
Abstract
Background
Mutation in several podocyte-specific genes have been noted to result in phenotypic heterogeneity. Herein, we report a novel, autosomal dominant TRPC6 mutation in a family with disease ranging from asymptomatic minimal change disease to end-stage kidney disease.
Case presentation
A 35 year old woman developed asymptomatic, nephrotic range proteinuria during pregnancy that did not resolve after delivery. Her mother had end-stage kidney disease of unknown etiology and her brother had asymptomatic proteinuria. Kidney biopsy revealed minimal change disease in both the proband and her brother. Genetic testing was performed in the proband and mother, revealing a novel frameshift mutation in TRPC6, D873fsX878. The proband continues to have subnephrotic range proteinuria and normal creatinine but her brother has since developed progressive chronic kidney disease.
Conclusions
The current case report underscores the heterogeneity of disease in podocytopathies and related genes. Genetic testing of podocyte genes is useful in order to understand the pathophysiologic processes underlying these overlapping diseases.
Similar content being viewed by others


Background
Diseases of the podocyte most commonly manifest as minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) in children and adults, respectively. A substantial proportion of both diseases are due to genetic aberrations of genes that are important to podocyte structure or function. The majority of the currently recognized genetic forms of MCD and FSGS follow simple Mendelian inheritance patterns [1]. Mutation in the podocyte-specific gene TRPC6 was first identified as causing autosomal dominant (AD) FSGS in 2005 [2, 3]. A total of 16 mutations in TRPC6 have since been cited in both familial and sporadic FSGS as well as in adult and childhood onset disease [2–11]. To date, only a single family has been identified as having kidney histopathology other than FSGS [8]. Herein, we add to this body of literature by reporting a novel TRPC6 mutation in a family with phenotypic heterogeneity ranging from asymptomatic minimal change disease to end-stage kidney disease (ESKD).
Case presentation
A 34 year-old Korean woman was in her first trimester of pregnancy when she developed proteinuria. She was not edematous and did not have hypertension. Her proteinuria became increasingly severe and by her third trimester of pregnancy her urine protein to creatinine ratio (UPC) was greater than 10 gm/gm. She was induced at 37 weeks gestation and delivered a healthy boy who had no complications. Her proteinuria did not resolve following delivery and percutaneous kidney biopsy was performed. This demonstrated 1 out of 20 globally sclerotic glomeruli with no other abnormalities on light microscopy. Immunofluorescent staining was negative. Electron microscopy revealed moderate podocyte effacement and segmental basement membrane thinning. She has persistently declined treatment, including use of angiotensin converting enzyme inhibitors and all forms of immunomodulatory therapy. Despite this, her UPC has been stable for several years at 1.0 gm/gm and her creatinine has consistently measured less than 1.0 gm/dl.
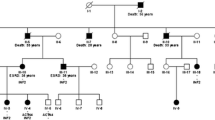
There was suspicion of a genetic component to her disease as her mother had developed end-stage kidney disease in her mid-50s and her brother had proteinuria progressing to chronic kidney disease (Figure 1). The mother presented with shrunken kidneys and had not been able to undergo biopsy; she underwent kidney transplantation at 55 years and has had no proteinuria since that time. The brother’s clinical history has not been confirmed due to lack of consent. Per his sister’s report, proteinuria was initially mild and he opted against kidney biopsy until his proteinuria worsened and he subsequently developed chronic renal insufficiency. His biopsy reportedly demonstrated minimal change disease and he was treated with oral steroids without any improvement.

Genealogical tree of a non-consanguineous, Korean family with minimal change disease and a novel TRPC6 deletion mutation in exon 12.
Genetic testing of TRPC6 was performed due to the apparent autosomal dominant inheritance pattern. She was found to be heterozygous for a novel TRPC6 frameshift mutation resulting from a 4 base pair GATA deletion in exon 12 [c. 2617–2620 del GATA, p. D873fsX878] (Figure 2). The reported mutation is novel, as it has not been seen in over 90 patients with nephrotic syndrome sequenced in our laboratory, is not reported in any dbSNP or exome sequencing databases of normal controls, and has not been previously reported in TRPC6. The resultant frameshift leads to a premature stop codon, and is predicted to result in a truncated TRPC6 protein lacking the C-terminal coiled coil domain. The proband’s mother also tested positive for the same mutation but her brother did not wish to be tested. DNA sequencing for mutations in ACTN4, INF2, and NPHS2 associated with adolescent or adult onset nephrotic syndrome revealed no other mutations in this family (data not shown). Due to segmental basement membrane thinning on the proband’s kidney biopsy and the possibility of X-linked transmission in this family, the collagen 4 alpha 5 (COL4A5) gene was also sequenced in the proband’s mother and was negative for mutations.

Sequence analysis of the novel TRPC6 mutation found in 3 family members. Shown is a partial electropherogram of Sanger DNA sequencing analysis of TRPC6 exon12 from the proband. The arrow shows the position of the heterozygous four base pair deletion resulting in a downstream frameshift. Shown above are the nucleotide and predicted amino acid sequences of the wild-type TRPC6 sequence (top line) and the heterozygous four base pair deletion (bottom line). The deleted nucleotides (GATA) are depicted in red font. The four base pair deletion results in a frame shift and premature protein truncation, five amino acids downstream [p.D873fsX878].
Conclusions
TRPC6 is a transient receptor potential (TRP) channel that plays a role in intracellular calcium signaling and is expressed in a signaling complex with nephrin and podocin in the podocyte slit diaphragm [1]. Although the reported TRPC6 frameshift mutation is novel, it encodes a truncated TRPC6 protein lacking the C-terminal coiled coil domain, a highly conserved domain thought to play an essential role in TRPC6 calcium channel function. This truncating mutation is likely to have a dominant gain-of-function effect similar to other known pathogenic TRPC6 mutations (Table 1) [3, 6]. Other TRPC6 mutations mapped to the coiled-coil domain at the C-terminus, such as R895C and E897K, have been demonstrated to result in increased calcium ion influx [3, 6]. The C-terminal K874X nonsense mutation and N-terminal mutations within the ankyrin repeats have demonstrated delayed calcium channel inactivation, resulting in increased channel opening time [5, 12]. The D873fsX878 mutation results in protein truncation only four amino acids downstream of the truncating K874X mutation and may have a similar effect on delaying calcium channel inactivation. The tracking of this mutation with nephrotic syndrome in this family supports an autosomal dominant gain of function effect. The mechanism whereby increased calcium current leads to the pathogenic manifestations of MCD or FSGS is unknown. TRPC6 functions as a mechanoreceptor of membrane stretch [1]. Other pathogenic mechanisms of TRPC6 mutation that have been hypothesized include altered channel regulation, altered interaction with other slit diaphragm proteins, and altered protein turnover [3]. It has been speculated that mutations in TRPC6 may result in apoptosis, podocyte detachment or an alteration in the ultrafiltration coefficient [12, 13].
To date, the vast majority of identified mutations in TRPC6 have been associated with FSGS, including collapsing glomerulopathy [2–8, 10, 11]. The current report underscores the phenotypic heterogeneity that can occur in podocyte-specific gene mutations. We have replicated the finding that TRPC6 mutation can result in MCD as well as FSGS [8]. Gigante et al. (2011) previously reported a family with a distinct TRPC6 mutation resulting in childhood onset nephrotic syndrome, with one sibling having MCD and another having IgAN with a membranoproliferative-like pattern. In some cases, the finding of MCD is due to unsampled FSGS and in other instances it may represent an earlier clinical stage of FSGS, which could be true in the present case. TRPC6 mutation has traditionally been thought to cause familial, adult onset disease; however, a significant number of sporadic and childhood onset FSGS cases have been identified [5–8]. Other podocyte-specific genes have also been found to result in multiple pathologic and clinical disease phenotypes. Mutations in podocin (NPHS2) have been reported in FSGS, steroid resistant MCD and diffuse mesangial sclerosis [14]. Phenotypic heterogeneity also exists for nephrin (NPHS1) mutations which have been reported to result in congenital nephrotic syndrome, steroid-resistant MCD and FSGS [15]. Given the heterogeneity in disease that can result from mutations within the same gene, it has been suggested that these disease entities be referred to as ‘podocytopathies’ rather than by their histologic phenotype [16].
This family demonstrates the significant heterogeneity in clinical manifestations that can occur from the same genetic mutation. Although genotype-phenotype correlation of this novel TRPC6 mutation is not known, all three affected adults presented with proteinuria at 32–34 years of age, with progressive chronic kidney disease subsequently developing in the proband’s mother and brother. However, the proband has thus far remained stable following pregnancy-induced proteinuria without any increase in creatinine. There are likely other genetic and/or environmental modifiers that play a significant role in disease penetrance and expressivity. Currently, there are no known genetic modifiers of TRPC6-associated disease. Modifiers for TRPC6-mediated podocytopathies can be speculated when considering that TRPC6 is partially regulated by podocin and is thought to be assembled in a complex with nephrin. The R229Q single nucleotide polymorphism in podocin (NPHS2) results in decreased nephrin binding and can result in both familial and seemingly sporadic FSGS when in a compound heterozygous state with another deleterious NPHS2 mutation [17]. Heterozygous lesions in collagen IV have also been hypothesized to modify susceptibility to FSGS [18]. Although sequence analysis of both NPHS2 and the X-linked collagen IV chain gene COL4A5 was negative in this family, other genetic or environmental factors may play a role in modifying disease severity within a family and in determining whether TRPC6 mutation presents as FSGS or MCD in different families.
This case report underscores the complexity in nomenclature, pathogenesis and genetics of podocytopathies. The clinical presentation, histological pattern, causative genes and specific mutations should all be taken into consideration when assessing a familial or even seemingly sporadic case of proteinuria. Documentation of atypical cases of familial proteinuric syndromes along with technological advances in genetic testing will facilitate a better understanding of diseases of the podocyte.
Consent
Written, informed consent was obtained from the patient for publication of this case report.
Abbreviations
- MCD:
-
Minimal Change Disease
- FSGS:
-
Focal Segmental Glomerulosclerosis
- ESKD:
-
End-Stage Kidney Disease.
References
Machuca E, Benoit G, Antignac C: Genetics of nephrotic syndrome: connecting molecular genetics to podocyte physiology. Hum Mol Genet. 2009, 18 (R2): R185-R194. 10.1093/hmg/ddp328.
Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB: A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005, 308 (5729): 1801-1804. 10.1126/science.1106215.
Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR: TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005, 37 (7): 739-744. 10.1038/ng1592.
Zhu B, Chen N, Wang ZH, Pan XX, Ren H, Zhang W, Wang WM: Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat Res. 2009, 664 (1–2): 84-90.
Heeringa SF, Moller CC, Du J, Yue L, Hinkes B, Chernin G, Vlangos CN, Hoyer PF, Reiser J, Hildebrandt F: A novel TRPC6 mutation that causes childhood FSGS. PLoS One. 2009, 4 (11): e7771-10.1371/journal.pone.0007771.
Santin S, Ars E, Rossetti S, Salido E, Silva I, Garcia-Maset R, Gimenez I, Ruiz P, Mendizabal S, Luciano Nieto J, Pena A, Camacho JA, Fraga G, Cobo MA, Bernis C, Ortiz A, de Pablos AL, Sanchez-Moreno A, Pintos G, Mirapeix E, Fernandez-Llama P, Ballarin J, Torra R, Zamora I, Lopez-Hellin J, Madrid A, Ventura C, Vilalta R, Espinosa L, Garcia C: TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2009, 24 (10): 3089-3096. 10.1093/ndt/gfp229.
Buscher AK, Kranz B, Buscher R, Hildebrandt F, Dworniczak B, Pennekamp P, Kuwertz-Broking E, Wingen AM, John U, Kemper M, Monnens L, Hoyer PF, Weber S, Konrad M: Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2010, 5 (11): 2075-2084. 10.2215/CJN.01190210.
Gigante M, Caridi G, Montemurno E, Soccio M, D’Apolito M, Cerullo G, Aucella F, Schirinzi A, Emma F, Massella L, Messina G, De Palo T, Ranieri E, Ghiggeri GM, Gesualdo L: TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011, 6( (7): 1626-1634.
Liakopoulos V, Huerta A, Cohen S, Pollak MR, Sirota RA, Superdock K, Appel GB: Familial collapsing focal segmental glomerulosclerosis. Clin Nephrol. 2011, 75 (4): 362-368.
Mir S, Yavascan O, Berdeli A, Sozeri B: TRPC6 gene variants in Turkish children with steroid-resistant nephrotic syndrome. Nephrol Dial Transplant. 2012, 27 (1): 205-209. 10.1093/ndt/gfr202.
Buscher AK, Konrad M, Nagel M, Witzke O, Kribben A, Hoyer PF, Weber S: Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin Nephrol. 2012, 78 (1): 47-53.
El Hindi S, Reiser J: TRPC channel modulation in podocytes-inching toward novel treatments for glomerular disease. Pediatr Nephrol. 2011, 26 (7): 1057-1064. 10.1007/s00467-010-1718-4.
Winn MP: 2007 Young Investigator Award: TRP'ing into a new era for glomerular disease. J Am Soc Nephrol. 2008, 19 (6): 1071-1075. 10.1681/ASN.2007121292.
Caridi G, Bertelli R, Di Duca M, Dagnino M, Emma F, Onetti Muda A, Scolari F, Miglietti N, Mazzucco G, Murer L, Carrea A, Massella L, Rizzoni G, Perfumo F, Ghiggeri GM: Broadening the spectrum of diseases related to podocin mutations. J Am Soc Nephrol. 2003, 14 (5): 1278-1286. 10.1097/01.ASN.0000060578.79050.E0.
Santin S, Garcia-Maset R, Ruiz P, Gimenez I, Zamora I, Pena A, Madrid A, Camacho JA, Fraga G, Sanchez-Moreno A, Cobo MA, Bernis C, Ortiz A, de Pablos AL, Pintos G, Justa ML, Hidalgo-Barquero E, Fernandez-Llama P, Ballarin J, Ars E, Torra R: Nephrin mutations cause childhood- and adult-onset focal segmental glomerulosclerosis. Kidney Int. 2009, 76 (12): 1268-1276. 10.1038/ki.2009.381.
Pollak MR: Inherited podocytopathies: FSGS and nephrotic syndrome from a genetic viewpoint. J Am Soc Nephrol. 2002, 13 (12): 3016-3023. 10.1097/01.ASN.0000039569.34360.5E.
Machuca E, Hummel A, Nevo F, Dantal J, Martinez F, Al-Sabban E, Baudouin V, Abel L, Grunfeld JP, Antignac C: Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. 2009, 75 (7): 727-735. 10.1038/ki.2008.650.
Voskarides K, Pierides A, Deltas C: COL4A3/COL4A4 mutations link familial hematuria and focal segmental glomerulosclerosis. glomerular epithelium destruction via basement membrane thinning?. Connect Tissue Res. 2008, 49 (3): 283-288.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2369/14/104/prepub
Acknowledgments
Only the authors contributed to this manuscript. Dr. Mottl is partially funded by the NIDDK 1K23DK093804-01 and CDC/NIDDK 1U18DP002708-01. Dr. Weck is partially funded by Susan Komen grant #KG100355, NIH/NCATS 5-UL1RR025747-04 and NIH/NHGR 5-U01HG006487-02. Dr. Lu is partially funded by NIH/NCATS 5-UL1RR025747-04.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AKM and CAF provided care to the proband. ML and KEW were responsible for genetic testing. AKM wrote the manuscript. All authors held discussions regarding the content of the manuscript and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mottl, A.K., Lu, M., Fine, C.A. et al. A novel TRPC6mutation in a family with podocytopathy and clinical variability. BMC Nephrol 14, 104 (2013). https://doi.org/10.1186/1471-2369-14-104
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2369-14-104