < 0.05, Table 2).
FVC
Mean baseline FVC values were comparable across treatment periods, ranging from 4.23 L to 4.30 L. Abediterol produced significantly greater change from baseline in peak FVC compared with placebo (p < 0.01 for all doses, Table 2). The change from baseline in peak FVC was similar to that observed with salbutamol. Additionally, abediterol significantly improved trough FVC from baseline compared with placebo (p < 0.05 for all doses, Table 2). All doses of abediterol significantly (p < 0.05) increased FVC versus placebo up to 24 h post-dose except for the 0.625 μg dose at 12 and 23 h.
Safety and tolerability
The percentage of patients experiencing TEAEs was similar across all treatment groups (Table 4). Most TEAEs were mild to moderate in intensity, no patient discontinued the study due to a TEAE, and there were no serious adverse events or deaths. The most common TEAEs were headache and nasopharyngitis, reported by 41.9 and 27.4% of patients, respectively. The percentage of patients that experienced at least one treatment-related TEAE was <10% for all doses of abediterol, and was similar to that of salbutamol and placebo. No clinically significant changes in serum glucose or potassium concentrations were observed over time in any of the treatments groups (Table 5). Minor variations in heart rate over time were observed for all treatment groups, including placebo; none were considered to be dose related or clinically relevant (Table 5). There were no clinically relevant changes in clinical laboratory tests, blood pressure, or electrocardiogram parameters.
Discussion
In this study, abediterol 0.313, 0.625, 1.25, and 2.5 μg provided clinically and statistically significant improvements in peak bronchodilation versus placebo in patients with persistent, stable asthma. Abediterol doses ≥0.625 μg also caused bronchodilation compatible with once a day administration, as improvements in trough FEV1 >250 mL compared with placebo were observed. The peak bronchodilatory effect of abediterol was similar to that of salbutamol 400 μg for all doses ≥0.625 μg.
A mean increase in peak FEV1 >200 mL is considered clinically relevant in asthma [1]. This magnitude of change was exceeded for all doses of abediterol, including the lowest dose, 0.313 μg. The primary objective of this study was to define abediterol doses that caused peak bronchodilation similar to salbutamol, the most widely prescribed SABA [21]. High doses of β2-agonists may increase the degree of bronchodilation but also can cause excessive systemic side effects through systemic absorption [22]. However, the current study demonstrates that the doses of abediterol from 0.625 to 2.5 μg induce acute bronchodilation similar to salbutamol, and therefore treatment with higher doses may not be warranted.
Significant bronchodilation compared with placebo was achieved with a range of abediterol doses from 0.313 to 2.5 μg, consistent with the high potency of abediterol demonstrated in preclinical studies that suggested a potential for significant bronchodilation in vivo[7]. Abediterol has higher potency than formoterol, salmeterol, indacaterol, olodaterol, and vilanterol in anesthetized guinea pigs [7, 10] as well as higher potency than salmeterol and indacaterol with comparable potency to formoterol in isolated human bronchi [7]. Based on the results of this preliminary dose-ranging study and the guidelines published by Chowdhury et al. [16], the clinically effective dose in asthma is considered most likely to be in the range of 0.625–2.5 μg.
Significant bronchodilation compared with placebo was observed for salbutamol and all doses of abediterol at the first-assessed time point (15 min) post-dose; this indicates a rapid onset of action for both salbutamol and abediterol. Salmeterol and formoterol, both twice-daily LABAs, are widely used as maintenance therapy in COPD and asthma due to their longer durations of action compared with salbutamol [6, 18]. In clinical studies in patients with asthma, the onset of action of formoterol 24 μg is consistently faster than that of salmeterol 50 μg, with a median onset of action of 3.6 min compared with 31.0 min in one study [23, 24]. In vitro experiments in isolated human bronchi suggest that the onset of action of abediterol is faster than salmeterol and is not significantly different from formoterol and indacaterol [7].
The prolonged duration of action of abediterol, with significant improvements in trough FEV1, normalized FEV1 AUC0–24, and trough FVC relative to placebo, and significant bronchodilation maintained up to 36 h post-dose is consistent with the results of previous clinical studies in healthy subjects and patients with COPD or asthma [11, 13, 25]. For example, a recent Phase II clinical study in patients with COPD demonstrated that abediterol 2.5, 5, and 10 μg achieved >200 mL increases in FEV1 at 24 h, and that this bronchodilatory effect was greater than that of indacaterol 150 μg, and the duration of action was similar [25]. Taken together, these results are compatible with a once-daily dosing regimen in asthma and COPD.
The convenience of once-daily dosing is likely to improve compliance in patients with asthma, leading to better patient outcomes [26, 27], as shown in various studies, including that of Wells et al. whereby once-daily ICS led to improved compliance in patients with asthma compared with ICS administered multiple times daily [28]. LABA monotherapy treatment is contra-indicated in asthma; co-administration with an ICS is mandated, preferably as a fixed-dose combination to reduce the risk of monotherapy administration. Development of once-daily LABAs for use in fixed-dose combinations with once-daily ICS is important for simplifying treatment regimens and, therefore, helping improve patient compliance and general asthma management.
The safety and tolerability profile of abediterol in this study is comparable to that reported in other studies in the abediterol development program [11–13, 25]. Single doses of abediterol 0.313–2.5 μg were safe and well tolerated, and the TEAEs were consistent with the known safety profile of β2-agonists.
Conclusion
Abediterol 0.625–2.5 μg demonstrated a peak bronchodilatory response similar to that of a high dose of salbutamol (400 μg) at very low doses, and showed a clear dose–response relationship. Abediterol is a promising new, highly potent, LABA with a sustained duration of action and a favorable safety and tolerability profile. These results suggest that further studies are warranted to further evaluate the efficacy and safety of abediterol in combination with ICS in both asthma and COPD.
Abbreviations
- ATS:
-
American Thoracic Society
- COPD:
-
Chronic obstructive pulmonary disease
- ERS:
-
European Respiratory Society
- FEV1 AUC:
-
Area under the FEV1 curve
- FEV1 :
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- GINA:
-
Global Initiative for Asthma
- ICS:
-
Inhaled corticosteroid
- IMP:
-
Investigational medicinal product
- LABA:
-
Long-acting β2-adrenergic agonist
- MDI:
-
Metered dose inhaler
- SABA:
-
Short-acting β2-adrenergic agonist
- TEAE:
-
Treatment-emergent adverse event.
References
Global Strategy for Asthma Management and Prevention. [http://www.ginasthma.org]
Ventolin Evohaler Summary of Product Characteristics. [http://www.medicines.org.uk/emc/medicine/97/SPC]
Serevent Accuhaler Summary of Product Characteristics. [http://www.medicines.org.uk/emc/medicine/91]
Formoterol Easyhaler Summary of Product Characteristics. [http://www.medicines.org.uk/emc/medicine/19503/SPC]
Relvar Ellipta Summary of Product Characteristics. [http://www.medicines.org.uk/emc/medicine/28496/SPC/]
Cazzola M, Matera MG: Novel long-acting bronchodilators for COPD and asthma. Br J Pharmacol. 2008, 155: 291-299.
Aparici M, Gómez-Angelats M, Vilella D, Otal R, Carcasona C, Viñals M, Ramos I, Gavaldá A, De Alba J, Gras J, Cortijo J, Morcillo E, Puig C, Ryder H, Beleta J, Miralpeix M: Pharmacological characterization of abediterol, a novel inhaled β(2)-adrenoceptor agonist with long duration of action and a favorable safety profile in preclinical models. J Pharmacol Exp Ther. 2012, 342: 497-509. 10.1124/jpet.112.193284.
Aparici M, Gómez-Angelats M, Vilella D, Cortijo J, Morcillo EJ, Carcasona C, Gavaldà A, Beleta J, Puig C, Ryder H, Miralpeix M: The in vitro pharmacological profile of LAS100977 – a potent, selective and long-acting beta-2 receptor agonist [abstract A5675]. Am J Respir Crit Care Med. 2010, 181: A5675-
Miralpeix M, Aparici M, Carcasona C, Ramos I, Puig C, Vilella D, Gavalda A: Abediterol (LAS100977) is A Potent and Selective β2-Agonist with Sustained Duration of Action and Favourable Selectivity in Vitro [poster]. 2014, San Diego, CA, USA: Presented at the American Thoracic Society Annual Congress
De Alba J, Otal R, Fernández-Blanco JA, Aparici M, Puig C, Gavaldà A, Miralpeix M: Abediterol (LAS100977) Demonstrates a Higher Potency and a Longer Duration of Action in Comparison with Other Long-Acting Β2-Adrenoceptor Agonists in a Guinea Pig Bronchoconstriction Model [poster]. 2014, San Diego, CA, USA: Presented at the American Thoracic Society Annual Congress
Beier J, Fuhr R, Massana È, Jiménez E, Seoane B, de Miquel G, Ruiz S: Efficacy and safety of single inhaled doses of LAS100977, a novel long acting β2-agonist, in patients with persistent asthma [abstract A5414]. Am J Respir Crit Care Med. 2010, 181: A5414-
Miralpeix M, Gomez-Angelats M, Aparici M, Vinals M, Beleta J, Gavalda A: LAS100977, a novel beta-2 receptor agonist, with a longer duration of action and more favourable safety margin than salmeterol in anesthetised dogs [abstract A5676]. Am J Respir Crit Care Med. 2010, 181: A5676-
Timmer W, Massana E, Jiménez E, Seoane B, de Miquel G, Ruiz S: Single doses of LAS100977, a novel long acting β2-agonist, show high activity and long duration in healthy subjects [abstract A5663]. Am J Respir Crit Care Med. 2010, 181: A5663-
Beier J, Fuhr R, Massana E, Jiménez E, Seoane B, de Miquel G, Ruiz S: Efficacy and Safety of Single Inhaled Doses of LAS100977, A Novel Long Acting β2-Agonist, in Patients with Persistent Asthma [poster]. 2010, New Orleans, Louisiana, USA: Presented at the American Thoracic Society International Conference
Beier J, Pujol H, Seoane B, Jiménez E, Astbury C, Massana E, Ruiz S, de Miquel G: Efficacy and Safety of Abediterol (LAS100977) in Patients with COPD: Phase III, Randomized, Crossover Study [poster]. 2013, Barcelona, Spain: Presented at the European Respiratory Society Annual Congress
Chowdhury B, Seymour SM, Michele TM, Durmowicz AG, Liu D, Rosebraugh CJ: The risks and benefits of indacaterol–the FDA’s review. N Engl J Med. 2011, 365: 2247-2249. 10.1056/NEJMp1109621.
Whale CI, Sovani MP, Mortimer KJ, Harrison TW, Tattersfield AE: Systemic and bronchodilator effects of inhaled rac-formoterol in subjects with chronic obstructive pulmonary disease: a dose–response study. Br J Clin Pharmacol. 2008, 65: 841-847. 10.1111/j.1365-2125.2007.03081.x.
Beeh KM, Beier J: The short, the long and the “ultra-long”: why duration of bronchodilator action matters in chronic obstructive pulmonary disease. Adv Ther. 2010, 27: 150-159. 10.1007/s12325-010-0017-6.
Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, Casaburi R, Coates A, van der Grinten CP, Gustafsson P, Hankinson J, Jensen R, Johnson DC, MacIntyre N, McKay R, Miller MR, Navajas D, Pedersen OF, Wanger J: Interpretative strategies for lung function tests. Eur Respir J. 2005, 26: 948-968. 10.1183/09031936.05.00035205.
Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF, Peslin R, Yernault JC: Lung volumes and forced ventilatory flows. Report Working Party Standardization of Lung Function Tests, European Community for Steel and Coal. Official Statement of the European Respiratory Society. Eur Respir J Suppl. 1993, 16: 5-40.
Crompton G: A brief history of inhaled asthma therapy over the last fifty years. Prim Care Respir J. 2006, 15: 326-331. 10.1016/j.pcrj.2006.09.002.
Cazzola M, Page CP, Calzetta L, Matera MG: Pharmacology and therapeutics of bronchodilators. Pharmacol Rev. 2012, 64: 450-504. 10.1124/pr.111.004580.
Palmqvist M, Persson G, Lazer L, Rosenborg J, Larsson P, Lötvall J: Inhaled dry-powder formoterol and salmeterol in asthmatic patients: onset of action, duration of effect and potency. Eur Respir J. 1997, 10: 2484-2489. 10.1183/09031936.97.10112489.
Bodzenta-Lukaszyk A, Dymek A, McAulay K, Mansikka H: Fluticasone/formoterol combination therapy is as effective as fluticasone/salmeterol in the treatment of asthma, but has a more rapid onset of action: an open-label, randomized study. BMC Pulm Med. 2011, 11: 28-10.1186/1471-2466-11-28.
Beier J, Pujol H, Seoane B, Jiménez E, Astbury C, Massana E, Ruiz S, de Miquel G: Efficacy and safety of abediterol (LAS100977) in patients with COPD: Phase II, randomized, crossover study [abstract 83]. Eur Respir J. 2013, 42 (Suppl 57): 183-
Chapman KR, Walker L, Cluley S, Fabbri L: Improving patient compliance with asthma therapy. Respir Med. 2000, 94: 2-9. 10.1053/rmed.1999.0667.
Makela MJ, Backer V, Hedegaard M, Larsson K: Adherence to inhaled therapies, health outcomes and costs in patients with asthma and COPD. Respir Med. 2013, 107: 1481-1490. 10.1016/j.rmed.2013.04.005.
Wells KE, Peterson EL, Ahmedani BK, Williams LK: Real-world effects of once vs greater daily inhaled corticosteroid dosing on medication adherence. Ann Allergy Asthma Immunol. 2013, 111: 216-220. 10.1016/j.anai.2013.06.008.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2466/14/176/prepub
Acknowledgments
The authors wish to thank the study investigators and PAREXEL International Ltd. Suzanne McAllister, PhD, from Complete Medical Communications provided medical writing assistance funded by Almirall S.A., Barcelona, Spain.
This study is registered with clinicaltrials.gov, trial registration number NCT01425801, and was funded by Almirall S.A., Barcelona, Spain.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
DS has received sponsorship to attend international meetings, honoraria for lecturing or attending advisory boards and research grants from various pharmaceutical companies including Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, CIPLA, Forest, Genetech, GlaxoSmithKline, Merck, Novartis, Pfizer and Takeda. HP, AR, BS, EM, CA, SR and GdM are all employees of Almirall. BS, SR and GdM have a patent pending for abediterol (Novel dosage form and formulation of abediterol, WO2013178742 A1). HP, AR, BS and GdM hold Almirall shares or stocks.
Authors’ contributions
All authors contributed to the conception and design of the study, data analysis/interpretation and revision of the manuscript for important intellectual content and provided final approval of the manuscript. DS was the coordinating investigator of the study and drafted the article. There are no other contributors who fulfill the authorship criteria.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Singh, D., Pujol, H., Ribera, A. et al. A dose-ranging study of the bronchodilator effects of abediterol (LAS100977), a long-acting β2-adrenergic agonist, in asthma; a Phase II, randomized study. BMC Pulm Med 14, 176 (2014). https://doi.org/10.1186/1471-2466-14-176
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2466-14-176