
Abstract
Background
Although muscle weakness is a hallmark of facioscapulohumeral muscular dystrophy (FSHD), the molecular mechanisms that lead to weakness in FSHD remain largely unknown. Recent studies suggest aberrant expression of genes involved in skeletal muscle development and sarcomere contractility, and activation of pathways involved in sarcomeric protein degradation. This study will investigate the contribution of sarcomeric protein dysfunction to the pathogenesis of muscle weakness in FSHD.
Methods/Design
Evaluation of sarcomeric function using skinned single muscle fiber contractile studies and protein analysis in muscle biopsies (quadriceps femoris and tibialis anterior) from patients with FSHD and age- and gender-matched healthy controls. Patients with other forms of muscular dystrophy and inflammatory myopathy will be included as disease controls to assess whether results are due to changes specific for FSHD, or a consequence of muscle disease in general. A total of 56 participants will be included. Extensive clinical parameters will be measured using MRI, quantitative muscle studies and physical activity assessments.
Discussion
This study is the first to extensively investigate muscle fiber physiology in FSHD following an earlier pilot study suggesting sarcomeric dysfunction in FSHD. The results obtained in this study will increase the understanding of the pathophysiology of muscle weakness in FSHD, and possibly identify novel targets for therapeutic intervention.
Similar content being viewed by others

Background
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common hereditary myopathy, affecting 1:15,000-20,000 people [1]. Age of onset and disease severity is variable, although in general presentation starts in the second decade of life [2]. FSHD derives its name from the characteristic early involvement of the muscles of the face, shoulder girdle and upper arm, which, in contrast to other muscular dystrophies, is often asymmetrical. Further in the disease course weakness progresses to the foot dorsiflexors, abdominal and proximal leg muscles [3]. About 20% of patients become wheelchair dependent [4].
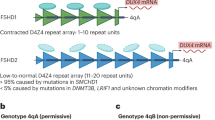
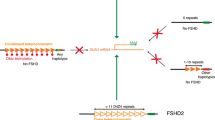
FSHD1 (OMIM #158900), the most common type of FSHD, is an autosomal dominant disorder caused by a contraction of the D4Z4 repeat array, a macrosatellite repeat array consisting of 3.3 kb large D4Z4 units located on chromosome 4q. FSHD patients have 1 – 10 repeat units, in contrast to unaffected individuals who have 11 – 100 units [5, 6]. Residual repeat length is roughly and inversely correlated to disease severity and onset. Patients having 1 – 3 repeats usually show infantile onset and rapid disease progression [7]. The repeat array contraction leads to a more open D4Z4 chromatin configuration, which is hypothesized to permit transcription of otherwise epigenetically silenced genes located on chromosome 4 [5, 6]. For FSHD to develop, there also needs to be a permissive genetic background of chromosome 4, as described below. In a minority of patients with a FSHD phenotype there is no D4Z4 contraction. These patients are categorized as FSHD2 and the majority of them have a mutation in the chromatin modifier SMCHD1 [8, 9]. Both FSHD1 and FSHD2 share the above-mentioned changes in D4Z4 chromatin configuration in somatic tissue on a FSHD-permissive chromosome 4 background.
FSHD candidate genes include Double Homeobox Protein 4 (DUX4) and FSHD Region Gene 1 (FRG1). Permissive chromosome 4 backgrounds contain a polymorphic DUX4 polyadenylation signal, which facilitates ectopic expression of DUX4 in skeletal muscle upon D4Z4 chromatin opening. DUX4 is currently considered the primary mediator of FSHD pathology, and activates the p53 and caspase-3 pathways when overexpressed in mice. Both of these pathways are involved in skeletal muscle differentiation, sarcomeric protein degradation and apoptosis [10, 11]. DUX4 may also activate ubiquitin-mediated protein degradation pathways including E3 ubiquitin ligases such as Atrogin-1 and MuRF1 [12, 13]. These muscle specific ligase enzymes specifically target sarcomeric proteins and have been implicated in the atrophic phenotype in FSHD myotubes [13, 14]. FRG1 is localized to the Z-disc of the sarcomere and functions as an actin-binding and bundling protein [15, 16]. Overexpression of FRG1 in mice causes muscular dystrophy and vascular abnormalities, both features of FSHD [17, 18]. Taken together, these findings suggest that expression of FSHD candidate genes such as DUX4 or FRG1 have downstream effects on sarcomere development and sarcomere turnover.
The sarcomere – the smallest contractile unit in muscle – is composed of several proteins that work together to enable muscle contraction. Upon activation with calcium, cross-bridges are formed between actin and myosin filaments. This induces filament sliding and enables muscle contraction. Other important constituents of the sarcomere include titin, which stabilizes the sarcomere and contributes to passive force, and nebulin, which plays an important role in the regulation of actin filament length.
We hypothesize that dysfunction of sarcomeric proteins contributes to the pathogenesis of muscle weakness in FSHD. A pilot study in 4 FSHD muscle biopsies performed in our laboratory supports this hypothesis [19]. The present study aims to confirm these data in a larger cohort and elucidate the causes of sarcomeric dysfunction in FSHD. Secondary objectives are:
-
To determine whether sarcomeric dysfunction is specific for FSHD, or part of a generalized pathology common to muscular dystrophy and/or inflammatory myopathy. To investigate this, patients with oculopharyngeal muscular dystrophy (OPMD) and sporadic inclusion body myositis (sIBM) will be included in the study as disease controls.
-
To explore other contractile properties of FSHD muscle fibers such as cross-bridge cycling kinetics, calcium sensitivity and passive force generation. With this, we aim to clarify the cause of sarcomeric dysfunction in FSHD.
-
Compare sarcomeric function in muscles affected early (tibialis anterior) and late (quadriceps femoris) in the disease course of FSHD to shed light on the remarkable distribution of weakness in FSHD. Also, by assessing these muscles within the same patient we will be able to determine when sarcomeric dysfunction occurs in the pathological process.
-
Correlate in vitro sarcomeric function to in vivo clinical parameters such as physical activity, muscle strength, disease severity, residual D4Z4 fragment length, muscle/fat fraction and quantitative muscle studies.
-
This study will also provide extensive new data on sarcomeric function in OPMD and sIBM. This will result in a better understanding of these disorders and generate new hypotheses for research and treatment.
Methods/Design
Study population
Genetically confirmed patients with FSHD1 will be recruited from the Radboud university medical center. Healthy individuals without a history of neuromuscular disease will be included as healthy controls. Two disease control groups (one with muscular dystrophy and one with inflammatory myopathy) will be included to determine whether the results obtained in this study are specific for FSHD, or a general feature of muscular dystrophy or inflammation – both histological features of FSHD. For the muscular dystrophy disease control group, patients with genetically confirmed OPMD (12–17 trinucleotide repeats in PABPN1) will be included. OPMD was chosen because it is a relatively common muscular dystrophy, has an adult age of onset and has no associated systemic features which might influence muscle fiber function. Also, patients are not likely to be wheelchair bound, which is an exclusion criterion for this study. For the inflammatory myopathy control group, patient with sIBM fulfilling the modified 2010 Griggs criteria will be included [20]. We have chosen sIBM because it is relatively common, has clearly delineated histological features and diagnostic criteria, and patients are not treated with corticosteroids, which is an exclusion criterion for this study. The full list of exclusion criteria is as follows:
-
Age <18 or >65 years
-
Diabetes mellitus
-
Chronic obstructive pulmonary disease
-
Chronic heart failure
-
Current malignancy
-
Previous treatment with chemotherapy and/or radiation therapy
-
Use of corticosteroids during more than two weeks in the past 5 years
Use of statins in the past year
-
Wheelchair bound
Contra-indications for MRI or muscle biopsy
-
Pregnancy
In each group, 14 participants will be included for a total of 56 participants. Age and gender matching will be applied on the group level. The Medical Ethics Review Committee region Arnhem-Nijmegen approved the study protocol.
In vivo study procedures
General
Information on medical history and current medication use will be collected. Body weight and length will be measured. Parameters of muscle injury will be assessed in serum (creatine kinase (CK), lactic dehydrogenase (LDH), aspartate transaminase (AST) and alanine transaminase (ALT)). In FSHD patients, disease severity will be assessed using the Ricci Clinical Severity Scale (CCS) and the CSS corrected for age: age-corrected CSS (ACCSS) [21–23].
Physical activity and muscle strength
Physical activity will be assessed in several ways. Participants will be equipped with an actometer, a device that is attached to the ankle to constantly measure physical activity during 12 days [24, 25]. Validated questionnaires will be administered to assess participants' daily living activities, disease impact and possible confounding factors such as pain: Health Assessment Questionnaire (HAQ) [26], International Physical Activity Questionnaire (IPAQ) [27], Sickness Impact Profile (SIP 68) [28], McGill’s Pain Questionnaire (MPQ) [29]. Muscle strength will be graded with the Medical Research Council (MRC) scale [30]. A trained examiner will obtain a Motor Function Measure [31]. In addition, a six-minute walk test and timed up and go test will be performed [32, 33].
Quantitative muscle studies
Isometric force recordings of maximal voluntary and electrical evoked contractions of the quadriceps femoris will be performed as described previously [34]. Participants will be seated in a custom-build chair with the knee angle set at 120°. To prevent compensatory movements, participants are strapped at the hips and upper body to maintain their position. Electrical pulses will be delivered through two self-adhesive surface electrodes placed proximally over the rectus femoris and distally over the vastus medialis portion of the quadriceps muscle. Two gel electrodes will be used to make a surface electromyogram (sEMG).
Maximum voluntary contraction
All measurements will be performed in the leg from which the muscle biopsy will be taken (i.e., the right leg, unless in the presence of asymmetrical weakness in which case the weakest leg will be biopsied). Participants will be asked to perform a Maximum Voluntary Contraction (MVC) during 3 seconds. The highest force from three contractions is used to represent the MVC. Next, the 100 Hz current that evokes 30% of MVC will be determined and used for all subsequent measurements.
Contractile properties
After a 5-minute rest period, the force–frequency relation will be determined with 1 Hz, 100 Hz, 1 Hz, 10 Hz, 20 Hz, 30 Hz, and 50 Hz pulse trains of 1 s duration. There will be a 1-minute rest period after each contraction. The 1 Hz stimulation will be performed multiple times to verify that the 100 Hz pulse train causes no fatigue or potentiation. The contraction time, rate of force rise, and early and late relaxation times will be derived from the 100 Hz contraction (positively filtered with a 30 Hz filter frequency). These parameters provide a measure for the rate of cross-bridge cycling and the rates of Ca2 reuptake by the sarcoplasmic reticulum.
Fatigue
After another 5-minute rest period, a series of 30 Hz 1 s on 1 s off pulses will be applied for a period of 2 minutes to determine muscle fatigability. Fatigue is determined as the relative decline of force between the first three contractions compared with that of the last three contractions.
MRI
A MR exam of the upper and lower leg will be performed in all participants using a 3 Tesla MR system (Tim TRIO, Siemens, Erlangen, Germany) with the spine array coil and two phased-array coils placed around the legs. A fish-oil marker will be placed on the site where the muscle biopsy will be taken from, to ensure that MRI findings correspond to the approximate site of muscle biopsy. Patients will be placed in the scanner feet first supine. The table will be positioned to have first the upper and subsequently the lower leg in the isocenter of the magnetic field for imaging of the thigh and calf muscles respectively.
Scout images will be acquired in three orthogonal directions for accurate positioning of the MRI slices. Thereafter 8 transversal slices (FOV 175×175 mm2, thickness 4 mm, gap 6 mm, baseresolution 256) will be acquired with a T2 multi spin echo sequence (TR: 3000 ms, 16 equally spaced echo times 7.7 – 123.2 ms). Subsequently followed by 23 transversal slices (thickness 4 mm, gap 0.4 mm) that will be obtained with a T1 turbo spin echo sequence (FOV 250×244.5 mm2, TR/TE 600 ms/13 ms, baseresolution 448), and with a Turbo Inversion Recovery sequence (TIRM) (FOV 175x175 mm2, TR/TE/TI 4100 ms/41 ms/220 ms, baseresolution 256). The same imaging protocol will be used for both the upper and lower leg.
Hyperintensities on the TIRM images will be detected visually as an indicator for edema and/or inflammation [35]. Muscle fraction at the slice corresponding to the approximate site of muscle biopsy will be quantified using the method described by Kan [36].
In vitro study procedures
Muscle biopsies
One needle muscle biopsy of the quadriceps femoris (vastus lateralis), and one of the tibialis anterior will be obtained from each participant. The tibialis anterior is frequently affected in FSHD, but early involvement of the quadriceps femoris is uncommon [3, 37]. In the presence of symmetrical muscle strength, the right side will be biopsied. In case of asymmetrical weakness based on neurological examination, the weakest muscle will be biopsied.
An experienced neurologist or neurology resident, taking routine antiseptic precautions, will perform the muscle biopsies. Biopsy material intended for histology will be frozen in isopentane and stored at −80°C. Material for protein analysis will be immediately frozen in liquid nitrogen and stored at −80°C. Specimens intended for contractile studies are stored at −20°C in a glycerinating solution containing half glycerol and half relaxing solution. For composition of this solution, see below.
Muscle fiber preparation
For contractile studies, biopsy material will be placed in a relaxing solution containing 1% (v/v) Triton X-100 at 4°C [38]. Triton is used to permeabilize the plasma membranes, resulting in “skinned” muscle fibers. The skinning process enables studies of sarcomeric function in isolation by eliminating the confounding effects of upstream processes of excitation-contraction coupling, such as sarcoplasmatic reticulum Ca2+ handling. Protease inhibitors will be added to the solution to prevent protein degradation. Skinned single muscle fibers will be isolated and fiber ends will be attached to aluminum t-clips. The clips are then mounted to a length motor on one end, and a force transducer on the other.
Muscle fiber mechanics
Maximum force generation and cross-bridge cycling kinetics
Skinned muscle fibers will be activated by sequentially bathing them in a relaxing solution, pre-activation solution and activation solution. The composition of these solutions has been described previously [39]. Force generating capacity will be measured at a sarcomere length of 2.5 μm. Maximal force generating capacity will be measured at pCa 4.5 and corrected for cross-sectional area. To determine whether changes in maximal force are due to changes in the kinetics of cross-bridge attachment and detachment, a rapid stretch-release protocol will be imposed on an activated fiber. The rate constant of tension redevelopment (Ktr) will be determined by fitting the rise of tension to the following equation: F = Fss(1 – e-Ktr·t) [40]. After force redevelopment reaches the steady-state maximal force again, active stiffness will be determined. Length changes of sequentially 0.3, 0.6, 0.9, -0.3, -0.6 and −0.9% will be applied, during which peak force is measured. Corrected peak force will be linearly plotted against the imposed length changes, with the slope representing the fraction of strongly attached cross bridges [41].
Calcium sensitivity of force generation
During daily-life muscle activation, individual fibers typically function at a submaximal level. Therefore, the calcium sensitivity of force generation provides additional information on contractile properties. The calcium sensitivity of force generation will be measured by bathing skinned muscle fibers in activation solutions with different pCas ranging from 4.5-7.0 and measuring steady-state force. The obtained force-pCa data will be fitted to the Hill equation, providing the pCa50 (the pCa giving 50% of maximal active tension) and the Hill coefficient, nH (an index of myofilament cooperativity) [42].
Passive force generation
Passive force generation will be measured by placing skinned muscle fibers in a chamber filled with relaxing solution and imposing length changes as previously described [43]. The fibers will be set to their slack length (i.e. the length at which passive force is zero) and from there they will be stretched with a constant velocity to a sarcomere length of 3.2 μm, held for 90 s and then released back to slack length. Tension development during stretch will be determined.
Muscle protein analysis
To study whether changes in sarcomere contractile function are a consequence of proteomic alterations, extensive protein analysis using SDS-PAGE and Western blotting will be performed as described previously [43, 44]. We will pay special attention to myosin heavy chains, actin, titin, nebulin and regulatory proteins such as troponin and tropomyosin. In addition we will determine oxidation/nitrosylation (i.e. posttranslational modifications in response to oxidative stress) of sarcomeric proteins by Oxyblot and Western blotting, as described previously [45].
To study whether decreased maximal force generation is a result of decreased contractile protein content, we will determine myosin content per half sarcomere and will relate the results to the force generated by those fibers as described previously [46]. In brief, myosin heavy chain (MHC) content and MHC isoform composition will be determined using SDS-PAGE. We will calculate fiber myosin concentration by dividing total MHC content by fiber volume (at a sarcomere length of 2.5 μm). This fiber myosin concentration will be multiplied by half-sarcomere volume to calculate MHC content per half sarcomere. Finally, MHC content per half sarcomere will be related to muscle fiber mechanics.
For analysis of titin and nebulin exon composition, we will use a titin/nebulin exon microarray that consists of 50-mer oligonucleotides representing all exons in the titin gene, spotted in triplicate on the array. The array contains human titin and nebulin genes as well as all known titin-binding proteins [47].
Histology
Histology will be used for light-microscopic evaluation of frozen sections. We will study size, shape, type and cytoarchitecture of muscle fibers, presence of internalized nuclei, destruction of muscle fibers and regeneration, as well as supporting connective tissue.
Outcome assessment
The primary outcome measurement of this study is the maximum force generating capacity (mN/mm2) of permeabilized single muscle fibers of FSHD patients and healthy control subjects. Secondary outcome measurements are:
-
The maximum force generating capacity of OPMD and sIBM fibers in relation to FSHD fibers.
-
Contractile properties of FSHD fibers such as cross-bridge cycling kinetics, calcium sensitivity and passive force generation in relation to fibers of healthy controls.
-
Maximum force generating capacity and contractile properties of muscle affected early (tibialis anterior) compared to muscle affected late (quadriceps femoris) in the disease course.
-
Analysis of sarcomeric proteins and titin and nebulin exon composition of FSHD muscle.
-
In vivo clinical parameters such as physical activity, muscle strength, disease severity, residual D4Z4 fragment length, muscle/fat fraction and quantitative muscle studies.
Statistical analysis and power calculation
Statistical analysis
Differences between the above-mentioned outcome measurements will be analyzed using analysis of covariance (ANCOVA). A p value <0.05 will be considered significant.
Power
Due to the exploratory nature of this study, there are virtually no previous studies to refer to for power calculations. Based on our pilot study, we aim to test the hypothesis that there is a difference in maximum force generating capacity of 7 mN/mm2 (~10% of FSHD maximum force generation), with standard deviations of 6 mN/mm2, α = 0.05 and β = 0.20. This hypothesis requires a sample size of 13 subjects per group. Because the muscle tissue can be damaged during the biopsy procedure, we anticipate that ~10% of the collected biopsies will not be useful for experiments. This brings the total sample size to 14 subjects per group.
Discussion
Over the past decades, the identification and understanding of the complex genetic and epigenetic mechanism underlying FSHD has been the primary focus of the scientific community. Recently, this effort resulted in a unifying genetic model for FSHD [5]. However, many questions concerning the molecular and physiological mechanisms underlying muscle weakness in FSHD remain unanswered. This study aims to look at the pathophysiology of FSHD from a different perspective, and is the first to extensively investigate the pathophysiology of muscle weakness in FSHD.
A major strength of this study is the combined analysis of extensive in vitro and in vivo data in the same cohort of patients and control subjects. This will provide the unique opportunity to increase our understanding of the relationship between pathophysiological mechanisms and the clinical condition of the patient. Biopsies of two muscles with either early or late involvement (tibialis anterior and quadriceps femoris, respectively) will be collected specifically for this study to provide insight into the consequences of disease progression and the distribution of weakness in FSHD. Studies of patients with other forms of muscular dystrophy and inflammatory myopathy will not only indicate whether the observed changes found are specific to FSHD or a consequence of neuromuscular disease in general, but will also provide unique insights into the pathophysiology of these disorders.
The study of muscle biopsies from FSHD patients means that the data acquired will reflect consequences of pathology in adult, differentiated human muscle cells. This prevents possible confounders that may arise with other frequently used methods in FSHD research such as myotubes, myoblasts or mouse models.
As in all studies that use biopsies, the sampled material represents only a small part of the muscle. This is a risk especially in FSHD, which is characterized by a marked difference in muscle pathology even within the same muscle. In this study, we will use MRI imaging of the area of muscle biopsy to provide information on atrophy, muscle/fat fraction and inflammation to correct for sampling error.
In conclusion, this study will provide new insights in sarcomeric function of patients suffering from FSHD, and correlate these to clinical measures in the same patients. This will be an important step in the understanding of the pathophysiology of muscle weakness in this disease, possibly identifying novel targets for therapeutic intervention.
References
Emery AE: Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord. 1991, 1 (1): 19-29. 10.1016/0960-8966(91)90039-U.
van der Maarel SM, Frants RR, Padberg GW: Facioscapulohumeral muscular dystrophy. Biochim Biophys Acta. 2007, 1772 (2): 186-194. 10.1016/j.bbadis.2006.05.009.
Padberg GW: Facioscapulohumeral dystrophy. 1982, Leiden, the, Netherlands: Leiden University
Pandya S, King WM, Tawil R: Facioscapulohumeral dystrophy. Phys Ther. 2008, 88 (1): 105-113. 10.2522/ptj.20070104.
Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camano P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, et al: A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010, 329 (5999): 1650-1653. 10.1126/science.1189044.
van der Maarel SM, Tawil R, Tapscott SJ: Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol Med. 2011, 17 (5): 252-258. 10.1016/j.molmed.2011.01.001.
Lunt PW, Jardine PE, Koch MC, Maynard J, Osborn M, Williams M, Harper PS, Upadhyaya M: Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD). Hum Mol Genet. 1995, 4 (5): 951-958. 10.1093/hmg/4.5.951.
de Greef JC, Lemmers RJ, Camano P, Day JW, Sacconi S, Dunand M, van Engelen BG, Kiuru-Enari S, Padberg GW, Rosa AL, et al: Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010, 75 (17): 1548-1554. 10.1212/WNL.0b013e3181f96175.
Lemmers RJ, Tawil R, Petek LM, Balog J, Block GJ, Santen GW, Amell AM, van der Vliet PJ, Almomani R, Straasheijm KR, et al: Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nature genetics. 2012, 44 (12): 1370-1374. 10.1038/ng.2454.
Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE: Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004, 113 (1): 115-123.
Wallace LM, Garwick SE, Mei W, Belayew A, Coppee F, Ladner KJ, Guttridge D, Yang J, Harper SQ: DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Annals Neurology. 2011, 69 (3): 540-552. 10.1002/ana.22275.
Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, van der Maarel SM, Ruzzo WL, Gentleman RC, Tawil R, et al: DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell. 2012, 22 (1): 38-51. 10.1016/j.devcel.2011.11.013.
Vanderplanck C, Ansseau E, Charron S, Stricwant N, Tassin A, Laoudj-Chenivesse D, Wilton SD, Coppee F, Belayew A: The FSHD atrophic myotube phenotype is caused by DUX4 expression. PLoS One. 2011, 6 (10): e26820-10.1371/journal.pone.0026820.
Lokireddy S, McFarlane C, Ge X, Zhang H, Sze SK, Sharma M, Kambadur R: Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting. Mol Endocrinol. 2011, 25 (11): 1936-1949. 10.1210/me.2011-1124.
Liu Q, Jones TI, Tang VW, Brieher WM, Jones PL: Facioscapulohumeral muscular dystrophy region gene-1 (FRG-1) is an actin-bundling protein associated with muscle-attachment sites. J Cell Sci. 2010, 123 (Pt 7): 1116-1123.
Hanel ML, Sun CY, Jones TI, Long SW, Zanotti S, Milner D, Jones PL: Facioscapulohumeral muscular dystrophy (FSHD) region gene 1 (FRG1) is a dynamic nuclear and sarcomeric protein. Differ res Biol Diversity. 2011, 81 (2): 107-118. 10.1016/j.diff.2010.09.185.
Gabellini D, D'Antona G, Moggio M, Prelle A, Zecca C, Adami R, Angeletti B, Ciscato P, Pellegrino MA, Bottinelli R, et al: Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature. 2006, 439 (7079): 973-977.
Wuebbles RD, Hanel ML, Jones PL: FSHD region gene 1 (FRG1) is crucial for angiogenesis linking FRG1 to facioscapulohumeral muscular dystrophy-associated vasculopathy. Disease Models Mechanisms. 2009, 2 (5–6): 267-274.
Lassche S, Stienen GJ, Irving TC, van der Maarel SM, Voermans NC, Padberg GW, Granzier H, van Engelen BG, Ottenheijm CA: Sarcomeric dysfunction contributes to muscle weakness in facioscapulohumeral muscular dystrophy. Neurology. 2013, 80 (8): 733-737. 10.1212/WNL.0b013e318282513b.
Hilton-Jones D, Miller A, Parton M, Holton J, Sewry C, Hanna MG: Inclusion body myositis: MRC Centre for Neuromuscular Diseases, IBM workshop, London, 13 June 2008. Neuromuscular Disord: NMD. 2010, 20 (2): 142-147. 10.1016/j.nmd.2009.11.003.
Mathieu J, Boivin H, Meunier D, Gaudreault M, Begin P: Assessment of a disease-specific muscular impairment rating scale in myotonic dystrophy. Neurology. 2001, 56 (3): 336-340. 10.1212/WNL.56.3.336.
Ricci E, Galluzzi G, Deidda G, Cacurri S, Colantoni L, Merico B, Piazzo N, Servidei S, Vigneti E, Pasceri V, et al: Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Annals Neurology. 1999, 45 (6): 751-757. 10.1002/1531-8249(199906)45:6<751::AID-ANA9>3.0.CO;2-M.
van Overveld PG, Enthoven L, Ricci E, Rossi M, Felicetti L, Jeanpierre M, Winokur ST, Frants RR, Padberg GW, van der Maarel SM: Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Annals Neurology. 2005, 58 (4): 569-576. 10.1002/ana.20625.
Kalkman JS, Schillings ML, Zwarts MJ, van Engelen BG, Bleijenberg G: The development of a model of fatigue in neuromuscular disorders: a longitudinal study. J Psychosom Res. 2007, 62 (5): 571-579. 10.1016/j.jpsychores.2006.11.014.
van der Werf SP, Prins JB, Vercoulen JH, van der Meer JW, Bleijenberg G: Identifying physical activity patterns in chronic fatigue syndrome using actigraphic assessment. J Psychosom Res. 2000, 49 (5): 373-379. 10.1016/S0022-3999(00)00197-5.
Fries JF, Spitz P, Kraines RG, Holman HR: Measurement of patient outcome in arthritis. Arthritis and rheumatism. 1980, 23 (2): 137-145. 10.1002/art.1780230202.
Craig CL, Marshall AL, Sjostrom M, Bauman AE, Booth ML, Ainsworth BE, Pratt M, Ekelund U, Yngve A, Sallis JF, et al: International physical activity questionnaire: 12-country reliability and validity. Med Sci Sports Exerc. 2003, 35 (8): 1381-1395. 10.1249/01.MSS.0000078924.61453.FB.
Jacobs HM, Luttik A, Touw Otten FW, De Melker RA: The sickness impact profile; results of an evaluation study of the Dutch version]. Nederlands tijdschrift voor geneeskunde. 1990, 134 (40): 1950-1954.
Vanderiet K, Adriaensen H, Carton H, Vertommen H: The McGill Pain Questionnaire constructed for the Dutch language (MPQ-DV). Preliminary data concerning reliability and validity. Pain. 1987, 30 (3): 395-408. 10.1016/0304-3959(87)90027-3.
Council MR: Aids to the examination of the peripheral nervous system, Memorandum no 45. edn. 1981, London, UK: Her Majesty's Stationery Office
Berard C, Payan C, Hodgkinson I, Fermanian J: A motor function measure for neuromuscular diseases, construction and validation study. Neuromuscul Disord. 2005, 15 (7): 463-470. 10.1016/j.nmd.2005.03.004.
Enright PL: The six-minute walk test. Respir Care. 2003, 48 (8): 783-785.
Podsiadlo D, Richardson S: The timed "Up & Go": a test of basic functional mobility for frail elderly persons. J Am Geriatr Soc. 1991, 39 (2): 142-148.
Degens H, Sanchez Horneros JM, Heijdra YF, Dekhuijzen PN, Hopman MT: Skeletal muscle contractility is preserved in COPD patients with normal fat-free mass. Acta physiologica Scandinavica. 2005, 184 (3): 235-242. 10.1111/j.1365-201X.2005.01447.x.
Wattjes MP, Kley RA, Fischer D: Neuromuscular imaging in inherited muscle diseases. Eur Radiol. 2010, 20 (10): 2447-2460. 10.1007/s00330-010-1799-2.
Kan HE, Scheenen TW, Wohlgemuth M, Klomp DW, van Loosbroek-Wagenmans I, Padberg GW, Heerschap A: Quantitative MR imaging of individual muscle involvement in facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2009, 19 (5): 357-362. 10.1016/j.nmd.2009.02.009.
The FSH-DY Group: A prospective, quantitative study of the natural history of facioscapulohumeral muscular dystrophy (FSHD): implications for therapeutic trials. Neurology. 1997, 48 (1): 38-46.
Ottenheijm CA, Hooijman P, DeChene ET, Stienen GJ, Beggs AH, Granzier H: Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2). J Struct Biol. 2010, 170 (2): 334-343. 10.1016/j.jsb.2009.11.013.
Stienen GJ, Kiers JL, Bottinelli R, Reggiani C: Myofibrillar ATPase activity in skinned human skeletal muscle fibres: fibre type and temperature dependence. J Physiol. 1996, 493 (Pt 2): 299-307.
Ottenheijm CA, Lawlor MW, Stienen GJ, Granzier H, Beggs AH: Changes in cross-bridge cycling underlie muscle weakness in patients with tropomyosin 3-based myopathy. Hum Mol Genet. 2011, 20 (10): 2015-2025. 10.1093/hmg/ddr084.
Chandra M, Mamidi R, Ford S, Hidalgo C, Witt C, Ottenheijm C, Labeit S, Granzier H: Nebulin alters cross-bridge cycling kinetics and increases thin filament activation: a novel mechanism for increasing tension and reducing tension cost. J Biol Chem. 2009, 284 (45): 30889-30896. 10.1074/jbc.M109.049718.
Ottenheijm CA, Witt CC, Stienen GJ, Labeit S, Beggs AH, Granzier H: Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum Mol Genet. 2009, 18 (13): 2359-2369. 10.1093/hmg/ddp168.
Ottenheijm CA, Knottnerus AM, Buck D, Luo X, Greer K, Hoying A, Labeit S, Granzier H: Tuning passive mechanics through differential splicing of titin during skeletal muscle development. Biophys J. 2009, 97 (8): 2277-2286. 10.1016/j.bpj.2009.07.041.
Warren CM, Krzesinski PR, Greaser ML: Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis. 2003, 24 (11): 1695-1702. 10.1002/elps.200305392.
Ottenheijm CA, Heunks LM, Geraedts MC, Dekhuijzen PN: Hypoxia-induced skeletal muscle fiber dysfunction: role for reactive nitrogen species. Am J Physiol Lung Cell Mol Physiol. 2006, 290 (1): L127-L135.
Ottenheijm CA, Heunks LM, Sieck GC, Zhan WZ, Jansen SM, Degens H, de Boo T, Dekhuijzen PN: Diaphragm dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005, 172 (2): 200-205. 10.1164/rccm.200502-262OC.
Lahmers S, Wu Y, Call DR, Labeit S, Granzier H: Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res. 2004, 94 (4): 505-513. 10.1161/01.RES.0000115522.52554.86.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2377/13/144/prepub
Acknowledgements
This study is supported by the Prinses Beatrix Spierfonds and Stichting Spieren voor Spieren (W.OR10-30). B. Janssen is supported by the Prinses Beatrix Spierfonds (WAR08-15).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BGME and CACO designed the study and obtained funding. HJW performed extensive preparative work prior to the start of this study and drafted the study protocol. SL is the primary investigator of this study and responsible for collection, analysis, and interpretation of the data, and wrote the manuscript. BHJ designed the MRI scanning protocol and supervises analysis and interpretation of MRI data. MTH supervises the collection and analysis of data for quantitative muscle studies. CACO and GJMS will supervise collection and analysis of data for contractile studies. CACO, NCV, BGME, GJMS, SMvdM and GWP will supervise the execution of the study and interpretation of the data. All authors read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lassche, S., Ottenheijm, C.A., Voermans, N.C. et al. Determining the role of sarcomeric proteins in facioscapulohumeral muscular dystrophy: a study protocol. BMC Neurol 13, 144 (2013). https://doi.org/10.1186/1471-2377-13-144
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2377-13-144