
Abstract
Background
Studies have shown that nonspecific interstitial pneumonitis (NSIP), even when initially diagnosed as an idiopathic form of the disease, might be associated with an autoimmune background that later reveals itself as an organ-specific or a systemic autoimmune disease.
Methods
NSIP patients were divided into three groups. The NSIP patients who met the criteria for having a systemic autoimmune disease (SAD) were defined as the systemic autoimmune disease-associated NSIP (SAD-NSIP) group. The NSIP patients who did not meet the criteria for a systemic autoimmune disease were defined as an antibody-positive group (i-NSIP-Ab + group) if their sera were positive for autoantibodies. The NSIP patients with negative serologic tests for auto-antibodies were defined as the antibody-negative group (i-NSIP-Ab- group). The clinical characteristics were analyzed and compared among the three groups.
Results
Ninety-seven NSIP patients were included. The mean age of the study population was 48 ± 11 years. The mean follow-up time was 54 ± 34 months. At the time of the surgical lung biopsies, 23/97 (23.7%) of the patients were classified as SAD-NSIP; 30/97 (30.9%) were in the i-NSIP-Ab + group; and 44/97 (45.4%) were in the i-NSIP-Ab- group. At the end of the follow-up period, three cases were diagnosed with polymyositis (one case from the i-NSIP-Ab + group, two cases from the i-NSIP-Ab- group), one with scleroderma (from the i-NSIP-Ab + group, scl-70 positive and skin biopsy) and another one with microscopic polyarteritis (from the i-NSIP-AB-group, p-ANCA and MPO positive, renal biopsy). Three cases in the i-NSIP-Ab- group were later found to be positive for autoantibodies. Due to these changes in classification, at the end of the follow-up period, the SAD-NSIP group consisted of 28/97 patients (28.9%), the i-NSIP-Ab + group of 31/97 (32.0%) and the i-NSIP-Ab- group of 38/97(39.1%). There were no significant differences in clinical manifestations, radiographic findings or pulmonary function tests among the three groups at the time of surgical lung biopsy or after reclassification after the follow-up period. SAD was an independent risk factor for the survival of the patients with NSIP after follow-up.
Conclusion
Follow-up is recommended because idiopathic NSIP may be the first manifestation of a systemic autoimmune disease.
Similar content being viewed by others


Background
The histopathologic pattern of nonspecific interstitial pneumonitis (NSIP) has been found in a wide variety of clinical contexts, including chronic hypersensitivity pneumonitis, drug-related interstitial pneumonia, and connective tissue disease (CTD) [1–5]. NSIP has been identified as one of the most common pathologic patterns in patients with CTD [6–11]. Recent studies have shown that NSIP, even when initially diagnosed as an idiopathic form of NSIP, might be associated with an autoimmune background that later reveals itself as an organ-specific or a systemic autoimmune disease [12–14]. In a cohort study of 27 idiopathic NSIP patients, more than 50% of the cases developed an autoimmune disease after a mean follow-up of 22 months [13]. Furthermore, an Asian study revealed that CTD developed in 10% of idiopathic NSIP patients during the follow-up period [14]. It has been suggested that Multidisciplinary Discussion and follow-up are especially important to establish the diagnosis of idiopathic NSIP [15, 16].
Compared with idiopathic interstitial lung disease (ILD), patients with CTD-associated ILD (CTD-ILD) had a better prognosis [17–21]. For patients with rheumatoid arthritis-associated usual interstitial pneumonia (UIP), the survival time was longer than that for patients with UIP without rheumatoid arthritis [18]. According to these studies, it might be deduced that CTD-NSIP had a better outcome than idiopathic NSIP (i-NSIP). However, the current literature is controversial. Additional studies showed that CTD did not affect the survival of patients with pathologically confirmed NSIP [22, 23]. Interestingly, the classification of undifferentiated CTD-NSIP conferred a minor prognostic advantage [23].
Given that CTD-NSIP is similar to i-NSIP on clinical and radiologic features [24–26], and if there are no differences in the disease outcomes for the two classifications, the question arises as to whether there is a value to differentiating between CTD-NSIP and i-NSIP. To answer this question, we reviewed the clinical, radiologic and physiologic findings in NSIP patients with systemic autoimmune disease and i-NSIP at the Peking Union Medical College Hospital. We wanted to evaluate whether there was a difference in prognosis for i-NSIP patients compared with those with systemic autoimmune disease in this Chinese cohort.
Methods
Study subjects and diagnostic criteria
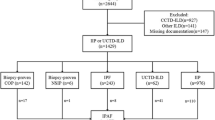
Between December 2002 and December 2011, 354 patients underwent surgical lung biopsies at the Peking Union Medical College Hospital, the biggest referral center in China. The ending date for the follow-up period was December 2012. The 354 cases in this study were clinical suspicion for diffuse infiltrates. NSIP was diagnosed in 101 cases; 4 cases of drug-induced NSIP were excluded. The remaining 97 cases were enrolled in this study. Their clinical features, radiological images and pathological findings were reviewed and analyzed. NSIP was diagnosed according to the American Thoracic Society (ATS)/European Respiratory Society consensus classification criteria [1, 15]. Informed consent for using the medical records was obtained from every patient and/or their guardian when the patient was admitted to the hospital. This study was approved by Peking Union Medical College Hospital Institutional Review Board (reference number for ethics approval: 2013-9-322).
Patients with a history of drug toxicity, airborne antigen or environmental exposures were excluded. Individual forms of systemic autoimmune disease including CTD, microscopic polyangiitis and Crohn's disease were diagnosed according to the criteria of the corresponding societies. The following were considered autoantibody positive (Ab+) in our study: an antinuclear antibody (ANA) titer greater than 1:320, a positive of anti-Sjogren's syndrome antigen A (SSA) or anti-Sjogren's syndrome antigen B (SSB), anti-Scl-70, anti-Sm, anti-Jo-1, anti-ribonucleoprotein antibody (anti-RNP), anti-keratin antibody (AKA), anti-perinuclear factor (APF), or anti-cyclic citrullinated peptide antibodies (anti-CCP) [27–32]. NSIP patients who meet the criteria for a systemic autoimmune disease (SAD) were defined as the SAD-NSIP group [30, 33–35]; NSIP patients who had at least one positive serologic antibody test were defined as the antibody positive group (i-NSIP-Ab + group); and NSIP patients with negative serologic antibodies tests were defined as the antibody negative group (i-NSIP-Ab- group).
Clinical characteristics
The patients in this study had the following clinical characteristics documented at the time of their first visit: age, sex, symptoms at the time of the surgical lung biopsy (cough, dyspnea, or wheeze), symptoms or signs of systemic autoimmune disease, smoking status, physical exam findings, arterial blood gas analysis (ABG), and serologic auto-antibody tests.
Pulmonary function tests
Spirometry, total lung capacity (TLC) by plethysmography, forced vital capacity (FVC) and diffusing capacity of the lung for carbon monoxide (DLCO) were measured according to the ATS recommendations [36–38], and the results were expressed as percentage of the normal predicted values.
Analysis of subsets of lymphocytes from bronchoalveolar lavage fluid (BALF)
The patients were examined by bronchoscopy with an electric video bronchoscope wedged into the segmental bronchus of the right middle lobe. One hundred milliliters of sterile saline was injected according to the guidelines for the measurement of the cellular components and standardization of BAL [39, 40]. To evaluate the cell subsets, the lymphocytes were stained with anti-CD3, anti-CD4 and anti-CD8 monoclonal antibodies coupled to fluorescein isothiocyanate, and the cellular fluorescence was measured with a FACS Calibur flow cytometer.
High-resolution CT scanning
High-resolution computed tomography (HRCT) chest scans were performed on all patients at the time of the initial evaluation. The films were reviewed in a blinded fashion by chest radiologists experienced in interpretation of diffuse lung diseases. The specific findings of the HRCT were documented for the index scan (the first scan that documented the presence of ILD). The extent of emphysema, ground-glass opacity (GGO), reticulation, consolidation, and honeycombing (HC) were scored on a scale of 5% for all lobes. HC was defined as clustered cystic airspaces, 3 to 10 mm in diameter, in the subpleural areas of the lungs with well-defined shared walls and layering.
Treatment and follow-up
After being diagnosed with NSIP, all patients received a course of oral prednisolone, starting at 0.5 mg/kg/d for one month, tapered every 3 weeks to 5-7.5 mg/d and then maintained in 5-7.5 mg/d. The total length of treatment was 12-18 months.
The patients underwent follow-up assessments at the Interstitial Lung Disease Clinic of Peking Union Medical College Hospital 3, 6 and 12 month after surgical lung biopsy, and then once a year. The patients in the SAD-NSIP group were treated combined with cytotoxic drugs.
Statistical analysis
All values are expressed as the mean ± SD. Student’s t-test was used for analysis of normally distributed data. The Wilcoxon test and Kruskal-Wallis test were used to compare two or more sets of non-normally distributed data, respectively. Fisher's exact test was used to determine group differences. Cox multivariate regression analysis was used to evaluate the survival risk factors. The overall survival durations were calculated by the Kaplan-Meier method. The correlation coefficients were calculated using Spearman’s rank method. The probability values were obtained from 2-sided tests, with a statistical significance of p < 0.05. SPSS 15.0 (SPSS for Windows, version 15.0 [SPSS Inc., Chicago, Illinois, USA]) was used for the statistical analyses.
Results and discussion
Ninety-seven NSIP patients were included. The mean age of the study population was 48 ± 11 years (median 48, range 16 to 69), and 73% were women. The mean follow-up time was 54 ± 34 months (median 45, range 2 to 120).
We divided the NSIP patients into three groups according the above criteria at the time of surgical lung biopsy. The SAD-NSIP group included 23/97(23.7%) of the patients; the i-NSIP-Ab + group included 30/97 (30.9%); and the i-NSIP-Ab- group included 44/97(45.4%). The underlying types of systemic autoimmune disease were rheumatoid arthritis (n = 3), scleroderma (n =3), Sjogren’s syndrome (n =4), polymyositis (n = 9), SLE (n = 1), MPA (n = 2) and ulcerative colitis (n = 1).
At the end of December 2012, typical clinic manifestations of CTD developed in five cases from i-NSIP-Ab + group and i-NSIP-Ab- group. Respiratory symptoms preceded other systemic manifestations by a median of 48 months (range 1-108). Additionally, 4 cases in the i-NSIP Ab- group were later found to have positive autoantibodies during the median follow-up period of 66 months (range 12-108). Three cases were diagnosed as having polymyositis (one case from i-NSIP-Ab + group, two cases from i-NSIP-Ab- group), one as scleroderma (scl-70 positive and skin biopsy) and another one as microscopic polyarteritis (from i-NSIP-AB-group, p-ANCA and MPO positive, renal biopsy). Additionally, three cases in the i-NSIP-Ab- group were later found to have positive autoantibodies. Two cases were ANA positive (one at 1:640; another at 1:1280), and another case was anti-Jo-1 positive.
At the end of December 2012, the underlying types of systemic autoimmune disease were rheumatoid arthritis (n = 3), scleroderma (n =4), Sjogren’s syndrome (n =4), polymyositis (n = 12), SLE (n = 1), MPA (n = 3) and ulcerative colitis (n = 1). We re-classified the NSIP patients according the follow-up results. The post-follow-up distribution of the subjects was: 28/97 patients (28.9%) in the SAD-NSIP group, 31/97 (32.0%) in the i-NSIP-Ab + group and 38/97 (39.1%) in the i-NSIP-Ab- group.
Comparison of clinical manifestations among the CTD-NSIP, i-NSIP-Ab + and i-NSIP-Ab- groups
The demographic characteristics are shown in Table 1. There were no differences among the three groups of the NSIP patients as classified at the time of surgical lung biopsy or after re-classification at the time of follow-up (a median of 45 months).
The clinical characteristics of study subjects are shown in Table 2. Skin rash was strongly associated with the SAD-NSIP and i-NSIP-Ab + groups relative to the i-NSIP-Ab- group as classified at the time of surgical lung biopsy or after re-classification at the time of follow-up.
The laboratory data are shown in Table 3. There were no differences among the three groups either at the time of the surgical lung biopsy or after the post-follow-up reclassification. Pulmonary function, BAL lymphocytes analysis and ABG were similar among the three groups at the time of surgical lung biopsy or after the post-follow-up reclassification.
Radiologic and pathologic findings in CTD-NSIP, i-NSIP-Ab + and i-NSIP-Ab- groups
The characteristic features of the HRCT for the three groups are shown in Table 4. No statistically significant differences were observed among the three groups of patients as classified at the time of surgical lung biopsy or after the post-follow-up reclassification.
The histological analysis is shown in Table 4. Additionally, no statistically significant differences were observed among the three subgroups of patients as classified at the time of surgical lung biopsy or after the post-follow-up reclassification.
Treatment and survival
The treatment and follow-up results are summarized in Table 5.Based on the classification at the time of surgical lung biopsy, there were no significant differences among the three groups, p = 0.511 (Kaplan-Meier survival curves are shown in Figure 1). The diagnosis of systemic autoimmune disease was not associated with poorer survival (HR, 0.368, 95% CI, 0.680-3.067; p = 0.339). However, based on the classification after the follow-up period, a marginally significant statistical difference could be observed between the ASD-NSIP and NSIP-Ab- groups (Figure 2, p = 0.059). Systemic autoimmune disease was an independent risk factor for the survival of patients with NSIP after follow-up (HR, 0.471; 95% CI, 0.246-0.901; p = 0.023).Pathological pattern was associated with the survival time, p = 0.010 (Kaplan-Meier survival curves are shown in Figure 3). There is a significant difference (p = 0.01) among three groups (Log Rank=13.391). Fibrotic pattern was an independent risk factor for the survival of patients with NSIP (HR, 0.316; 95% CI, 0.115-0.870; p = 0.026).

Kaplan-Meier survival curve for NSIP patients divided by clinical status at intital: SAD-NSIP, i- NSIP-AB + and i-NSIP-Ab- patients at the time of surgical lung biopsy. There is no significant difference (p = 0.511) among three groups (Log Rank = 1.342).

Kaplan-Meier survival curve for NSIP patients re-divided by clinical status after follow up 54 ± 34 months. ASD-NSIP vs i-NSIP-Ab- =0.059; ASD-NSIP vs i-NSIP-Ab + =0.232; i-NSIP-Ab + vs i-NSIP-Ab- =0.456.

Kaplan-Meier survival curve for NSIP patients divided by pathological pattern: cellular pattern vs mixed pattern =0.001; cellular pattern vs fibrotic pattern =0.000; Mixed pattern vs fibrotic pattern =0.451.
Smoking (HR, 0.964; 95% CI, 0.348-2.679; p = 0.943), TLC (HR, 1.013; 95% CI, 0.991-1.036; p = 0.260), FVC (HR, 1.032; 95% CI, 0.997-1.067; p = 0.764) and DLCO (HR, 0.978; 95% CI, 0.945-1.012; p = 0.206) were not the risk factor for survival.
Our study revealed a total of 5/74 (6.8%) cases were diagnosed as having systemic autoimmune disease after follow-up. Romagnoli’s study showed that 3 of 27 (11%) i-NSIP patients were diagnosed as having CTD after a follow-up of 59.7 months [13]. Park reported that 8/87 cases (10%) of i-NSIP patients developed CTD during a median follow-up of 53 months [14]. The prevalence in these previous studies was higher than in our study. One reason for the difference could be that the median follow-up time in our study was 45 months, but the typical systemic autoimmune disease symptoms, signs and serum biomarkers were generally first observed at times between 48 and 66 months of follow-up. It is therefore possible that some of the i-NSIP patients might have developed typical systemic autoimmune disease after our follow-up period.
We reviewed the published papers that compared survival between CTD-ILD and IIP and found that all of the studies classified the patients according to the patient’s presentations at the time of the first visit. The results indicated that the clinical and radiologic features of CTD-NSIP were similar to idiopathic NSIP [22, 23], and CTD did not affect survival in NSIP patients [24, 26]. In our study, we followed the methods of the previous studies and divided the patients according the clinical manifestation at the time of surgical lung biopsy. The results were consistent with the previous studies in showing no differences in survival time among those three NSIP classifications [18, 41, 42]. i-NSIP-Ab + was also not associated with a survival benefit according to several studies [18, 23, 43]. Then, we re-classified the NSIP patents according the follow-up results and compared the patients’ clinical manifestations, radiographic findings and pathological features. There were still no differences among three groups after follow-up. The patients who were defined as having systemic autoimmune disease associated NSIP could not be distinguished from those who were defined as i-NSIP with or without antibodies based on pulmonary manifestations or respiratory physiology even after follow-up. This result indicates that recognition of systemic autoimmune disease is particularly challenging in NSIP [25, 26, 44, 45].
Using Cox multivariate analysis, we found that systemic autoimmune disease was a risk factor for survival. Furthermore, we found that the survival times for the SAD-NSIP patients were shorter than for those classified as i-NSIP because some i-NSIP patients with poor prognoses were eventually diagnosed as having systemic autoimmune disease and were re-assigned to the SAD-NSIP group. Lee’s study showed that the patients with NSIP and various systemic conditions had worse prognoses. However, no statistically significant relationship was found between the systemic conditions and poor prognosis in that study [42]. Felício [24] detected significantly greater collagen and elastic fiber production in the lungs of patients with CTD-NSIP compared with those with idiopathic NSIP [24]. The increased elastin content may have been caused by major repair and remodeling processes following septal inflammation and consequent fiber fragmentation in CTD-NSIP. These processes might be responsible for the worse prognosis. However, the specific mechanism is currently uncertain. More prospective studies with larger numbers of subjects are required.
Our study was limited by its retrospective nature, and because this was a retrospective study, the subjects were restricted to the patients who had undergone a surgical lung biopsy. In addition, the follow-up period might not have been long enough to clearly show differences in the prognosis between SAD-NSIP and i-NSIP. Although the number of subjects in our study was relatively large, it might not have been enough to clearly show the differences between SAD-NSIP and i-NSIP. Further studies of larger numbers of subjects, perhaps through multicenter cooperation, will be required to overcome this limitation. Nevertheless, this is the first time that the NSIP patients were re-classified after the follow-up period. We believe that the data presented here remain valid despite these shortcomings.
Conclusion
Based on the results of our study, we concluded that some idiopathic NSIP cases may represent the first manifestation of an underlying systemic autoimmune disease. Long-term follow-up of patients with idiopathic NSIP is recommended.
Abbreviations
- AKA:
-
Anti-keratin antibody
- ANA:
-
Anti-nuclear antibody
- APF:
-
Anti-perinuclear factor
- ATS:
-
American Thoracic Society
- BALF:
-
Bronchoalveolar lavage Fluid
- CCP:
-
Anti-cyclic citrullinated peptide antibodies
- CTD:
-
Connective tissue disease
- CBC:
-
Complete blood count
- DLCO:
-
Diffusing capacity of the lung for carbon monoxide
- ERS:
-
European Respiratory Society
- ESR:
-
Erythrocyte sedimentation rate
- FVC:
-
Forced vital capacity
- HRCT:
-
High-resolution computed tomography
- ILD:
-
Interstitial lung diseases
- IIP:
-
Idiopathic interstitial pneumonitis
- NSIP:
-
Nonspecific interstitial pneumonitis
- RA:
-
Rheumatoid arthritis
- RNP:
-
Anti-ribonucleoprotein antibody
- SAD:
-
Systemic autoimmune disease
- SSA:
-
Anti-Sjogren's syndrome antigen A
- SSB:
-
Anti-Sjogren's syndrome antigen B
- UIP:
-
Usual interstitial pneumonia.
References
American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias: This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002, 165 (2): 277-304.
Jacobs RL, Andrews CP: Hypersensitivity pneumonia-nonspecific interstitial pneumonia/fibrosis histopathologic presentation: a study in diagnosis and long-term management. Ann Allergy Asthma Immunol. 2003, 90 (2): 265-270. 10.1016/S1081-1206(10)62153-9.
Churg A, Muller NL, Flint J, Wright JL: Chronic hypersensitivity pneumonitis. Am J Surg Pathol. 2006, 30 (2): 201-208. 10.1097/01.pas.0000184806.38037.3c.
Rossi SE, Erasmus JJ, McAdams HP, Sporn TA, Goodman PC: Pulmonary drug toxicity: radiologic and pathologic manifestations. Radiographics. 2000, 20 (5): 1245-1259. 10.1148/radiographics.20.5.g00se081245.
de Lauretis A, Veeraraghavan S, Renzoni E: Review series: Aspects of interstitial lung disease: connective tissue disease-associated interstitial lung disease: how does it differ from IPF? How should the clinical approach differ?. Chron Respir Dis. 2011, 8 (1): 53-82.
Tafti SF, Mokri B, Mohammadi F, Bakhshayesh-Karam M, Emami H, Masjedi MR: Comparison of clinicoradiologic manifestation of nonspecific interstitial pneumonia and usual interstitial pneumonia/idiopathic pulmonary fibrosis: a report from NRITLD. Ann Thorac Med. 2008, 3 (4): 140-145. 10.4103/1817-1737.43081.
Nagai S, Handa T, Tabuena R, Kitaichi M, Izumi T: Nonspecific interstitial pneumonia: a real clinical entity?. Clin Chest Med. 2004, 25 (4): 705-715. 10.1016/j.ccm.2004.04.009. vi
Saketkoo LA, Matteson EL, Brown KK, Seibold JR, Strand V: Developing disease activity and response criteria in connective tissue disease-related interstitial lung disease. J Rheumatol. 2011, 38 (7): 1514-1518. 10.3899/jrheum.110281.
Shi JH, Xu WB, Liu HR, Tian XL, Feng RE, Xu ZJ, Zhu YJ: Clinico-pathological manifestations in interstitial lung diseases associated with polymyositis-dermatomyositis. Zhonghua Jie He He Hu Xi Za Zhi. 2008, 31 (4): 250-254.
Ito I, Nagai S, Kitaichi M, Nicholson AG, Johkoh T, Noma S, Kim DS, Handa T, Izumi T, Mishima M: Pulmonary manifestations of primary Sjogren's syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med. 2005, 171 (6): 632-638. 10.1164/rccm.200403-417OC.
Kim DS, Yoo B, Lee JS, Kim EK, Lim CM, Lee SD, Koh Y, Kim WS, Kim WD, Colby TV, Kitiaichi ML: The major histopathologic pattern of pulmonary fibrosis in scleroderma is nonspecific interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis. 2002, 19 (2): 121-127.
Kinder BW, Collard HR, Koth L, Daikh DI, Wolters PJ, Elicker B, Jones KD, King TE: Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease?. Am J Respir Crit Care Med. 2007, 176 (7): 691-697. 10.1164/rccm.200702-220OC.
Romagnoli M, Nannini C, Piciucchi S, Girelli F, Gurioli C, Casoni G, Ravaglia C, Tomassetti S, Gurioli C, Gavelli G, Carloni A, Dubini A, Cantini F, Chilosi M, Poletti V: Idiopathic nonspecific interstitial pneumonia: an interstitial lung disease associated with autoimmune disorders?. Eur Respir J. 2011, 38 (2): 384-391. 10.1183/09031936.00094910.
Park IN, Jegal Y, Kim DS, Do KH, Yoo B, Shim TS, Lim CM, Lee SD, Koh Y, Kim WS, Kim WD, Jang SJ, Kitaichi M, Nicholson AG, Colby TV: Clinical course and lung function change of idiopathic nonspecific interstitial pneumonia. Eur Respir J. 2009, 33 (1): 68-76. 10.1183/09031936.00158507.
Travis WD, Costabel U, Hansell DM, King TE, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, et al: An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013, 188 (6): 733-748. 10.1164/rccm.201308-1483ST.
Travis WD, Hunninghake G, King TE, Lynch DA, Colby TV, Galvin JR, Brown KK, Chung MP, Cordier JF, du Bois RM, Flaherty KR, Franks TJ, Hansell DM, Hartman TE, Kazerooni EA, Kim DS, Kitaichi M, Koyama T, Martinez FJ, Nagai S, Midthun DE, Müller NL, Nicholson AG, Raghu G, Selman M, Wells A: Idiopathic nonspecific interstitial pneumonia: report of an American Thoracic Society project. Am J Respir Crit Care Med. 2008, 177 (12): 1338-1347. 10.1164/rccm.200611-1685OC.
Bouros D, Wells AU, Nicholson AG, Colby TV, Polychronopoulos V, Pantelidis P, Haslam PL, Vassilakis DA, Black CM, du Bois RM: Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002, 165 (12): 1581-1586. 10.1164/rccm.2106012.
Park JH, Kim DS, Park IN, Jang SJ, Kitaichi M, Nicholson AG, Colby TV: Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med. 2007, 175 (7): 705-711. 10.1164/rccm.200607-912OC.
Lee HK, Kim DS, Yoo B, Seo JB, Rho JY, Colby TV, Kitaichi M: Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest. 2005, 127 (6): 2019-2027. 10.1378/chest.127.6.2019.
Flaherty KR, Colby TV, Travis WD, Toews GB, Mumford J, Murray S, Thannickal VJ, Kazerooni EA, Gross BH, Lynch JP, Martinez FJ: Fibroblastic foci in usual interstitial pneumonia: idiopathic versus collagen vascular disease. Am J Respir Crit Care Med. 2003, 167 (10): 1410-1415. 10.1164/rccm.200204-373OC.
Agusti C, Xaubet A, Roca J, Agusti AG, Rodriguez-Roisin R: Interstitial pulmonary fibrosis with and without associated collagen vascular disease: results of a two year follow up. Thorax. 1992, 47 (12): 1035-1040. 10.1136/thx.47.12.1035.
Nakamura Y, Chida K, Suda T, Hayakawa H, Iwata M, Imokawa S, Tsuchiya T, Ida M, Gemma H, Yasuda K, Yagi T, Shirai T, Tamura R, Nakano Y, Hirata T, Nakamura H, Colby TV: Nonspecific interstitial pneumonia in collagen vascular diseases: comparison of the clinical characteristics and prognostic significance with usual interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis. 2003, 20 (3): 235-241.
Corte TJ, Copley SJ, Desai SR, Zappala CJ, Hansell DM, Nicholson AG, Colby TV, Renzoni E, Maher TM, Wells AU: Significance of connective tissue disease features in idiopathic interstitial pneumonia. Eur Respir J. 2012, 39 (3): 661-668. 10.1183/09031936.00174910.
Felicio CH, Parra ER, Capelozzi VL: Idiopathic and collagen vascular disease nonspecific interstitial pneumonia: clinical significance of remodeling process. Lung. 2007, 185 (1): 39-46. 10.1007/s00408-006-0104-2.
Strange C, Highland KB: Interstitial lung disease in the patient who has connective tissue disease. Clin Chest Med. 2004, 25 (3): 549-559. 10.1016/j.ccm.2004.05.009. vii
Fujita J, Ohtsuki Y, Yoshinouchi T, Yamadori I, Bandoh S, Tokuda M, Miyawaki H, Kishimoto N, Ishida T: Idiopathic non-specific interstitial pneumonia: as an "autoimmune interstitial pneumonia". Respir Med. 2005, 99 (2): 234-240. 10.1016/j.rmed.2004.07.001.
Lonzetti LS, Joyal F, Raynauld JP, Roussin A, Goulet JR, Rich E, Choquette D, Raymond Y, Senecal JL: Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum. 2001, 44 (3): 735-736. 10.1002/1529-0131(200103)44:3<735::AID-ANR125>3.0.CO;2-F.
Smolen JS, Steiner G: Mixed connective tissue disease: to be or not to be?. Arthritis Rheum. 1998, 41 (5): 768-777. 10.1002/1529-0131(199805)41:5<768::AID-ART3>3.0.CO;2-Z.
Hochberg MC: Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997, 40 (9): 1725-
Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Ménard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, et al: 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62 (9): 2569-2581. 10.1002/art.27584.
Miller FW, Rider LG, Plotz PH, Isenberg DA, Oddis CV: Diagnostic criteria for polymyositis and dermatomyositis. Lancet. 2003, 362 (9397): 1762-1763. 10.1016/S0140-6736(03)14862-3. author reply 1763
Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, Daniels TE, Fox PC, Fox RI, Kassan SS, Pillemer SR, Talal N, Weisman MH: Classification criteria for Sjogren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002, 61 (6): 554-558. 10.1136/ard.61.6.554.
Magro F, Langner C, Driessen A, Ensari A, Geboes K, Mantzaris GJ, Villanacci V, Becheanu G, Borralho Nunes P, Cathomas G, Fries W, Jouret-Mourin A, Mescoli C, de Petris G, Rubio CA, Shepherd NA, Vieth M, Eliakim R: European consensus on the histopathology of inflammatory bowel disease. J Crohns Colitis. 2013, 7 (10): 827-851. 10.1016/j.crohns.2013.06.001.
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen EC, Hoffman GS, Jayne DR, Kallenberg CG, Lamprecht P, Langford CA, Luqmani RA, Mahr AD, Matteson EL, Merkel PA, Ozen S, Pusey CD, Rasmussen N, Rees AJ, Scott DG, Specks U, Stone JH, Takahashi K, Watts RA: 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65 (1): 1-11. 10.1002/art.37715.
Annese V, Daperno M, Rutter MD, Amiot A, Bossuyt P, East J, Ferrante M, Gotz M, Katsanos KH, Kiesslich R, Ordás I, Repici A, Rosa B, Sebastian S, Kucharzik T, Eliakim R: European evidence based consensus for endoscopy in inflammatory bowel disease. J Crohns Colitis. 2013, 7 (12): 982-1018. 10.1016/j.crohns.2013.09.016.
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP, Gustafsson P, Jensen R, Johnson DC, MacIntyre N, McKay R, Navajas D, Pedersen OF, Pellegrino R, Viegi G, Wanger J: Standardisation of spirometry. Eur Respir J. 2005, 26 (2): 319-338. 10.1183/09031936.05.00034805.
Wanger J, Clausen JL, Coates A, Pedersen OF, Brusasco V, Burgos F, Casaburi R, Crapo R, Enright P, van der Grinten CP, Gustafsson P, Hankinson J, Jensen R, Johnson D, Macintyre N, McKay R, Miller MR, Navajas D, Pellegrino R, Viegi G: Standardisation of the measurement of lung volumes. Eur Respir J. 2005, 26 (3): 511-522. 10.1183/09031936.05.00035005.
Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, Burgos F, Casaburi R, Coates A, Enright P, Gustafsson P, Hankinson J, Jensen R, McKay R, Miller MR, Navajas D, Pedersen OF, Pellegrino R, Wanger J: Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J. 2005, 26 (4): 720-735. 10.1183/09031936.05.00034905.
Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, Drent M, Haslam PL, Kim DS, Nagai S, Rottoli P, Saltini C, Selman M, Strange C, Wood B: An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012, 185 (9): 1004-1014. 10.1164/rccm.201202-0320ST.
Haslam PL, Baughman RP: Report of ERS Task Force: guidelines for measurement of acellular components and standardization of BAL. Eur Respir J. 1999, 14 (2): 245-248.
Douglas WW, Tazelaar HD, Hartman TE, Hartman RP, Decker PA, Schroeder DR, Ryu JH: Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med. 2001, 164 (7): 1182-1185. 10.1164/ajrccm.164.7.2103110.
Lee JY, Jin SM, Lee BJ, Chung DH, Jang BG, Park HS, Lee SM, Yim JJ, Yang SC, Yoo CG, Han SK, Shim YS, Kim YW: Treatment response and long term follow-up results of nonspecific interstitial pneumonia. J Korean Med Sci. 2012, 27 (6): 661-667. 10.3346/jkms.2012.27.6.661.
Suda T, Kono M, Nakamura Y, Enomoto N, Kaida Y, Fujisawa T, Imokawa S, Yasuda K, Hashizume H, Yokomura K, Toyoshima M, Koshimizu N, Suganuma H, Shirai T, Hashimoto D, Inui N, Colby TV, Chida K: Distinct prognosis of idiopathic nonspecific interstitial pneumonia (NSIP) fulfilling criteria for undifferentiated connective tissue disease (UCTD). Respir Med. 2010, 104 (10): 1527-1534. 10.1016/j.rmed.2010.04.022.
Fischer A, Meehan RT, Feghali-Bostwick CA, West SG, Brown KK: Unique characteristics of systemic sclerosis sine scleroderma-associated interstitial lung disease. Chest. 2006, 130 (4): 976-981. 10.1378/chest.130.4.976.
Tzelepis GE, Toya SP, Moutsopoulos HM: Occult connective tissue diseases mimicking idiopathic interstitial pneumonias. Eur Respir J. 2008, 31 (1): 11-20. 10.1183/09031936.00060107.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2466/14/175/prepub
Acknowledgments
The authors express their appreciation to all staff and physicians in Division of Pulmonary Disease PUMCH who cared for these patients between 2003 and 2012.
Appreciation is extended to Dr. Cartin-ceba Ridrigo, Division of Pulmonary and Critical Care Medicine, Mayo Clinic for his helpful suggestions on discussion and Dr. Guangxi Li, Division of Pulmonary and Critical Care Medicine, Mayo Clinic (e-mail: Li.guangxi@mayo.edu) for his double check on the statistic date.
Funding
This study was supported by the Ministry of Science and Technology of China “Key Projects in the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period” 2011BA11B17.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Study conceptualization and design: Dr. JS; Dr. JS takes responsibility for the content of the manuscript, including the data and analysis. Data collection: Drs WBX, YX and JS. Pathological review: Dr. HL. Statistical analyses: Dr. YX. Manuscript preparation: Drs JS and YX. A special thanks to Dr. WZ for her guide in the aspect of rheumatologic disease and Dr. MWQ for his radiology experience. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Xu, W., Xiao, Y., Liu, H. et al. Nonspecific interstitial pneumonia: clinical associations and outcomes. BMC Pulm Med 14, 175 (2014). https://doi.org/10.1186/1471-2466-14-175
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2466-14-175