
Abstract
Background
Hemophilia A represents the most common and severe inherited hemorrhagic disorder. It is caused by mutations in the F8 gene, which leads to a deficiency or dysfunctional factor VIII protein, an essential cofactor in the factor X activation complex.
Methods
We used long-distance polymerase chain reaction and denaturing high performance liquid chromatography for mutation scanning of the F8 gene. We designed the competitive multiplex PCR to identify the carrier with exonal deletions. In order to facilitate throughput and minimize the cost of mutation scanning, we also evaluated a new mutation scanning technique, high resolution melting analysis (HRM), as an alternative screening method.
Results
We presented the results of detailed screening of 122 Taiwanese families with hemophilia A and reported twenty-nine novel mutations. There was one family identified with whole exons deletion, and the carriers were successfully recognized by multiplex PCR. By HRM, the different melting curve patterns were easily identified in 25 out of 28 cases (89%) and 15 out of 15 (100%) carriers. The sensitivity was 93 % (40/43). The overall mutation detection rate of hemophilia A was 100% in this study.
Conclusion
We proposed a diagnostic strategy for hemophilia A genetic diagnosis. We consider HRM as a powerful screening tool that would provide us with a more cost-effective protocol for hemophilia A mutation identification.
Similar content being viewed by others
Background
Hemophilia represents the most common and severe inherited hemorrhagic disorder. Hemophilia A(HA) is caused by mutations in the F8 gene, leading to a deficiency or dysfunctional III protein, an essential cofactor in the factor X activation complex. The F8 gene is 186 kb long; it has 26 exons and encodes a 9-kb mRNA transcript [1, 2]. The mutations causing hemophilia A are spread throughout the gene and are mostly represented by point alterations. However, the inversion of intron 22 was found in 40–50% of patients with severe HA [3] and the inversion of intron 1 was reported with a prevalence of about 5% in the UK [4].
Patients suffering from the disorder, along with their families, bear great financial and social burden; therefore, it is very important to prevent recurrence of the disease. For the sake of genetic counselling and prenatal diagnosis of hemophilia A, it is necessary to establish a sensitive, rapid and economic genetic diagnostic system. However, comprehensive analysis of mutations in the F8 gene is difficult to conduct due to the large gene size, its many scattered exons, and the high frequency of de novo mutation. The most direct strategy for mutation detection would be to amplify these regions from genomic DNA using PCR [5]. However, it would necessitate almost 30 amplifications of genomic DNA to cover all the essential regions.
The first systematic analysis of the complete coding sequence of the F8 gene was performed by applying denaturing gradient gel electrophoresis (DGGE) after PCR amplification in 1991. The analysis demonstrated a 90% mutation detection rate [6]. Since then, a wide range of different mutations have been identified, providing the genetic basis for the extensive variability observed in the clinical phenotypes. Mutation detection in the F8 gene is so challenging that it is only partially met by conventional screening methods such as single stranded conformational polymorphism (SSCP), conformational sensitive gel electrophoresis (CSGE) and chemical mismatch cleavage (CMC), each with varying applicability and efficiency; however, they all suffer from incomplete detection rates in the range of 70–85% [7–11]. Moreover, each method places variable demands on the technical skills and time investment of the investigator. In contrast, the recently introduced denaturing high performance liquid chromatography (DHPLC) offers a promising new method for a fast and sensitive analysis(96.2%) of PCR-amplified DNA segment [12–15].
We have established a diagnostic strategy, consisting of screening for most common mutations in the F8 gene, using long-distance polymerase chain reaction (LD-PCR) and DHPLC. We reported the result of detailed screening of 122 Taiwan families with hemophilia A. In order to facilitate throughput and minimize the cost of mutation scanning, we also evaluated a new mutation scanning technique, high resolution melting analysis (HRM). This alternative screening technique detects sequence variation by the use of a saturating double-stranded DNA dye.
Methods
Patients
This study was approved by the Ethic Institute Review Board of National Taiwan University Hospital and included 122 families, in which 329 samples had hemophilia A family history. We obtained consent from each subject. The patients presented varying degrees of severity of the disease. The mild and moderate hemophilia A was diagnosed following the familial transmission analysis and after the elimination of von Willebrand disease type 2N either by FVIII:vWF binding test or sequencing of exon 18 to 24 of the von Willebrand gene.
DNA extraction
According to the manufacturer's instructions, genomic DNA was extracted from 3 ml of peripheral blood cell samples with a Puregene DNA Isolation Kit (Gentra Systems, Minneapolis, MN).
DNA mutation numbering is based on cDNA sequence and nucleotide +1 corresponds to A of the ATG translation initiation codon. The nomenclature of this study follows the Nomenclature for Description of Genetic Variations approved by the Human Genome Variation Society and differs in 19 amino acids from the reference mutation database because the first 19 amino acids compose a signal peptide.
Mutation identification
The PCR assay for intron 22 inversion
The PCR mixture contained a total volume of 50 μl: 1 mM 10× buffer, 76 mM DMSO, 0.5 mM of dNTP and 0.3 mM deaza-Dgtp(Amershan Biosciences; Freiburg Germany), 0.12 mM of Expand long Template DNA polymerase (Roches) and 0.36 mM each of primer P, Q, 0.06 mM each of primer A, B as described by Qiang Liu et al. [16, 17]. Subcycling multiplex long distance PCR(LD-PCR) was carried out as described by Qiang Liu et al. [16, 17] with slight modifications in the reaction temperatures. The conditions for LD-PCR for the first 10 cycles were set at 94°C for 12s, followed by four subcycles of annealing/elongation carried out at 62°C for 120s and 65°C for 120s. The remaining 20 cycles were modified by the addition of an extra 5s per cycle for each of the annealing/elongation step. 10 μl of PCR product were mixed with TAE buffer and electrophoresed for one hour on a 0.7% agarose gel, then stained with ethidium bromide and visualized under UV Light.
The PCR assay for intron 1 inversion
The PCR amplification of the F8 intron 1 region and the intron 1 h repeats were performed on 100 ng of genomic DNA, in a reaction volume of 25 μl containing 0.1 mM dNTP, 2 mM MgCl2, 10 mM buffer and 0.5 units Taq polymerase (Perkin Elmer, ABI Foster City, CA). The primers used for the amplification of the F8 intron 1 region were 0.12 mm of 9F, 9CR and int1h-2F each. Primers int1h-2F, int1h-2R and 9F were used for the amplification of intron 1 h repeats as previously described by Tizzano and Banagnall et al. [18, 19]. After the initial denaturing step at 95°C for 5 min, thirty cycles of PCR were carried out designated at 94°C for 30s, 65°C for 30s and 72°C for 2 min. Finally, 5 μl of PCR products were mixed with TAE buffer and electrophoresed for one hour on a 0.7% agarose gel, then stained with ethidium bromide and visualized under UV light.
PCR assay and DHPLC for entire coding region
If there was no inversion in intron 1 or intron 22, we perform mutation analysis of the entire F8 coding region, including flanking splicing sites, by DHPLC using appropriate primers (Table 1). The DHPLC allows for the automated detection of single base DNA substitutions as well as small insertions and deletions. When using heteroduplex analysis by DHPLC under partially-denaturing conditions, heteroduplexes are retained less than their corresponding homoduplexes on a unique DNA separation matrix. DHPLC uses unpurified PCR products that are subjected to a final denaturing/reannealing step to ensure adequate formation of the heteroduplex. The cycling parameters of the amplification reactions were optimized for the fragment. Equal volumes of the polymerase chain reaction (PCR) product from the patient and from a wild-type control male were mixed, denatured at 95°C for 5 min and then incubated at 65°C for 30 min to obtain heteroduplexes. Mutational screening, performed on all amplified fragments from each patient, was carried out via denaturing high-performance liquid chromatography (DHPLC) on a Wave® DNA Fragment Analysis System (Transgenomic Inc., San Jose, CA) with a DNASep column (Transgenomic). The DNASep column contains proprietary 2-mm nonporous alkylated poly (styrene divinylbenzene) particles. The DNA molecules eluted from the column are detected by scanning with a UV detector at 260 nm. DHPLC-grade acetonitrile (9017-03; J.T. Baker, Phillipsburg, NJ) and triethylammonium acetate (TEAA; Transgenomic, Crewe, UK) constituted the mobile phase [14, 20]. For DHPLC analysis, heterozygous profiles were identified by visual inspection of the chromatograms.
Samples showing an abnormal peak pattern were sequenced using a fluorescent ABI Prism BigDye terminator kit (Applied Biosystems, UK), and the mutations were characterized. Each mutation was confirmed on a second, independent, amplified PCR sample. In order to rule out the possibility of exonic polymorphisms, 150 DNA samples from healthy, unrelated controls were screened and found negative for the novel missense mutations. All mutations found in the patients' family members were confirmed by sequencing.
High Resolution Melting Analysis
Melting curve analysis with the high-resolution melting instrument is a simple, high-performance, reliable, high-resolution, time-saving, and low labor-intensive technique that has shown promise as a sensitive and specific tool for the detection of variations in DNA. The thermal stability of a PCR product is determined by its base sequence [21, 22]. When the PCR product sequence is altered, duplex stability is changed, leading to different melting behavior. During high resolution melting analysis, melting curves are produced using dyes that fluoresce in the presence of double-stranded DNA and specialized instruments designed to monitor fluorescence during heating; as the temperature increases, the fluorescence decreases, producing a characteristic melting profile. The melting curve data were normalized, temperature-shifted, and converted to melting peaks by plotting the negative derivative of the fluorescence with respect to temperature against temperature (-dF/dT vs. temperature). This melting data can be analyzed to detect sequence variations such as single nucleotide polymorphisms and small insertions and deletions.
PCR amplification mixture included 2.5 mM MgCl2, 5 μl 2× Master Mix, 3.1 μl H2O (from Roche Reaction Mix Kit, Mannheim, Germany), 0.2 mM primers. And we added 25 ng DNA template. PCR amplification began with the first denaturation step at 95°C for 10 min, followed by 25 cycles of denaturation at 94°C for 30 s, annealing at 53°C for 45 s, extension at 72°C for 45 s, and then a final extension step at 72°C for 10 min.
Pipet 10 μl PCR products into each well of the LightCycler® 480 (Roche Applied Science) Multiwell Plate. Seal Multiwell Plate with LightCycler® 480 Sealing Foil. Load the multiwall Plate in LightCycler® 480 instrument and start the melting program. High resolution melting was performed at 95°C for 1 min, 40°C for 1 min, 6.5°C for 1 s and acquisitions at 95°C. The data was evaluated using the LightCycler® 480 Gene Scanning Software.
In order to improve the melting analysis, we redesigned the primers for some exons to shorten the PCR product and to avoid DNA-dimer formation (Table 2).
Multiplex PCR by Capillary Electrophoresis
In one family, the band of exon 4 to exon 10 by electrophoresis after PCR was visible in female cases but not in affected male patients. Large deletion was suspected, so we designed multiplex PCR in order to differentiate normal female from female carriers.
Multiplex PCRs were used to optimize the system and allowed the amplification of exon 3 to exon11. The KRIT and FGFR2 genes were used as internal controls. Each multiplex PCR for the DNA fragments was performed in a total volume of 25 μL containing the following: 100 ng of genomic DNA; 0.1 to 0.5 μM each primer; 200 μM dNTPs; 1 unit of AmpliTaq Gold enzyme (PE Applied Biosystems); and 2.5 μL of GeneAmp 10× buffer II (10 mmol/L Tris-HCl, pH 8.3, 50 mmol/L KCl) in 2 mM MgCl2 as provided by the manufacturer. PCR amplification was carried out with an initial denaturation step at 95°C for 10 min, followed by 25 cycles of denaturation at 94°C for 30 s, annealing at 53°C for 45 s, extension at 72°C for 45 s, and then a final extension step at 72°C for 10 min.
The HDA system with GCK-5000 cartridge kit (eGene) was used to analyze exons of the F8 gene. The gel-matrix in the gel cartridge consists of proprietary linear polymer with ethidiumbromide (EtBr) dye. The PCR products were diluted 20-fold by deionized water and placed in the sample chamber of the instrument. The DNA samples were then automatically injected into the capillary channel and subjected to electrophoresis according to the manufacturer's operation protocol. BioCalculator Graphing software (eGene) enabled automatic labelling of the peak sizes.
Results
Genomic DNA from 122 HA unrelated families were analyzed for mutations in the F8 gene (Table 3). There were 128 affected patients identified. This resulted in the identification of 61 different F8 gene mutations. The mutation detection rate was 100%.
Screening of the F8 intron inversion
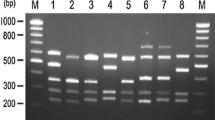
There were fifty-three (42.7%) families with intron 22 inversion (Fig. 1a) and one (0.8%) family with intron 1 inversion (Fig. 1b &1c) in this study. Since the intron 1 and the intron 22 inversion were well established causative mutations [3], there would be no need to search for other mutations in the coding regions in these families.

Result of inversion detection of intron 1 and intron 22. a) LD-PCR result of intron 22 inversion. Wild type has two band with PQ segment (12 KB) and AB segment (10 KB). With intron 22 rearrangement, PQ segment will be interrupted so that PB and AQ (both 11 KB) form. That is why we can see all three bands in carrier. Patient will show only 11 KB and 10 KB bands. Patient 154 is proved to be an affected case. M, marker. C, carrier. P, known affected male patient. WT, wild type. b) PCR result of intron 1 inversion. Set 1 is the product of primer 9F, int-2F and 9CR. In wild type, the only segment amplified is 1908 bp. In the affected male patient with intron 1 inversion, the amplified segment is 1323 bp in length. c) Set 2 is the product of primer 9F, int-2F and 2R. 93-2, 104-1 and 104-2 are normal. 93-1 is proved to be an affected case. 93-3 is a carrier with heterogenous components.
Screening of the F8mutation by DHPLC and direct sequencing
We performed DHPLC for mutation screening in those families who didn't have intron 1 and intron 22 inversions. By DHPLC, 100% genetic mutation was detected in the 119 HA families and confirmed by sequencing. The two X chromosomes from the carriers are responsible for the heteroduplex formation of the PCR amplicons. These stutter peaks (heteroduplex) were eluted much earlier in the chromatographic process than the main peaks (homoduplex). Yet, the affected male patients with one X chromosome would not form a heteroduplex until it is mixed together with the wild type DNA (Fig. 2).

Result of DHPLC. DHPLC results of two families were illustrated. a) Family 85. b) Family 139. The wild type DNA showed homoduplex. The carrier DNA would show heteroduplex peak just as affected male after mixing with normal DNA.
Evaluating of High-Resolution Melting Analysis
In order to evaluate the HRM analysis, 25 families, including a total of 43 samples with known mutation and 341 normal controls were tested. The mutations consisted of 13 different missense mutations, 5 deletions, 4 insertions, 2 nonsense mutations, one splicing site nucleotide substitution and one duplication.
In order to avoid DNA-dimer formation, we redesigned the primers for the HRM PCR reaction. The resulting melting curves were easier to differentiate from normal controls and the sensitivity was elevated. Figure 3 compares the melting curves of the original and modified primers, respectively. As temperature increased, the double strand DNA was unwound and the fluorescence was released (Fig 3a and 3b). The melting curves were normalized by calculation of the "line of best fit" in between two normalization regions before and after the major fluorescence decrease representing the melting of the PCR product using the software the LightCycler® Gene Scanning Software 4.0. The samples with high signal difference would present different peaks from the wild type (Fig 3d).

Comparison of the results of different HRM primers. a) and c) are HRM plots for exon 14-5 with first-designed primer. We are unable to see the grouping in the normalized, temperature-shifted plot. So we redesigned the primer pairs by LightCycler Probe Design Software 2.0. The result is shown in b) and d). The carrier and the patient are easy to be distinguished in the latter.
The different melting curve patterns were easily identified in 25 out of 28 cases (89%) and 15 out of 15 (100%) carriers. The sensitivity of HRM in this study was 93 % (40/43).
In addition, we performed HRM for whole coding region scanning in three families with unknown mutations. Three abnormal melting curves were identified. After these exons were confirmed by direct sequencing, we were able to identify the mutation in these three families. The specificity of HRM in this pilot study was high but it required further investigation. A larger sample sizes was needed for validation.
Multiplex PCR
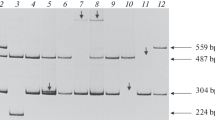
There were two families, which we were unable to see the exon 4 to exon 10 band and the Exon1 band from electrophoresis after PCR, respectively. For the former, we designed the multiplex PCR that showed a total deletion from exon 4 to exon 10 (Fig 4). The ratio of the defective exon to the control gene in peak heights (the exon 4 to exon 10/the KRIT and the exon 4 to exon 10/the FGFR2) in the female carrier was half of that of the wild type. That is, the relative gene dosage in exon 4 to the exon 10 of the wild type was two times to that of the female carrier. As for the other family, we were unable to perform the multiplex PCR without having the DNA of the obligate carrier.

Multiplex PCR in a family with exon 4 to exon 10 deletion. FGFR2 (*) and KRIT (#) gene are used as internal control. a) are from wild type and b) are from female carrier. The X axis was time, which also represented the size of the amplicon. The Y axis was the fluorescence intensity of each amplicon, which also represented the relative gene dosage. The relative peak height of the female carrier with arrow is half to that of the wild type in exon 4, 5, 8,9 but not in exon 11.
In this study we identified twenty-eight missense mutations, thirteen nonsense mutations, one four- base pair duplication, five insertions, nine small deletions, one splicing site nucleotide substitution and two large segment deletions in the F8 gene.
We identified twenty-nine novel mutations of the F8 gene in hemophilia A patients, in which eleven were missense mutations, one was splicing site substitution, seven were nonsense mutation, which lead to premature terminating of the protein, and ten were frameshift mutations including one four-base pair duplication, four deletions and five insertions, resulting in truncations of the F8 protein. None of the novel missense mutations were presented in the 150 normal DNA samples.
Discussion
The most common mutation, found in approximately 40% of patients with severe hemophilia A, was the intron 22 inversion mutation [23]. This mutation happens when homologous recombination occurs between the Int22h-1 in intron 22 and 1 of 2 homologous regions (Int22h-2 or Int22h-3) telomeric to the F8 gene. An additional inversion of intron 1 of the factor VIII gene that affects up to 5% of patients with severe hemophilia A has been reported [18]. The large size of the F8 gene predisposes to the occurrence of deletions, which account for approximately 5% of characterized mutations. Single base pair changes (resulting in missense, frameshift, or splice junction mutations), insertions, or duplications account for the balance of cases of hemophilia A that are spread throughout the F8 gene [24]. Although the structure-function relationships of some of the missense mutations are known or can be deduced (eg, alteration of vWF binding site, thrombin cleavage site, etc), the structural consequences of most such mutations remain undefined [25].
Our data showed a mutation detection rate at 100% out of 122 hemophilia A families. Since the intron 22 inversion and intron 1 inversion present in 45.1 % in this Taiwanese database, 47.5 to 53% in India population [8, 26] and 40–50% in European [3, 18], it is rational to perform the inversion detection of intron 1 and intron 22 at first, and then genetic scan if no intron 1 or 22 inversion were found. In our cohort, 54.5% severe HA families had intron 1 and intron 22 inversion; 65.5% cases with intron 1 and intron 22 inversion belonged to severe hemophilia A patients. It is also suggested to check for intron 1, intron 22 inversion and big deletions in cases having the severe phenotype; on the other hand, it is recommended to go directly to the F8 gene mutation scanning in cases having the moderate/mild phenotype [25, 27]. However, we are a referred center and the clinical data is not always available. Because the inversion detection of intron 1 and intron 22 is cheaper and more time-saving than other techniques, our strategy seemed to be economical under this situation.
The B domain accounts for 40% of the F8 cDNA. Apart from large exon or intron deletions, fourteen (14/52, 26.9%) different mutations and one SNP were found to be located in the B domain of the F8 cDNA. All mutations belonging to deletion, insertion, or nonsense mutations, lead to different protein products. This may imply the low functional importance of this region since no phenotype would be identified unless the protein product is changed or truncated. The SNP identified in the B domain was located in c. 3780 on exon 14, which changed aspartic acid to glutamic acid. Through family studies in a Caucasian population, Machiah et al. hypothesized that Asp1241Glu influences about 5% F8 levels and modifies thrombosis risk by modulating the rate of secretion into the circulation by family studies [28]. Scanavini et al. also conducted a case-control study and demonstrated that the F8 D1241E polymorphism was associated with decreased F8 activity [29].
Affected males with large deletions can be readily detected by the absence of an amplification product in PCR. Up to 98% of all frequent deletions can be detected, but in carriers, the nondeleted X chromosome hampers detection, making identification of hemophilia A carrier status difficult. In one particular family, the PCR product of an affected male patient failed to show exon 4 to exon 10. But of the same family, the same exon region in the female cases was successfully amplified by PCR and sequenced. Large deletion was suspected, but we were unable to distinguish the female carriers from the wild type. We designed the multiplex PCR [12, 15] so that we could differentiate between them by examining different peak heights with the use of different gene dosages(Fig 4). In addition, the absence of peaks from exon 4 to exon 10 confirmed the deletion in the affected male. Multiplex PCR is an in-house designed reaction, which compares the dosage of several genes. This system may also be adapted for diagnostic use in other genetic diseases involving deletion and duplication mutations.
We had two heterozygous females from two unrelated families. Hemophilia A is transmitted through heterozygous females denoted as carriers, who are generally asymptomatic. This is because random X inactivation results in approximately equal proportions of somatic cells, in which either the normal X or the mutated X chromosome is active [23, 30]. Because females have two X chromosomes and only 5% of FVIII is enough for the body to maintain hemostasis, it is very rare to have a female HA patient with very low FVIII activity and severe bleeding symptoms [31]. Even though the disease is extremely rare in females, a few cases have been documented as a result of different pathophysiologic mechanisms [32–34]. The skewed X-chromosome inactivation is the most common possible mechanism for the phenomenon [31, 35–39].
The hemophilia A mutation screening is laborious and expensive, even when employing DHPLC as a screening method. In order to facilitate throughput and minimize the cost of hemophilia A mutation screening, we evaluated a new mutation scanning technique, high resolution melting system(HRM). We performed high resolution melting analysis on 43 samples, including carriers and affected cases. All the carriers were easily to be distinguished (100%). Nevertheless, we were unable to identify three out of 28 affected cases. The detection rate was 93%. We had a reason to believe that it is easier for the heteroduplex to release double-stranded DNA dye, so that HRM is more sensitive to carrier detection. DHPLC is not ideal in detecting affected case until mixed together with normal DNA. On the other hand, even though HRM was able to distinguish the affected male from the wild type, the sensitivity did not reach 100% despite optimizing condition. Therefore, mixing up the affected male DNA with normal control would improve the detection rate for both DHPLC and HRM. Besides, we proved that the analysis was greatly enhanced by good primer design that avoided DNA-dimer formation. The sensitivity of the analysis was not only influenced by the size of PCR products but also the primers and the PCR condition. We recommend the software, LightCycler Probe Design Software 2.0, as the tool for primer design.
As for clinical application, once the intron 1 and 22 inversion has been ruled out, HRM becomes a good initial screening method, especially for those families with obligate carriers. Comparing with our familiar method, DHPLC, HRM costs less ($0.6 versus $3.0 per sample per amplicon, respectively), takes less time (3 hours versus 10 hours) and works more efficiently (96 or 384 wells at one time). The analysis of HRM takes only fifteen minutes. With this highly sensitive and specific tool, we will be able to manage a large quantity of clinical samples and do mutation screening.
Conclusion
In conclusion, we are capable of detecting 100% of mutations in the F8 gene; also our study identified a number of novel mutations in the largest cohort of Taiwanese patients yet to be reported. Furthermore, we validate HRM for hemophilia A mutation screening. Mutation detection is best started with obligate carriers. We proposed a cost-effective screening protocol with sequential combination of different genetic diagnostic tools. HRM saves time and money, and we have proven the use of such technique to be effective for mutation screening.
Abbreviations
- DHPLC:
-
Denaturing High Performance Liquid Chromotography
- F8 gene:
-
FVIII gene
- HA:
-
Hemophilia A
- HRM:
-
High resolution Melting
- LD-PCR:
-
Long-distance PCR
References
Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, Vehar GA, Capon DJ, Lawn RM: Characterization of the human factor VIII gene. Nature. 1984, 312 (5992): 326-330. 10.1038/312326a0.
Wood WI, Capon DJ, Simonsen CC, Eaton DL, Gitschier J, Keyt B, Seeburg PH, Smith DH, Hollingshead P, Wion KL, et al: Expression of active human factor VIII from recombinant DNA clones. Nature. 1984, 312 (5992): 330-337. 10.1038/312330a0.
Lakich D, Kazazian HH, Antonarakis SE, Gitschier J: Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet. 1993, 5 (3): 236-241. 10.1038/ng1193-236.
Saenko EL, Ananyeva N, Kouiavskaia D, Schwinn H, Josic D, Shima M, Hauser CA, Pipe S: Molecular defects in coagulation Factor VIII and their impact on Factor VIII function. Vox Sang. 2002, 83 (2): 89-96. 10.1046/j.1423-0410.2002.00183.x.
Vidal F, Farssac E, Altisent C, Puig L, Gallardo D: Rapid hemophilia A molecular diagnosis by a simple DNA sequencing procedure: identification of 14 novel mutations. Thromb Haemost. 2001, 85 (4): 580-583.
Higuchi M, Antonarakis SE, Kasch L, Oldenburg J, Economou-Petersen E, Olek K, Arai M, Inaba H, Kazazian HH: Molecular characterization of mild-to-moderate hemophilia A: detection of the mutation in 25 of 29 patients by denaturing gradient gel electrophoresis. Proc Natl Acad Sci U S A. 1991, 88 (19): 8307-8311. 10.1073/pnas.88.19.8307.
Pieneman WC, Deutz-Terlouw PP, Reitsma PH, Briet E: Screening for mutations in haemophilia A patients by multiplex PCR-SSCP, Southern blotting and RNA analysis: the detection of a genetic abnormality in the factor VIII gene in 30 out of 35 patients. Br J Haematol. 1995, 90 (2): 442-449.
Jayandharan G, Shaji RV, Baidya S, Nair SC, Chandy M, Srivastava A: Identification of factor VIII gene mutations in 101 patients with haemophilia A: mutation analysis by inversion screening and multiplex PCR and CSGE and molecular modelling of 10 novel missense substitutions. Haemophilia. 2005, 11 (5): 481-491. 10.1111/j.1365-2516.2005.01121.x.
Lin SW, Lin SR, Shen MC: Characterization of genetic defects of hemophilia A in patients of Chinese origin. Genomics. 1993, 18 (3): 496-504. 10.1016/S0888-7543(05)80367-2.
Schwaab R, Oldenburg J, Schwaab U, Johnson DJ, Schmidt W, Olek K, Brackman HH, Tuddenham EG: Characterization of mutations within the factor VIII gene of 73 unrelated mild and moderate haemophiliacs. Br J Haematol. 1995, 91 (2): 458-464. 10.1111/j.1365-2141.1995.tb05322.x.
Becker J, Schwaab R, Moller-Taube A, Schwaab U, Schmidt W, Brackmann HH, Grimm T, Olek K, Oldenburg J: Characterization of the factor VIII defect in 147 patients with sporadic hemophilia A: family studies indicate a mutation type-dependent sex ratio of mutation frequencies. Am J Hum Genet. 1996, 58 (4): 657-670.
Hung CC, Chen CP, Lin SP, Chien SC, Lee CN, Cheng WF, Hsieh WS, Liu MS, Su YN, Lin WL: Quantitative assay of deletion or duplication genotype by capillary electrophoresis system: Application in Prader-Willi syndrome and Duchenne muscular dystrophy. Clin Chem. 2006, 52 (12): 2203-2210. 10.1373/clinchem.2006.071118.
Hung CC, Su YN, Chien SC, Liou HH, Chen CC, Chen PC, Hsieh CJ, Chen CP, Lee WT, Lin WL, Lee CN: Molecular and clinical analyses of 84 patients with tuberous sclerosis complex. BMC Med Genet. 2006, 7: 72-10.1186/1471-2350-7-72.
Hung CC, Su YN, Lin CY, Yang CC, Lee WT, Chien SC, Lin WL, Lee CN: Denaturing HPLC coupled with multiplex PCR for rapid detection of large deletions in Duchenne muscular dystrophy carriers. Clin Chem. 2005, 51 (7): 1252-1256. 10.1373/clinchem.2004.046144.
Hung CC, Su YN, Tsao PN, Chen PC, Lin SJ, Lin CH, Mu SC, Liu CA, Chang YC, Lin WL, Hsieh WS, Hsu SM: Unequal crossover recombination - population screening for PHOX2B gene polyalanine polymorphism using CE. Electrophoresis. 2007, 28 (6): 894-899. 10.1002/elps.200600383.
Liu Q, Nozari G, Sommer SS: Single-tube polymerase chain reaction for rapid diagnosis of the inversion hotspot of mutation in hemophilia A. Blood. 1998, 92 (4): 1458-1459.
Liu Q, Sommer SS: Subcycling-PCR for multiplex long-distance amplification of regions with high and low GC content: application to the inversion hotspot in the factor VIII gene. Biotechniques. 1998, 25 (6): 1022-1028.
Bagnall RD, Waseem N, Green PM, Giannelli F: Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. 2002, 99 (1): 168-174. 10.1182/blood.V99.1.168.
Tizzano EF, Cornet M, Baiget M: Inversion of intron 1 of the factor VIII gene for direct molecular diagnosis of hemophilia A. Haematologica. 2003, 88 (1): 118-120.
Chien SC, Lee CN, Hung CC, Tsao PN, Su YN, Hsieh FJ: Rapid prenatal diagnosis of X-linked chronic granulomatous disease using a denaturing high-performance liquid chromatography (DHPLC) system. Prenat Diagn. 2003, 23 (13): 1092-1096. 10.1002/pd.761.
Montgomery J, Wittwer CT, Palais R, Zhou L: Simultaneous mutation scanning and genotyping by high-resolution DNA melting analysis. Nat Protoc. 2007, 2 (1): 59-66. 10.1038/nprot.2007.10.
Ririe KM, Rasmussen RP, Wittwer CT: Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem. 1997, 245 (2): 154-160. 10.1006/abio.1996.9916.
Antonarakis SE, Rossiter JP, Young M, Horst J, de Moerloose P, Sommer SS, Ketterling RP, Kazazian HH, Negrier C, Vinciguerra C, Gitschier J, Goossens M, Girodon E, Ghanem N, Plassa F, Lavergne JM, Vidaud M, Costa JM, Laurian Y, Lin SW, Lin SR, Shen MC, Lillicrap D, Taylor SA, Windsor S, Valleix SV, Nafa K, Sultan Y, Delpech M, Vnencak-Jones CL, Phillips JA, Ljung RC, Koumbarelis E, Gialeraki A, Mandalaki T, Jenkins PV, Collins PW, Pasi KJ, Goodeve A, Peake I, Preston FE, Schwartz M, Scheibel E, Ingerslev J, Cooper DN, Millar DS, Kakkar VV, Giannelli F, Naylor JA, Tizzano EF, Baiget M, Domenech M, Altisent C, Tusell J, Beneyto M, Lorenzo JI, Gaucher C, Mazurier C, Peerlinck K, Matthijs G, Cassiman JJ, Vermylen J, Mori PG, Acquila M, Caprino D, Inaba H: Factor VIII gene inversions in severe hemophilia A: results of an international consortium study. Blood. 1995, 86 (6): 2206-2212.
Ensenauer RE, Reinke SS, Ackerman MJ, Tester DJ, Whiteman DA, Tefferi A: Primer on medical genomics. Part VIII: Essentials of medical genetics for the practicing physician. Mayo Clin Proc. 2003, 78 (7): 846-857.
Pruthi RK: Hemophilia: a practical approach to genetic testing. Mayo Clin Proc. 2005, 80 (11): 1485-1499.
Ahmed R, Kannan M, Choudhry VP, Saxena R: Mutation reports: intron 1 and 22 inversions in Indian haemophilics. Ann Hematol. 2003, 82 (9): 546-547. 10.1007/s00277-003-0704-3.
Bogdanova N, Markoff A, Eisert R, Wermes C, Pollmann H, Todorova A, Chlystun M, Nowak-Gottl U, Horst J: Spectrum of molecular defects and mutation detection rate in patients with mild and moderate hemophilia A. Hum Mutat. 2007, 28 (1): 54-60. 10.1002/humu.20403.
Abstracts of the American Society of Hematology 45th Annual Meeting. December 6-9, 2003. San Diego, California, USA. Blood. 2003, 102 (11 Pt 2): 1b-512b.
Scanavini D, Legnani C, Lunghi B, Mingozzi F, Palareti G, Bernardi F: The factor VIII D1241E polymorphism is associated with decreased factor VIII activity and not with activated protein C resistance levels. Thromb Haemost. 2005, 93 (3): 453-456.
Puck JM, Willard HF: X inactivation in females with X-linked disease. N Engl J Med. 1998, 338 (5): 325-328. 10.1056/NEJM199801293380511.
Cai XH, Wang XF, Dai J, Fang Y, Ding QL, Xie F, Wang HL: Female hemophilia A heterozygous for a de novo frameshift and a novel missense mutation of factor VIII. J Thromb Haemost. 2006, 4 (9): 1969-1974. 10.1111/j.1538-7836.2006.02105.x.
Gilchrist GS, Hammond D, Melnyk J: Hemophilia A in a phenotypically normal female with XX-XO mosaicism. N Engl J Med. 1965, 273 (26): 1402-1406.
Seeler RA, Vnencak-Jones CL, Bassett LM, Gilbert JB, Michaelis RC: Severe haemophilia A in a female: a compound heterozygote with nonrandom X-inactivation. Haemophilia. 1999, 5 (6): 445-449. 10.1046/j.1365-2516.1999.00352.x.
Coleman R, Genet SA, Harper JI, Wilkie AO: Interaction of incontinentia pigmenti and factor VIII mutations in a female with biased X inactivation, resulting in haemophilia. J Med Genet. 1993, 30 (6): 497-500.
Favier R, Lavergne JM, Costa JM, Caron C, Mazurier C, Viemont M, Delpech M, Valleix S: Unbalanced X-chromosome inactivation with a novel FVIII gene mutation resulting in severe hemophilia A in a female. Blood. 2000, 96 (13): 4373-4375.
David D, Morais S, Ventura C, Campos M: Female haemophiliac homozygous for the factor VIII intron 22 inversion mutation, with transcriptional inactivation of one of the factor VIII alleles. Haemophilia. 2003, 9 (1): 125-130. 10.1046/j.1365-2516.2003.00704.x.
Renault NK, Dyack S, Dobson MJ, Costa T, Lam WL, Greer WL: Heritable skewed X-chromosome inactivation leads to haemophilia A expression in heterozygous females. Eur J Hum Genet. 2007, 15 (6): 628-637. 10.1038/sj.ejhg.5201799.
Redonnet-Vernhet I, Ploos van Amstel JK, Jansen RP, Wevers RA, Salvayre R, Levade T: Uneven X inactivation in a female monozygotic twin pair with Fabry disease and discordant expression of a novel mutation in the alpha-galactosidase A gene. J Med Genet. 1996, 33 (8): 682-688.
Kristiansen M, Knudsen GP, Bathum L, Naumova AK, Sorensen TI, Brix TH, Svendsen AJ, Christensen K, Kyvik KO, Orstavik KH: Twin study of genetic and aging effects on X chromosome inactivation. Eur J Hum Genet. 2005, 13 (5): 599-606. 10.1038/sj.ejhg.5201398.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/9/53/prepub
Acknowledgements
The authors thank miss Mei-Ping Tu for acquisition of data. This work was supported by grants from the National Taiwan University Hospital in study design and sample collection(NTUH 94S186 and NTUH 97S913).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
S–YL and C–CH carried out the molecular genetic studies. WT and S–SC participated in the clinical diagnosis, sequence alignment and sample collecting. S–YL and C–TC drafted the manuscript. C–CH, H–NH and Y–NS participated in the design of the study and performed the statistical analysis. C–NL conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lin, SY., Su, YN., Hung, CC. et al. Mutation spectrum of 122 hemophilia A families from Taiwanese population by LD-PCR, DHPLC, multiplex PCR and evaluating the clinical application of HRM. BMC Med Genet 9, 53 (2008). https://doi.org/10.1186/1471-2350-9-53
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-9-53