
Abstract
Background
Previous results showed that over-expression of the WTH3 gene in MDR cells reduced MDR1 gene expression and converted their resistance to sensitivity to various anticancer drugs. In addition, the WTH3 gene promoter was hypermethylated in the MCF7/AdrR cell line and primary drug resistant breast cancer epithelial cells. WTH3 was also found to be directly targeted and up regulated by the p53 gene. Furthermore, over expression of the WTH3 gene promoted the apoptotic phenotype in various host cells.
Methods
To further confirm WTH3's drug resistant related characteristics, we recently employed the small hairpin RNA (shRNA) strategy to knockdown its expression in HEK293 cells. In addition, since the WTH3 promoter's p53-binding site was located in a CpG island that was targeted by methylation, we were interested in testing the possible effect this epigenetic modification had on the p53 transcription factor relative to WTH3 expression. To do so, the in vitro methylation method was utilized to examine the p53 transgene's influence on either the methylated or non-methylated WTH3 promoter.
Results
The results generated from the gene knockdown strategy showed that reduction of WTH3 expression increased MDR1 expression and elevated resistance to Doxorubicin as compared to the original control cells. Data produced from the methylation studies demonstrated that DNA methylation adversely affected the positive impact of p53 on WTH3 promoter activity.
Conclusion
Taken together, our studies provided further evidence that WTH3 played an important role in MDR development and revealed one of its transcription regulatory mechanisms, DNA methylation, which antagonized p53's positive impact on WTH3 expression.
Similar content being viewed by others


Background
Multidrug resistance (MDR) is a fatal event encountered during cancer chemotherapy [1–7]. To better understand MDR development, we employed the Methylation Sensitive-Representational Difference Analysis (MS-RDA) technique [8–10] to study DNA hypermethylation events in a human MDR breast cancer cell line, MCF7/AdrR, and its parental line, MCF7/WT. As a result, the WTH3 gene was discovered. WTH3 gene's product is homologous to the Rab6 and Rab6c genes that encode small G proteins and belong to the ras super family [9–14]. Similar to the Rab6s, WTH3 is a house-keeping gene and its product is capable of binding to GTP molecules [15]. However, unlike the Rab6s that reside in the Golgi network, most of WTH3 is located in the cytoplasm and to a lesser degree in the nuclei. This disparity could be due to WTH3's lack of a cysteine at its C-terminus for geranyl-geranylation, a necessary post-translational modification for membrane attachment [16]. Previous studies found that the WTH3 gene was down regulated in MDR cell lines, and by introducing it back into those lines caused down regulation of MDR1 gene expression that reversed their MDR phenotypes to various anti-cancer drugs [9, 15]. Our research revealed that hypermethylation (an epigenetic modification event in mammals, which represses gene expression) [17–22] of the WTH3 promoter and transcription factor modulation were involved in its differential expression in MCF7/AdrR versus MCF7/WT cells [15]. Furthermore, the hypermethylation event was also observed in primary drug resistant breast cancer cells [23].
Recently, we identified a p53-binding motif (p53M) in the WTH3 gene promoter, which was located in a CpG island that was targeted by DNA methylation [15, 23, 24]. The p53 gene product is a transcription factor that functions as a tumor suppressor and plays a pivotal role in apoptosis and cell cycle arrest [25–27]. In addition, various mutations of p53 were found to be associated with human cancers and the onset of MDR in a broad field of solid and hematological malignancies [28–34]. By performing the electrophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP) assays, we demonstrated that the WTH3 gene was a direct target of the p53 protein [24]. This relationship led us to evaluate the possible participation of WTH3 in promoting apoptosis via different approaches. Our findings suggested that over expression of WTH3 stimulated cell death [24]. As a result, we believed that this gene played an important role in MDR development.
To further understand WTH3's involvement in MDR, we carried out shRNA knockdown experiments to see if reduced WTH3 expression would increase tolerance of host cells to the anti-cancer drug, Doxorubicin (Dox). In addition, considering the physical interaction of p53 and the sequences subjected to DNA methylation, and a current observation that treating MCF7/AdrR cells with 5-aza-2'-deoxycytidine (5-aza), a DNA methylation inhibitor, further elevated p53 transgene activity, which in turn increased endogenous WTH3 expression in host cells, we explored the possible interplay between epigenetic modification and the p53 transcription factor regarding their influence on WTH3 gene expression.
Methods
Cell Lines and Treatment
MCF7/AdrR and MCF7/WT were grown at 37°C with 5% CO2 in DMEM medium with 10% FCS, 100 μg/ml streptomycin and 100 U/ml penicillin. HEK293 (human primary embryonic kidney cells, ATCC.) and Hela cells were grown at 37°C with 5% CO2 in RPMI 1640 culture medium with 10% FCS, 100 μg/ml streptomycin and 100 U/ml penicillin. To determine the influence of DNA methylation on p53 activity as it pertains to endogenous WTH3 gene expression, MCF7/AdrR cells were treated with 5-aza at 50 μM (this high concentration used was due to that the MCF7/Adr cell line was extremely drug resistant, whose IC50 was 975 nM, while MCF7/WT's IC50 was 1.25 nM[10]) for 24 and 72 hours, while Hela cells were treated with 5-aza at 5 μM for 24 hours.
Construction of Recombinant DNA
Detailed information about generating the full length WTH3 promoter in pGL3 to obtain the pGL/WTH3P construct and its deletion mutant with activity in pGL3 to generate the pGL/WTH3d3 construct were previously described [9, 15]. Wild type p53 in pcDNA/P53 and the mutated p53 gene in pcDNA/P53R249S, which did not contain trans-element-activity, were gifts from Dr. Moll M. Ute.
shRNA Knockdown
The WTH3 shRNA oligos, GATCCGTCAGGCAATAATTGGCATTGATTCAAGAGATCAATGCCAATTATTGCCTGACTTTTTTACGCGTG (sense) and AATTCACGCGTAAAAAAGTCAGGCAATAATTGGCATTGATCTCTTGAATCAATGCCAATTATTGCCTGACG (anti-sense) ending with BamH I and EcoR I restriction enzyme ends, were cloned into the pSIEN-RetroQ vector to obtain pSIEN-RetroQWTH3 according to the Clontech Knockout RNAi User Manual (PT3739-1). The infection procedure was performed based on the Clontech Retroviral Gene Transfer and Expression User Manual (PT3132-1). Briefly, pSIEN-RetroQWTH3 or pSIEN-RetroQ (negative control) along with the envelope vector, pAmpho were co-transfected into the GP2-293 packaging cell line via the phosphate calcium method. Viral supernatant was collected and centrifuged at 500 × g for 10 min to remove the cellular debris. The supernatant was then used to infect HEK293 cells. The cells with integrated WTH3 shRNA sequences were selected by adding 0.5 μg/ml puromycin into the medium for 2 weeks. The limiting dilution procedure was performed as previously described [9] to obtain individual HEK293 clones that were either integrated with pSIEN-RetroQWTH3 or pSIEN-RetroQ.
SQRT-PCR
Total RNAs were isolated from cell lines treated with 5-aza, transfectants and the corresponding negative controls by the High Pure RNA Isolation Kit (Roche). SQRT-PCR was performed using the Titan One Tube RT-PCR system based on the manufacturer's protocol (Roche). The sense and anti-sense primers for WTH3, MDR1 and GAPDH were previously described [9]. The sense and anti-sense primers for WTH3 were 5'-GATGGAACAATCGGGCTTCG-3' and 5'-GCTGCTACACGTCGAAAGAGC-3'. The sense and anti-sense primers for MDR1 were 5'-CCTATCATTGCAATAGCAGG-3' and 5'-GTTCAAACTTCTGCTCCTGA-3'. The length of the WTH3 and MDR1 PCR product was 341 and 167 bps, respectively. The SQRT-PCR assay for each gene of interest was performed more than three times. The PCR and quantification of PCR products were performed as noted [9, 10, 15, 23].
MTT Assay
MTT assays were carried out as described [9, 10]. Briefly, 2 × 103 cells/well were seeded in a 96-well plate and grown overnight. The cells were treated with serial concentrations of Dox (0 to 1 μg). In 6 days, the cells were then stained with 3- [4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT). IC50 (IC50 values represent Dox concentrations that cause 50% cell death) was quantitatively measured at 595 nm by the program software, EZ-ED50 Version 1.1 (Perrella Scientific Inc,) in a micro-plate spectrophotometer (Benchmark Plus, BIO-RAD).
In VitroDNA Methylation
To methylate the CpG sites in pGL/WTH3P and pGL/WTH3d3 plasmids, 2 μg of each construct was incubated with 12 U CpG methylase Sss I and 640 μM of S-adenosylmethionine (SAM) (New England Biolab) at 37°C for 3 hr. The temperature was then increased to 65°C for 20 min to terminate the reactions. The methylated plasmids, pGL/WTH3Pm and pGL/WTH3Pd3m, were purified with the Qiaquick PCR Purification Kit (Qiagen). The success of Sss I methylation was verified by methylation sensitive restrictive enzymes, Hha I and Hpa II.
Transient Transfection and Luciferase Assays
To determine whether the methylated WTH3 promoter influenced p53 transcriptional activity, pGL/WTH3Pm versus pGL/WTH3P and pGL/WTH3Pd3m versus pGL/WTH3Pd3 were co-transfected with pcDNA/P53, pcDNA/P53R249S (negative control) or pcDNA/3.1 (negative control) into HEK293 cells. In brief, 0.2 μg of each construct were transfected along with 0.1 μg of pCMV/β-galactosidase when the cells (seeded onto 24-well plates) reached 50–70% confluence. After 24 hrs, luciferase and β-galactosidase activity was measured using the Luciferase Assay System and Beta-Glo™Assay System (Promega) according to the manufacturer's instruction. Luciferase activities of transfectants were compared after normalizing their β-galactosidase activities and protein concentrations.
Results
The HEK293 cells with WTH3 knockdown resulted in higher resistance to Dox and stimulated MDR1gene expression
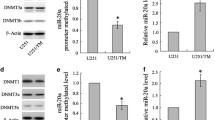
Earlier studies suggested that WTH3 was involved in MDR development[9, 23]. To further confirm this possibility, we decided to knockdown the WTH3 gene in HEK293 cells to see if reduced gene expression could increase the host cells' resistance to an anti-cancer drug, such as Dox. To do so, we employed the Clontech Knockout RNAi system to integrate a DNA sequence, which specifically interfered with the endogenous WTH3 transcripts, into the host cells' genome (see Materials and Methods for details). After antibiotic selection, RNAs were extracted from the cells infected with pSIEN-RetroQWTH3 or pSIEN-RetroQ (negative control), and quantified by SQRT-PCR. The results obtained from the three individual measurements showed that WTH3 transcripts in the cells infected with pSIEN-RetroQWTH3 (293/WTH3RNAi-P) were about 2 times lower than that in the control cells containing the empty vector (Fig. 1A, 1B). Therefore, the WTH3 gene was successfully knockdown in the HEK293 cell population. We then selected several individual cell clones via limiting dilution procedures. Consequently, we obtained 7 clones whose WTH3 expression was significantly lower due to the knockdown and 6 clones whose genome was integrated with the empty vector, but did not change WTH3 expression levels (Fig. 1A, 1B). The populations and three cloned cell lines, 293/WTH3RNAi-2, -3, -6, which were randomly picked, were used to perform MTT assays to obtain IC50s (the concentration of a drug that causes 50% cell death) to Dox. The results were an average of three individual experiments for each group. The IC50 value of the 293/WTH3RNAi-P cells was about 39 nM, while that of the control was about 13 nM. Clearly, the reduction of WTH3 gene expression made the host 3 times more resistance to Dox than its control, 293-V (Fig. 2A, 2B). Next, IC50s of the three cloned lines, 293/WTH3RNAi-2, -3 and -6 were also evaluated. The IC50 values to Dox were 44, 93, and 52 nM for 293/WTH3RNAi-2, -3 and -6, respectively, which were approximately 3.4, 7 and 4 times more resistance than the 293-V control (Fig. 2A, 2B, Table 1). Moreover, due to past findings that suggested a reverse correlation between WTH3 and MDR1 gene expression levels [23], we examined MDR1 gene expression levels in those cells by SQRT-PCR. As we expected, the results showed that the MDR1 gene was re-activated in the 293/WTH3RNAi-P, 293/WTH3RNAi-2, -3 and -6 cells, while MDR1 transcripts were undetectable in the corresponding control cells (Fig. 3A, 3B).

SQRT-PCR results for WTH3 expression in the knockdown cells, population (293/WTH3RNAi-P), 3 cloned cell lines (293/WTH3RNAi-2, -3, and -6), and 293 control cells (293-V) that were integrated with the empty vector. A. PCR products are displaced in an electrophoresis gel, while GAPDH served as positive control. B. Results of quantitative analysis for WTH3 expression in the cells presented in A. GAPDH served as quantitative reference.

MTT results obtained from the knockdown population and cloned cell lines, which were treated with Dox. A. Pharmacologic responsive plot for the knockdown population, 293/WTH3RNAi-P, versus control 293-V. B. Pharmacologic responsive plot for the cloned knockdown cell line, 293/WTH3RNAi-3 versus control 293-V. C. IC50 values of WTH3 knockdown 293 cells versus 293-V control cells to Dox.

SQRT-PCR results for MDR1 expression in population and cloned cell lines. A. The MDR1 gene's PCR products in various cell lines are displaced in an electrophoresis gel, while MDR1 expression in MCF7/AdrR and GAPDH served as positive controls. B. Results of quantitative analysis for MDR1 expression in the cells presented in A. GAPDH served as quantitative reference.
5-aza treatment promoted p53's positive impact on WTH3expression
Prior studies demonstrated that the CpG island found in the WTH3 gene promoter was targeted by DNA methylation in MCF7/AdrR cells [15, 23]. It was also discovered that this island contained p53M, which was directly bound by the p53 protein, a trans-element for activating WTH3 gene expression [24]. Based on these facts we speculated that DNA methylation could play an antagonistic role influencing the p53 transcription factor. To test this hypothesis, we transfected the pcDNA/P53 or pcDNA3.1 (negative control) construct into MCF7/AdrR cells (whose CpG island in the WTH3 promoter was methylated and p53 defective due to a mini-deletion) [32], and then treated the transfectants with 5-aza to see if the p53 transgene preferentially activated the WTH3 gene expression, but not in the untreated cells. After 24 and 72 hours of treatment, endogenous WTH3 transcript levels were evaluated by SQRT-PCR. We found that the p53 transgene's positive effect on the WTH3 gene expression of the cells treated with 5-aza was about 2 times stronger than that in the control cells who either contained the transgene or the empty vector, but were not treated with 5-aza (Fig. 4A, 4B). This suggested that DNA methylation could negatively affect p53 transactivity. In addition, as we expected, the 5-aza treat alone increased WTH3 transcript. Considering that Hela cells express a relatively low level of WTH3 and a relatively high IC50 value to Dox (data not shown), we introduced p53 transgene into those cells or treated them with 5-aza. It was observed that the p53 transgene and 5-aza positively affected the WTH3 expression of the cells compared to the untreated cells (Fig. 4C, 4D). These results further indicated that epigenetic modification and p53 regulated the WTH3 gene. To verify if DNA methylation could negatively affect p53 transactivity, the in vitro DNA methylation approach was employed to modify full length and deleted WTH3 promoters to analyzed p53's influence.

SQRT-PCR results for endogenous WTH3 gene expression in MCF7/AdrR and Hela cells. A. Electrophoresis gel display of the endogenous WTH3 gene expression in MCF7/AdrR cells that were transfected with pcDNA3.1 or p53, and transfected with pcDNA3.1 or p53 and treated with 5-aza for 24 or 72 hr. GAPDH served as positive control. B. Results of quantitative analysis for MDR1 expression in the cells presented in A. GAPDH served as quantitative reference.
P53 failed to activate the methylated WTH3promoter
To methylate full length and deleted WTH3 promoters, pGL/WTH3P and pGL/WTH3d3 were incubated with Sss I methylases and SAM. The resulting construct, pGL/WTH3Pm and pGL/WTH3d3m, their non-methylated controls, as well pCMV/β-galactosidase (transfection efficiency control) were then introduced into HEK293 cells. As we assumed, luciferase activities driven by both methylated promoters were 5 times lower than the corresponding controls (Fig. 5). Clearly, DNA methylation inhibited the promoters' function. We then co-transfected pGL/WTH3Pm versus pGL/WTH3P and pGL/WTH3d3m versus pGL/WTH3d3 with pcDNA/P53, or pcDNA/P53R249S (negative control), or pcDNA3.1 (negative control) into HEK293 cells to see if DNA methylation could antagonize p53 transactivity. The results showed that the p53 transgene only activated the non-methylated WTH3P and WTH3d3 promoters but had no effect on their methylated counterparts (Fig. 5). However, mutated p53, p53R249S, was unable to influence non-methylated WTH3P and WTH3d3 promoters. These findings indicated that there was interplay between DNA methylation and the p53 transcription factor regarding WTH3 gene expression regulation.

Luciferase activity driven by the methylated and non-methylated full length and deleted WTH3 promoters, pGL/WTH3P m , pGL/WTH3P, pGL/WTH3Pd3 m and pGL/WTH3Pd3 when they were co-transfected with pcDNA/P53, pcDNA/P53R249S, or pcDNA3.1 into HEK293 cells.
Discussion
Earlier studies suggested that WTH3 could be an important gene involved in the cellular MDR phenotype development. This idea was based on three observations, 1) its expression was reduced in cells exhibiting drug resistant traits [9, 15, 23]; 2) its restoration in MDR cell lines not only increased sensitivity to a variety of drugs, but also decreased endogenous MDR1 gene expression [9, 15, 23]; and 3) it was a direct target of p53 whose role during the onset of MDR has been well documented [26, 30, 32, 35–38]. To further verify WTH3's importance related to drug resistance, we recently employed the small hairpin RNA interference technique to permanently reduce its expression in non-MDR HEK293 cells to see if this could increase the host's tolerance to Dox. The reason for utilizing HEK293 was that it expressed normal amounts of the WTH3 gene, but undetectable MDR1 RNA, and was quite sensitive to Dox. The knockdown procedure was considered successful because WTH3 gene expression in the population and cloned host lines was 2 or more times below the original level. After measuring their IC50 values to Dox we found that all those cells with the WTH3 knockdown were 2 to 4 times more resistant to Dox than their controls. In addition, the knockdown was accompanied by significant MDR1 gene re-activation. These results were consistent with previous observations and supported the notion that there was a direct link between WTH3 gene function and MDR development. This relationship would be even more obvious if the WTH3 gene was completely knocked out by a traditional targeting knockout approach. This prediction is based on the fact that WTH3 gene expression was much lower (about 10 times) in MCF7/AdrR than in MCF7/WT cells, the former line processing a much stronger (about 100 times) MDR phenotype than the later one.
Considering that the WTH3 gene could play an important role in regards to the on set of MDR, we were interested in understanding the detailed mechanisms by which its transcription was regulated by DNA methylation and the p53 transcription factor. In the past, we presented data that the WTH3 gene promoter was hypermethylated not only in MCF7/AdrR but also drug resistant primary breast cancer epithelial cells [15, 23] and was physically targeted by p53 proteins [24]. We also found that the p53-binding site, p53M, in the gene promoter resided in a CpG island that was subjected to epigenetic modification. This information indicated that DNA methylation, whose role is to repress gene expression [39] could have a direct impact on p53 activated WTH3 expression. To test this hypothesis, two approaches were employed. The first included evaluating the p53 transgene's influence on WTH3 expression in MCF7/AdrR cells that were treated with 5-aza. The second was to study WTH3 promoter activity that was modified by methylation. The resulting information pointed out that DNA methylation significantly diminished promoter activity by at least one mechanism, by antagonizing p53 transactivity. Since, DNA methylation usually attracts methyl-binding-proteins (MBPs) and other epigenetic modification factors (EMFs), one could image that the methylated WTH3 promoter was bound by some MBPs and EMFs, which might change the chromatin structures and prevent p53 from recognizing its targeting structures.
Conclusion
Taken together, our studies provided solid evidence supporting the important role played by the WTH3 gene in MDR development and uncovered one of the mechanisms regulating its expression. Therefore, restoring or increasing this gene's activity could be another valuable strategy for easing MDR encountered during cancer chemotherapy. This could be achieved by introducing demethylation reagents if attenuated WTH3 gene activity in a patient was caused by DNA methylation.
References
Robinson LJ, Roberts WK, Ling TT, Lamming D, Sternberg SS, Roepe PD: Human MDR 1 protein overexpression delays the apoptotic cascade in Chinese hamster ovary fibroblasts. Biochemistry. 1997, 36 (37): 11169-11178. 10.1021/bi9627830.
Chen CJ, Chin JE, Ueda K, Clark DP, Pastan I, Gottesman MM, Roninson IB: Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986, 47 (3): 381-389. 10.1016/0092-8674(86)90595-7.
Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU, Duncan AM, Deeley RG: Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992, 258 (5088): 1650-1654. 10.1126/science.1360704.
Gros P, Ben Neriah YB, Croop JM, Housman DE: Isolation and expression of a complementary DNA that confers multidrug resistance. Nature. 1986, 323 (6090): 728-731. 10.1038/323728a0.
Gros P, Croop J, Housman D: Mammalian multidrug resistance gene: complete cDNA sequence indicates strong homology to bacterial transport proteins. Cell. 1986, 47 (3): 371-380. 10.1016/0092-8674(86)90594-5.
Johnstone RW, Cretney E, Smyth MJ: P-glycoprotein protects leukemia cells against caspase-dependent, but not caspase-independent, cell death. Blood. 1999, 93 (3): 1075-1085.
Smyth MJ, Krasovskis E, Sutton VR, Johnstone RW: The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc Natl Acad Sci USA. 1998, 95 (12): 7024-7029. 10.1073/pnas.95.12.7024.
Yuan L, Shan J, De Risi D, Broome J, Lovecchio J, Gal D, Vinciguerra V, Xu HP: Isolation of a novel gene, TSP50, by a hypomethylated DNA fragment in human breast cancer. Cancer Res. 1999, 59 (13): 3215-3221.
Shan J, Yuan L, Budman DR, Xu HP: WTH3, a new member of the Rab6 gene family, and multidrug resistance. Biochim Biophys Acta. 2002, 1589 (2): 112-123. 10.1016/S0167-4889(02)00164-7.
Shan J, Mason JM, Yuan L, Barcia M, Porti D, Calabro A, Budman D, Vinciguerra V, Xu H: Rab6c, a new member of the rab gene family, is involved in drug resistance in MCF7/AdrR cells. Gene. 2000, 257 (1): 67-75. 10.1016/S0378-1119(00)00395-4.
Echard A, Jollivet F, Martinez O, Lacapere JJ, Rousselet A, Janoueix-Lerosey I, Goud B: Interaction of a Golgi-associated kinesin-like protein with Rab6. Science. 1998, 279 (5350): 580-585. 10.1126/science.279.5350.580.
Echard A, Opdam FJ, de Leeuw HJ, Jollivet F, Savelkoul P, Hendriks W, Voorberg J, Goud B, Fransen JA: Alternative splicing of the human Rab6A gene generates two close but functionally different isoforms. Mol Biol Cell. 2000, 11 (11): 3819-3833.
Goud B, Zahraoui A, Tavitian A, Saraste J: Small GTP-binding protein associated with Golgi cisternae. Nature. 1990, 345 (6275): 553-556. 10.1038/345553a0.
Zahraoui A, Touchot N, Chardin P, Tavitian A: The human Rab genes encode a family of GTP-binding proteins related to yeast YPT1 and SEC4 products involved in secretion. J Biol Chem. 1989, 264 (21): 12394-12401.
Tian K, Jurukovski V, Yuan L, Shan J, Xu H: WTH3, which encodes a small G protein, is differentially regulated in multidrug-resistant and sensitive MCF7 cells. Cancer Res. 2005, 65 (16): 7421-7428. 10.1158/0008-5472.CAN-05-0658.
Barbacid M: ras genes. Annu Rev Biochem. 1987, 56: 779-827. 10.1146/annurev.bi.56.070187.004023.
Bird AP: CpG-rich islands and the function of DNA methylation. Nature. 1986, 321 (6067): 209-213. 10.1038/321209a0.
Antequera F, Bird A: CpG islands. Exs. 1993, 64: 169-185.
Doerfler W, Kruczek I, Eick D, Vardimon L, Kron B: DNA methylation and gene activity: the adenovirus system as a model. Cold Spring Harb Symp Quant Biol. 1983, 47 (Pt 2): 593-603.
Doerfler W: DNA methylation and gene activity. Annu Rev Biochem. 1983, 52: 93-124. 10.1146/annurev.bi.52.070183.000521.
Siegfried Z, Cedar H: DNA methylation: a molecular lock. Curr Biol. 1997, 7 (5): R305-307. 10.1016/S0960-9822(06)00144-8.
Kass SU, Landsberger N, Wolffe AP: DNA methylation directs a time-dependent repression of transcription initiation. Curr Biol. 1997, 7 (3): 157-165. 10.1016/S0960-9822(97)70086-1.
Tian K, Jurukovski V, Wang XP, Kaplan MH, Xu H: Epigenetic regulation of WTH3 in primary and cultured drug-resistant breast cancer cells. Cancer Res. 2005, 65 (21): 10024-10031. 10.1158/0008-5472.CAN-05-1944.
Tian K, Wang Y, Xu H: WTH3 is a direct target of the p53 protein. Br J Cancer. 2007, 96 (10): 1579-1586. 10.1038/sj.bjc.6603724.
Lowe SW: Cancer therapy and p53. Curr Opin Oncol. 1995, 7 (6): 547-553. 10.1097/00001622-199511000-00013.
Aas T, Borresen AL, Geisler S, Smith-Sorensen B, Johnsen H, Varhaug JE, Akslen LA, Lonning PE: Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat Med. 1996, 2 (7): 811-814. 10.1038/nm0796-811.
Righetti SC, Della Torre G, Pilotti S, Menard S, Ottone F, Colnaghi MI, Pierotti MA, Lavarino C, Cornarotti M, Oriana S, et al: A comparative study of p53 gene mutations, protein accumulation, and response to cisplatin-based chemotherapy in advanced ovarian carcinoma. Cancer Res. 1996, 56 (4): 689-693.
Schmitt CA, Lowe SW: Apoptosis and therapy. J Pathol. 1999, 187 (1): 127-137. 10.1002/(SICI)1096-9896(199901)187:1<127::AID-PATH251>3.0.CO;2-T.
Kim R: Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer. 2005, 103 (8): 1551-1560. 10.1002/cncr.20947.
Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW: Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene. 2004, 23 (16): 2934-2949. 10.1038/sj.onc.1207515.
Norbury CJ, Zhivotovsky B: DNA damage-induced apoptosis. Oncogene. 2004, 23 (16): 2797-2808. 10.1038/sj.onc.1207532.
Ogretmen B, Safa AR: Expression of the mutated p53 tumor suppressor protein and its molecular and biochemical characterization in multidrug resistant MCF-7/Adr human breast cancer cells. Oncogene. 1997, 14 (4): 499-506. 10.1038/sj.onc.1200855.
Smith ND, Rubenstein JN, Eggener SE, Kozlowski JM: The p53 tumor suppressor gene and nuclear protein: basic science review and relevance in the management of bladder cancer. J Urol. 2003, 169 (4): 1219-1228. 10.1097/01.ju.0000056085.58221.80.
Steele RJ, Lane DP: P53 in cancer: a paradigm for modern management of cancer. Surgeon. 2005, 3 (3): 197-205.
Xu H, Shan J, Jurukovski V, Yuan L, Li J, Tian K: TSP50 encodes a testis-specific protease and is negatively regulated by p53. Cancer Res. 2007, 67 (3): 1239-1245. 10.1158/0008-5472.CAN-06-3688.
Johnson RA, Shepard EM, Scotto KW: Differential regulation of MDR1 transcription by the p53 family members. Role of the DNA binding domain. J Biol Chem. 2005, 280 (14): 13213-13219. 10.1074/jbc.M414646200.
Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, Williams J, Lengauer C, Kinzler KW, Vogelstein B: Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999, 104 (3): 263-269. 10.1172/JCI6863.
Bartke T, Siegmund D, Peters N, Reichwein M, Henkler F, Scheurich P, Wajant H: p53 upregulates cFLIP, inhibits transcription of NF-kappaB-regulated genes and induces caspase-8-independent cell death in DLD-1 cells. Oncogene. 2001, 20 (5): 571-580. 10.1038/sj.onc.1204124.
Antequera F, Bird A: Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci USA. 1993, 90 (24): 11995-11999. 10.1073/pnas.90.24.11995.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/8/327/prepub
Acknowledgements
We thank for Dr. U. M. Moll and Dr. J. Cao for the DNA constructs and cell lines. We thank J. C. Duffy for preparation of the manuscript. This work was supported in part by the National Cancer Institute (Grant# 1R01CA090443-01A2) and American Cancer Society (Grant# RSG-03-137-01-CDD) awards.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
KT participated experiments designing and carried out shRNA Knockdown, DNA methylation and luciferase assay. YW participated cell culture and molecular cloning. YH carried out MTT assay. BS did the PCR and SQRT-PCR. YL carried out shRNA construction. HX conceived of the study, participated in its design and coordination, and drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tian, K., Wang, Y., Huang, Y. et al. Methylation of WTH3, a possible drug resistant gene, inhibits p53 regulated expression. BMC Cancer 8, 327 (2008). https://doi.org/10.1186/1471-2407-8-327
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-8-327