
Abstract
Background
DNA methylation at specific CpG sites within gene promoter regions is known to regulate transcriptional activity in vitro. In human adipose tissue, basal transcription of the aromatase (CYP19A1) gene is driven primarily by the I.4 promoter however the role of DNA methylation in regulating expression in ex vivo mature adipocytes is unknown. This observational study reports the correlation of DNA methylation within the I.4 promoter region of human mature subcutaneous and omental adipocytes with aromatase expression and body composition measures.
Methods
Omental and subcutaneous adipose tissue were collected from 25 obese subjects undergoing bariatric surgery and the mature adipocyte fraction purified. DNA methylation status of 5 CpG sites within a 550 base pair region encompassing the transcription start site (TSS) of promoter I.4 was determined using pyrosequencing. Relative aromatase and I.4 promoter specific mRNA expression was determined by qRT-PCR and whole body DXA performed in 25 participants.
Results
Site-specific DNA methylation varied from 21 ± 10% to 81 ± 11%. In omental adipocytes percentage methylation at the I.4.1 and I.4.2 CpG sites, but not other nearby sites, was negatively correlated with relative aromatase mRNA expression (R = - 0.52, P = 0.017 and R = - 0.52, P = 0.015). In contrast subcutaneous adipocytes percentage DNA methylation at the I.4.3 and I.4.5 sites were positively correlated with relative aromatase mRNA expression (R = 0.47, P = 0.022 and R = 0.55, P = 0.004). In a small subset of patients DNA methylation at the I.4.5 site was also positively correlated with whole body lean mass, bone mineral content and density.
Conclusions
In conclusion in mature adipocytes, the primary source of estradiol after menopause, increasing DNA methylation was correlated with aromatase mRNA expression and thus estradiol biosynthesis. These findings support a tissue-specific epigenetic regulation of the basal promoter activity in mature adipocytes; the mechanisms influencing this regulation and its physiological role remain to be elucidated.
Similar content being viewed by others


Background
Estrogen biosynthesis is catalysed by the aromatase enzyme which converts androstendione to estrone and testosterone to estradiol [1, 2]. Aromatase is a cytochrome P450 enzyme produced by the human CYP19A1 gene located on chromosome 15q21.2 [3]. The gene comprises of an upstream regulatory region spanning approximately 93 kb and a downstream coding region consisting of 9 coding exons, II-X, spanning 30 kb [4]. Aromatase expression has been detected in the ovary, placenta, breast, testes, adipose tissue, bone, peripheral blood leucocytes and brain [4, 5]. However adipose tissue is the primary source of estrogen in women after menopause and is important in maintaining bone remodelling and regulating adipose mass. Tissue specific aromatase gene expression is determined in part by tissue-specific promoters in the regulatory regions upstream of the untranslated first exon which gives rise to transcripts with unique 5′ non-coding termini [6–8].
In human adipose tissue, basal transcription of aromatase has been demonstrated to be driven primarily through the I.4 promoter [7, 9–11] with a 500 bp region of the aromatase I.4 promoter located 330 bp upstream to 170 bp downstream of the transcription start site (TSS) been shown to regulate glucocorticoid responsiveness [9]. DNA methylation is an epigenetic mark which involves the addition of a methyl group to the fifth carbon of the cytosine ring, forming 5-methylcytosine. The reaction is catalysed by DNA methyltransferases and primarily occurs in a 5′ CpG dinucleotide context [12]. Typically DNA methylation is associated with repressing gene expression [13] and activation of genes has been attributed to the demethylation of critical CpG loci both in vitro and in vivo[12, 14–17] however intragenic DNA methylation paradoxically has been shown to be associated with increased gene expression [18–20]. DNA methylation has also been suggested to regulate placental [21] and ovarian [22] aromatase expression in cattle, while a recent in vitro study [11] suggested an inverse association between DNA methylation in the aromatase I.4 promoter region and aromatase gene expression in human breast adipose fibroblasts and breast cell lines however the relationship between DNA methylation and other cell types has not been investigated.
In men and after menopause in women pre-adipocytes and mature adipocytes are considered the primary source of circulating levels of estradiol after menopause [23]. Endogenous production of estradiol and therefore its regulation in adipose tissue has important clinical implications for a range of diseases including breast cancer [24], Alzheimer’s disease [25] and fracture [26]. Previous studies by our group and others have investigated the association of genetic polymorphisms within the aromatase gene with estradiol levels, bone mineral density, and fracture risk [26–31], however little is known regarding the role of epigenetic regulation of the aromatase gene.
We therefore used human ex vivo mature adipocytes from the subcutaneous and omental sites to test the association of DNA methylation within the I.4 promoter and gene expression.
Methods
Subjects
Patients undergoing bariatric surgery at Sir Charles Gairdner Hospital were invited to participate in the study. Twenty five participants were recruited for the study and adipose tissue samples collected at surgery. Each participant gave written informed consent and the study was approved by the institutional ethics committee of Sir Charles Gairdner Hospital.
Body composition and bone mineral density
Whole body dual x-ray absorptiometry (DXA) was performed in 10 participants during a clinic visit post-surgery and headless bone area, mineral content and density as well as whole body bone free lean mass and fat mass on a fan-beam densitometer (Hologic Acclaim 4500A; Hologic Corp, Waltham, Mass) were measured.
Adipocyte isolation
Omental and subcutaneous adipose tissue was taken during bariatric surgery. Adipose tissue was rapidly processed after surgery using a modified protocol [32]. Briefly, connective tissue, blood vessels and fibrous materials were removed from the adipose tissue samples. The tissue were then homogenised and incubated in collagenase digest solution (25 mM HEPES, 5 mM glucose, 120 mM NaCl, 50 mM KCl, 7.5 mM CaCl2, 3 mg/ml Type II Collagenase, Sigma) with a ratio of 4:1 at 37°C in a 180 rpm shaker for 1.5 hours. The digest was then passed through a 500 μm nylon mesh and spun at 700 g for 10 min. After gentle agitation of the culture for 30 seconds the floating layer of cells that resurfaced (mature adipocytes) were then collected and the procedure repeated twice more. This method of isolating adipocytes has been shown to result in ~3-4 contaminating pre-adipocytes per 1,000 mature adipocytes, as described by others [33]. Cells were stored at −80°C in RNAlater prior to extraction. RNA samples were extracted using the AllPrep DNA/RNA and protein kit (Qiagen, VIC, Australia) according to the manufacturer’s instructions. RNA concentrations were then determined by spectrophotometry (ND-1000, Nanodrop Technologies, USA) with a 260/280 of 1.8 or greater. Where available mature adipocytes from 36 of the original 50 samples were re-extracted using the AllPrep Mini Kit (QIAGEN) for PI.3 and PI.4 transcript expression analysis.
Pyrosequencing primer design
Pyrosequencing assays were designed using the PyroMark assay design software (version 2.0, QIAGEN). The most recent version of the genomic DNA sequence was imported into the assay design software (accession number L21982). The region of interest for this study was identified as being 350 bp upstream of the transcription start site to 200 bp downstream as this region has previously been identified to be responsible for glucocorticoid stimulated aromatase gene expression in primary hepatocytes and adipose derived stromal cells [9]. This region encompasses a glucocorticoid response element from −133/119 bp and an SP1 binding site +151/158 responsible for glucocorticoid responsiveness [9] as well as a RUNX2 binding site from −231/204 bp [34] and a interferon-gamma activation site element from −282/272 bp [35] which are involved in activation of I.4 promoter specific transcription (Figure 1). Primers were designed for the 5 CpG sites within this region. In silico bisulfite conversion was performed by converting all CG’s in the gDNA sequence to YG’s and any remaining C’s to T’s. Sites I.4.1 (−350 bp), I.4.2 (−316 bp), I.4.3 (−174 bp), I.4.4 (−85 bp) and I.4.5 (+152 bp) correspond to CpG sites 4, 5, 6, 7 and 8 respectively, described by Knower et al. [11]. The primer sets which resulted in the highest primer score based on the predictive likelihood of primers forming duplexes and hairpins, mispriming or the amplicon looping for sites 1, 2 and 5 were selected (Table 1).

Location of the 5 CpG sites analysed within the I.4 promoter region of the aromatase gene. Numbers indicate positions of CpG sites analysed relative to the transcription start site (TSS, +1) of I.4, sites 1- 5n refer to I.4.1 – 5 respectively. Cis-acting elements are shown in circles (5, 14, 37). GAS, interferon γ activation site; GRE, glucocorticoid response element; Runx2, Runt-related transcription factor 2.
Pyrosequencing assay
Unmethylated cytosines were converted to uracil using the EpiTect bisulfite kit (QIAGEN) according to the manufacturer’s instructions. Bisulfite converted DNA was determined by spectrophotometry (ND-1000, Nanodrop Technologies, USA) and 40 ng was amplified using the PyroMark PCR kit (QIAGEN) as per the manufacturer’s instructions. Cycling conditions were as follows: 95°C for 15 min, 45 cycles of 94°C for 30 s, 56°C for 30 s and 72°C for 30 s, followed by a final extension step at 72°C for 10 mins. 2 μl of well-mixed streptavidin sepharose high performance slurry (GE Healthcare) and 40 μl of binding buffer (QIAGEN) was diluted in 18 μl of water in each well, followed by addition of 20 μl of PCR product generated previously. The mixture was vortexed for 5 mins at 1,400 rpm and the 24-well plate containing the PCR product-streptavidin mix was placed on the PyroMark Vacuum station. With the vacuum on, the filter probes were placed in sterile water for 10 s and inserted into the 24-well plate containing the PCR product-streptavidin mix to capture the double stranded PCR (dsPCR) product. The dsPCR product was washed in 70% ethanol solution for 5 s and then denaturation solution (QIAGEN) for 5 s. The single stranded biotinylated template was washed in 1 × wash buffer (QIAGEN) for 10 s and released by agitation. The template DNA was annealed to the sequencing primer by incubating the PyroMark Q24 plate at 80°C for 2 min. The PyroMark Q24 cartridge was loaded with PyroGold Q24 reagents, according to pre-run information (PyroMark Q24 software). Each run contained at least 1 internal bisulfite conversion control whereby C bases not followed by a G (typically not methylated) should be fully converted to T after bisulfite treatment. All runs also included a no template control. The proportion of DNA molecules methylated at each CpG site was automatically calculated by the PyroMark Q24 software and expressed as a percentage. As previously reported, the quantitative reproducibility of technical replicates amplified by the same PCR is approximately 2% and variation induced by different bisulfite treatments and/or separate PCR amplifications is approximately 5% [36].
Relative aromatase mRNA expression
mRNA from the adipocytes was reverse transcribed using the QuantiTect reverse transcriptase kit (QIAGEN), according to manufacturer’s instructions, including a genomic DNA elimination step and adding the maximum amount of template mRNA possible. Total aromatase mRNA expression was then determined using the QuantiFast SYBR green PCR kit (QIAGEN) and QuantiTect primer assay (Hs_CYP19A1_2_SG; QIAGEN) according to the manufacturer’s instructions, in an iQ5 real time PCR thermal cycler (BioRad). Briefly, each reaction consisted of 1× QuantiFast Master mix, 1× QuantiTect primer assay, 4 μl of cDNA previously diluted 1:2 with RNase-free H2O and made up to 15 μl with RNase-free H2O. The primers amplified an 82 bp region of aromatase (accession numbers: NM_000103, NM_031226). Aromatase mRNA expression was normalised to 18S rRNA gene expression, which was determined using the same conditions as above but with a different QuantiTect Primer Assay (Hs_RRNA18S_1_SG; QIAGEN). Where available (n = 36) excess adipocytes were stored in RNAlater® (QIAGEN) were re-extracted and relative promoter expression was calculated using RT-PCR using I.4 and I.3 promoter specific forward primers with a common reverse primers as described by Demura et al. [37].
Statistical analysis
Differences between groups were investigated using independent samples T-Tests and tissue depots by paired sample T-Tests. Spearman’s Rank correlation coefficient (R) was used to investigate the relationships between percentage DNA methylation and relative aromatase mRNA expression, age, body mass index, lean mass, fat mass and bone phenotypes. All correlations observed were tested with and without outliers and with and without men to account for potential gender differences. P values < 0.05 were considered statistically significant. Results were analysed using SPSS (PASW Statistics, version 18). Results are given as mean ± standard deviation.
Results
Cohort characteristics
The subjects consisted of 22 women and 3 men undergoing bariatric surgery. The mean age was 46.1 ± 13.8 years, height 166.7 ± 8.8 cm, weight 121.3 ± 24.2 kg and body mass index (BMI) 43.4 ± 8.1 kg/m2.
DNA methylation of CpG sites within the regulatory region of the I.4 promoter
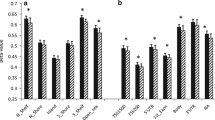
The four CpG sites upstream of the TSS identified by the PyroMark assay design software were I.4.1 and I.4.2, I.4.3 and I.4.4 while the CpG site I.4.5 was located downstream of the TSS (Figure 1). The percentage DNA methylation of the mature adipocytes from the omental and subcutaneous depots for the 5 CpG sites studied is shown in Figure 2. Three of the upstream CpG sites; (I.4.2 – I.4.4) had significantly different percentage DNA methylation between omental and subcutaneous adipocytes.

Mean ± SEM of percentage DNA methylation at the 5 CpG sites within the I.4 promoter of subcutaneous adipocytes compared to omental adipocytes. *** represents significantly different from omental adipocytes P < 0.001.
Site specific methylation and relative total mRNA expression
Omental adipocytes
Total relative aromatase expression in omental adipocytes was 0.42 ± 0.15 in omental adipocytes. In omental adipocytes percentage DNA methylation at CpG sites I.4.1 and I.4.2 was negatively correlated with relative total aromatase mRNA expression (R = −0.516, P = 0.017 and R = −0.522, P = 0.015 respectively). DNA methylation was not significantly correlated with total aromatase expression at sites I.4.3 (R = −0.203, P = 0.391), I.4.4 (R = −0.131, P = 0.582) or I.4.5 (R = −0.263, P = 0.237). The association between DNA methylation and total aromatase expression became non-significant at the I.4.1 site after the exclusion of men from the analysis (R = −0.462, P = 0.054). Similarly the correlation between the I.4 promoter specific mRNA expression and 1.4.1 and 1.4.2 DNA methylation failed to reach significance (R = −0.357, p = 0.199 and −0.262, P = −0.311).
Subcutaneous adipocytes
Total relative aromatase expression in subcutaneous adipocytes was 0.42 ± 0.15in subcutaneous adipocytes. In subcutaneous adipocytes percentage DNA methylation at the I.4.3 and I.4.5 CpG sites were positively correlated with relative total aromatase mRNA expression (Figure 3). DNA methylation was not significantly correlated with relative total aromatase expression at sites I.4.1 (R = 0.302, P = 0.142), I.4.2 (R = 0.047, P = 0.824) or I.4.4 (R = −0.063, P = 0.774). Similarly a positive correlation was also seen between DNA methylation and the I.4 promoter specific relative gene expression (Figure 3). The correlation between the I.4.5 CpG site and PI.4 aromatase expression remained significant after the exclusion of men (R = 0.473, P = 0.047). There were no correlations between percentage DNA methylation of the I.4 promoter CpG sites and relative PI.3 transcript expression.

Correlation between percentage DNA methylation at CpG sites 3 and 5 with total relative aromatase mRNA expression (n = 25) and I.4 promoter transcript aromatase expression (n = 19) in human subcutaneous adipocytes. Top left: Correlation between methylation percentage at the I.4.3 CpG site and total aromatase expression in (R = 0.474, P = 0.022). Top right: Correlation between methylation percentage at the I.4.5 CpG site and total aromatase expression (R = 0.549, P = 0.004). Bottom left: Correlation between methylation percentage at the I.4.3 CpG site and PI.4 aromatase expression (R = 0.449, P = 0.062). Bottom right: Correlation between methylation percentage at the I.4.5 site and PI.4 aromatase expression (R = 0.476, P = 0.040).
Site specific methylation, gene expression and phenotype
Methylation levels at the 5 CpG sites were not associated with age, weight or BMI in either tissue type. In subcutaneous adipocytes CpG methylation at the I.4.4 methylation was significantly lower in males than females (21% vs. 39%), there were no other differences between males and females at other sites. There was no difference in total or I.4 promoter specific relative gene expression in either depot. In 10 participants with whole body DXA (9 women and 1 man) subcutaneous adipocytes I.4.5 methylation percentage was correlated with increased headless whole body bone mineral content (R = 0.783, P = 0.007), bone mineral density (R = 0. 0.636, P = 0.048) and lean mass (R = 0.661, P = 0.038), but not whole body bone area, lean or fat mass (data not shown).
Discussion
In this study the correlation between DNA methylation determined by pyrosequencing within the regulatory region of the I.4 promoter region and total aromatase gene expression is extended to specific CpG sites from ex vivo human subcutaneous and omental mature adipocytes. The use of these primary fully differentiated cells allows a “snapshot” of the actual tissue specific relationship between DNA methylation and gene expression. Specifically we identified a negative correlation between DNA methylation at 2 CpG sites (I.4.1 and I 4.2) upstream of the aromatase promoter with aromatase mRNA expression in omental adipocytes. While in subcutaneous adipocytes percentage DNA methylation at I.4.3 site upstream of the transcription start site (TSS) and the I.4.5 site downstream of the TSS within the transcribed region of the gene were positively correlated with total aromatase mRNA expression. This “snapshot” of the DNA methylation status and gene expression in mature adipocytes expressing aromatase extends upon the findings of others using in vitro studies and supports the hypothesis that DNA methylation may play a role in tissue specific estrogen production.
Many epigenetic studies of DNA methylation and gene expression focus on genes with large CpG islands spanning the promoter regions of transcriptional initiation sites [38] and negative correlations between DNA methylation of CpG islands in the region of promoters and gene expression have been well described. However “non-classical” mechanisms are poorly understood. Given that the promoter regions of the aromatase gene does not contain CpG islands and is relatively CpG poor in comparison to other genomic regions [21, 39–41] it is unlikely that “classical” epigenetic regulation is occurring at this site. An example of epigenetic regulation of a CpG poor promoter is that of the cytochrome p450 27B1 (CYP27B1) gene promoter where 13 CpG sites around the transcription start site are either hypermethylated by vitamin D binding to an upstream regulatory site or demethylated in the presence of parathyroid hormone in MCT cells [42].
Two DNA methylation sites (I.4.1, I.4.2) that are 34 bp upstream from the glucocorticoid response element were negatively correlated with total aromatase gene expression in omental adipocytes. These findings are similar to those reported by Knower et al. [11].
Unlike omental adipocytes a strong positive correlation was observed between DNA methylation I.4.5 CpG sites and a somewhat weaker correlation with the I.4.3 site and aromatase mRNA expression in subcutaneous adipocytes. While DNA methylation within transcribed regions has previously been correlated with increased gene expression the mechanisms still remain uncertain [18–20]. A number of other potential models that may explain the correlation between DNA methylation within the transcribed gene regions and aromatase expression in mature adipocytes exist, including regulation of alternative promoter initiation [43], heterogeneity amongst the mature adipocyte population purified or splicing [44, 45] which is particularly likely given the tissue specific promoter usage of the aromatase gene [6] and the proximity of the I.4.5 CpG site to the splice junction. DNA methylation at the I.4.5 CpG site in subcutaneous adipocytes was also significantly positively associated with bone and body composition phenotypes. However caution must be taken in interpreting these findings given the nature of the cohort and the small number of participants with whole body composition and bone mineral density.
The strongest correlation between DNA methylation similarly was observed at the intragenic I.4.5 site located within a SP1 binding site that has previously been demonstrated to be important regulatory site for glucocorticoid stimulated aromatase gene expression in primary fetal hepatocytes and adipose derived stromal cells [9]. SP1 itself is an important regulator of CpG methylation status and when bound is thought to lead to hypomethylation enhanced transcription [46]. Interestingly, DNA methylation within the SP1 binding site does not appear to block SP1 binding [47], however it may be possible that DNA methylation within the SP1 binding site may block transcriptional repressors such as R1(RAM2/CDCA7L/JPO2) or E2F-associated phosphoprotein (EAPP) that compete with SP1 to bind to the SP1 binding site and have been reported to repress glucocorticoid dependant activation in other genes [48] further supporting the findings of others that adipocytes from subcutaneous tissue have greater responsiveness to glucocorticoids than omental tissue-derived adipocytes [23]. However further studies are needed to confirm the mechanism influencing these findings and its physiological role in estrogen production.
The strengths of this study was the use of matched omental and subcutaneous adipocytes, a purified homogenous ex vivo cell type to determine the relationship of DNA methylation within the I.4 promoter region with aromatase expression from a large number of human ex vivo samples not exposed to environmental hypomethylating agents in cell culture or extended passages. This model is therefore likely to represent endogenous relationships in mature adipocytes which are the primary extra-glandular source of estrogen in both men and women. The study also used pyrosequencing which is considered the “gold standard” for determining DNA methylation, in particular the inbuilt quality controls for bisulfite treatment [49] gives highly reproducible results [50] thus enabling quantitation of DNA methylation percentage at several CpG sites within close proximity [36].
An obvious limitation of the study is its observational nature however as yet no method of altering site specific DNA methylation status exists in primary human cells and hypo-methylation agents such as 5-aza-dC do not appear to hypomethylate CpG sites within the I.4 region of the aromatase gene in vitro[11]. Therefore currently the technology is not available to determine whether site specific DNA methylation actually alters gene expression in these cells.
Conclusions
These data demonstrate that DNA methylation of specific CpG sites across the I.4 promoter region is variable exhibiting both positive and negative correlations with aromatase expression in mature ex vivo adipocytes depending on the tissue depot. In addition DNA methylation at one CpG site within the transcribed region in subcutaneous adipocytes was correlated with body composition and bone mineral density as well as gene expression. These data further support the concept that epigenetic mechanisms may be involved in the regulation of aromatase expression and may be important in the determination of body composition by mechanisms yet to be identified.
References
Ryan KJ: Biological aromatization of steroids. J Biol Chem. 1959, 234 (2): 268-272.
Thompson EA, Siiteri PK: Utilization of oxygen and reduced nicotinamide adenine dinucleotide phosphate by human placental microsomes during aromatization of androstenedione. J Biol Chem. 1974, 249 (17): 5364-5372.
Chen SA, Besman MJ, Sparkes RS, Zollman S, Klisak I, Mohandas T, Hall PF, Shively JE: Human aromatase: cDNA cloning, Southern blot analysis, and assignment of the gene to chromosome 15. DNA. 1988, 7 (1): 27-38. 10.1089/dna.1988.7.27.
Bulun SE, Sebastian S, Takayama K, Suzuki T, Sasano H, Shozu M: The human CYP19 (aromatase P450) gene: update on physiologic roles and genomic organization of promoters. J Steroid Biochem Mol Biol. 2003, 86 (3–5): 219-
Sebastian S, Takayama K, Shozu M, Bulun SE: Cloning and characterization of a novel endothelial promoter of the human CYP19 (aromatase P450) gene that is up-regulated in breast cancer tissue. Mol Endocrinol. 2002, 16 (10): 2243-2254. 10.1210/me.2002-0123.
Sebastian S, Bulun SE: A highly complex organization of the regulatory region of the human CYP19 (aromatase) gene revealed by the Human Genome Project. J Clin Endocrinol Metab. 2001, 86 (10): 4600-4602. 10.1210/jc.86.10.4600.
Mahendroo MS, Mendelson CR, Simpson ER: Tissue-specific and hormonally controlled alternative promoters regulate aromatase cytochrome P450 gene expression in human adipose tissue. J Biol Chem. 1993, 268 (26): 19463-19470.
Toda K, Shizuta Y: Molecular cloning of a cDNA showing alternative splicing of the 5′-untranslated sequence of mRNA for human aromatase P-450. Eur J Biochem. 1993, 213 (1): 383-389. 10.1111/j.1432-1033.1993.tb17772.x.
Zhao Y, Mendelson CR, Simpson ER: Characterization of the sequences of the human CYP19 (aromatase) gene that mediate regulation by glucocorticoids in adipose stromal cells and fetal hepatocytes. Mol Endocrinol. 1995, 9 (3): 340-349. 10.1210/me.9.3.340.
Chen D, Reierstad S, Lu M, Lin Z, Ishikawa H, Bulun SE: Regulation of breast cancer-associated aromatase promoters. Cancer Lett. 2009, 273 (1): 15-27. 10.1016/j.canlet.2008.05.038.
Knower KC, To SQ, Simpson ER, Clyne CD: Epigenetic mechanisms regulating CYP19 transcription in human breast adipose fibroblasts. Mol Cell Endocrinol. 2010, 321 (2): 123-130. 10.1016/j.mce.2010.02.035.
Singal R, Ginder GD: DNA methylation. Blood. 1999, 93 (12): 4059-4070.
Dannenberg LO, Edenberg HJ: Epigenetics of gene expression in human hepatoma cells: expression profiling the response to inhibition of DNA methylation and histone deacetylation. BMC Genomics. 2006, 7: 181-10.1186/1471-2164-7-181.
Ho SM, Tang WY: Techniques used in studies of epigenome dysregulation due to aberrant DNA methylation: an emphasis on fetal-based adult diseases. Reprod Toxicol. 2007, 23 (3): 267-282. 10.1016/j.reprotox.2007.01.004.
Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, Pardhasaradhi K, McCann PP: S-Adenosylmethionine and methylation. FASEB J. 1996, 10 (4): 471-480.
Pogribny IP, Pogribna M, Christman JK, James SJ: Single-site methylation within the p53 promoter region reduces gene expression in a reporter gene construct: possible in vivo relevance during tumorigenesis. Cancer Res. 2000, 60 (3): 588-594.
Riviere G, Lienhard D, Andrieu T, Vieau D, Frey BM, Frey FJ: Epigenetic regulation of somatic angiotensin-converting enzyme by DNA methylation and histone acetylation. Epigenetics. 2011, 6 (4): 478-489. 10.4161/epi.6.4.14961.
Jones PA: The DNA methylation paradox. Trends Genet. 1999, 15 (1): 34-37. 10.1016/S0168-9525(98)01636-9.
Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM: Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009, 27 (4): 361-368. 10.1038/nbt.1533.
Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al: Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20 (3): 320-331. 10.1101/gr.101907.109.
Vanselow J, Selimyan R, Furbass R: DNA methylation of placenta-specific Cyp19 promoters of cattle and sheep. Exp Clin Endocrinol Diabetes. 2008, 116 (7): 437-442. 10.1055/s-2008-1058083.
Spitschak M, Vanselow J: Bovine large luteal cells show increasing de novo DNA methylation of the main ovarian CYP19A1 promoter P2. Gen Comp Endocrinol. 2012, 178 (1): 37-45. 10.1016/j.ygcen.2012.04.001.
Blouin K, Nadeau M, Mailloux J, Daris M, Lebel S, Luu-The V, Tchernof A: Pathways of adipose tissue androgen metabolism in women: depot differences and modulation by adipogenesis. Am J Physiol Endocrinol Metab. 2009, 296 (2): E244-E255.
Hankinson SE, Willett WC, Manson JE, Colditz GA, Hunter DJ, Spiegelman D, Barbieri RL, Speizer FE: Plasma sex steroid hormone levels and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 1998, 90 (17): 1292-1299. 10.1093/jnci/90.17.1292.
Paganini-Hill A, Henderson VW: Estrogen deficiency and risk of Alzheimer’s disease in women. Am J Epidemiol. 1994, 140 (3): 256-261.
Prince RL, Dick IM, Beilby J, Dhaliwal SS, Devine A: A cohort study of the effect of endogenous estrogen on spine fracture risk and bone structure in elderly women and an assessment of its diagnostic usefulness. Bone. 2007, 41 (1): 33-38. 10.1016/j.bone.2007.03.014.
Dick IM, Devine A, Prince RL: Association of an aromatase TTTA repeat polymorphism with circulating estrogen, bone structure, and biochemistry in older women. Am J Physiol Endocrinol Metab. 2005, 288 (5): E989-E995.
Mullin BH, Carter KW, Lewis JR, Ingley E, Wilson SG, Prince RL: Significant association between common polymorphisms in the aromatase gene CYP19A1 and bone mineral density in postmenopausal women. Calcif Tissue Int. 2011, 89 (6): 464-471. 10.1007/s00223-011-9535-8.
Wang JZ, Deogan MS, Lewis JR, Chew S, Mullin BH, McNab TJ, Wilson SG, Ingley E, Prince RL: A non-synonymous coding change in the CYP19A1 gene Arg264Cys (rs700519) does not affect circulating estradiol, bone structure or fracture. BMC Med Genet. 2011, 12 (1): 165-10.1186/1471-2350-12-165.
Riancho JA, Valero C, Naranjo A, Morales DJ, Sanudo C, Zarrabeitia MT: Identification of an aromatase haplotype that is associated with gene expression and postmenopausal osteoporosis. J Clin Endocrinol Metab. 2007, 92 (2): 660-665.
Riancho JA, Sanudo C, Valero C, Pipaon C, Olmos JM, Mijares V, Fernandez-Luna JL, Zarrabeitia MT: Association of the aromatase gene alleles with BMD: epidemiological and functional evidence. J Bone Miner Res. 2009, 24 (10): 1709-1718. 10.1359/jbmr.090404.
Dubois SG, Floyd EZ, Zvonic S, Kilroy G, Wu X, Carling S, Halvorsen YD, Ravussin E, Gimble JM: Isolation of human adipose-derived stem cells from biopsies and liposuction specimens. Methods Mol Biol. 2008, 449: 69-79.
Shigematsu M, Watanabe H, Sugihara H: Proliferation and differentiation of unilocular fat cells in the bone marrow. Cell struct Funct. 1999, 24 (2): 89-100. 10.1247/csf.24.89.
Jeong JH, Jung YK, Kim HJ, Jin JS, Kim HN, Kang SM, Kim SY, van Wijnen AJ, Stein JL, Lian JB, et al: The gene for aromatase, a rate-limiting enzyme for local estrogen biosynthesis, is a downstream target gene of Runx2 in skeletal tissues. Mol Cell Biol. 2010, 30 (10): 2365-2375. 10.1128/MCB.00672-09.
Agarwal VR, Ashanullah CI, Simpson ER, Bulun SE: Alternatively spliced transcripts of the aromatase cytochrome P450 (CYP19) gene in adipose tissue of women. J Clin Endocrinol Metab. 1997, 82 (1): 70-74. 10.1210/jc.82.1.70.
Tost J, Gut IG: DNA methylation analysis by pyrosequencing. Nat Protoc. 2007, 2 (9): 2265-2275. 10.1038/nprot.2007.314.
Demura M, Reierstad S, Innes JE, Bulun SE: Novel promoter I. 8 and promoter usage in the CYP19 (aromatase) gene. Reprod Sci. 2008, 15 (10): 1044-1053. 10.1177/1933719108322441.
Deaton AM, Bird A: CpG islands and the regulation of transcription. Genes Dev. 2011, 25 (10): 1010-1022. 10.1101/gad.2037511.
Furbass R, Selimyan R, Vanselow J: DNA methylation and chromatin accessibility of the proximal Cyp 19 promoter region 1.5/2 correlate with expression levels in sheep placentomes. Mol Reprod Dev. 2008, 75 (1): 1-7. 10.1002/mrd.20756.
Furbass R, Said HM, Schwerin M, Vanselow J: Chromatin structure of the bovine Cyp19 promoter 1.1. DNaseI hypersensitive sites and DNA hypomethylation correlate with placental expression. Eur J Biochem. 2001, 268 (5): 1222-1227. 10.1046/j.1432-1327.2001.01988.x.
Vanselow J, Pohland R, Furbass R: Promoter-2-derived Cyp19 expression in bovine granulosa cells coincides with gene-specific DNA hypo-methylation. Mol Cell Endocrinol. 2005, 233 (1–2): 57-64.
Kim MS, Kondo T, Takada I, Youn MY, Yamamoto Y, Takahashi S, Matsumoto T, Fujiyama S, Shirode Y, Yamaoka I, et al: DNA demethylation in hormone-induced transcriptional derepression. Nature. 2009, 461 (7266): 1007-1012. 10.1038/nature08456.
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al: Conserved role of intragenic DNA methylation in regulating alternative promoters. Nat Geosci. 2010, 466 (7303): 253-257.
Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S: CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nat Geosci. 2011, 479 (7371): 74-79.
Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T: Epigenetics in alternative pre-mRNA splicing. Cell. 2011, 144 (1): 16-26. 10.1016/j.cell.2010.11.056.
Brandeis M, Frank D, Keshet I, Siegfried Z, Mendelsohn M, Nemes A, Temper V, Razin A, Cedar H: Sp1 elements protect a CpG island from de novo methylation. Nat Geosci. 1994, 371 (6496): 435-438.
Harrington MA, Jones PA, Imagawa M, Karin M: Cytosine methylation does not affect binding of transcription factor Sp1. Proc Natl Acad Sci USA. 1988, 85 (7): 2066-2070. 10.1073/pnas.85.7.2066.
Chen K, Ou XM, Wu JB, Shih JC: Transcription factor E2F-associated phosphoprotein (EAPP), RAM2/CDCA7L/JPO2 (R1), and simian virus 40 promoter factor 1 (Sp1) cooperatively regulate glucocorticoid activation of monoamine oxidase B. Mol Pharmacol. 2011, 79 (2): 308-317. 10.1124/mol.110.067439.
Tost J, Dunker J, Gut IG: Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing. Biotechniques. 2003, 35 (1): 152-156.
Narasimhan S, Falkenberg VR, Khin MM, Rajeevan MS: Determination of quantitative and site-specific DNA methylation of perforin by pyrosequencing. BMC Res Notes. 2009, 2: 104-10.1186/1756-0500-2-104.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/14/87/prepub
Acknowledgements
We would like to thank all the participants and surgery staff for providing the samples for this study. We acknowledge the Australian Genome Research Facility (AGRF) for additional pyrosequencing. This work was supported by National Health and Medical Research Council (NHMRC) grant (Project grant 513472) and a Sir Charles Gairdner Hospital Research Advisory Committee Grant. The salary of JRL is supported by a Raine Medical Research Foundation Priming Grant. These funding agencies had no input into any aspect of the design and management of this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have read the journal’s policy and have the following conflicts: JRL, TJM, LJL, JZW, JT and RLP have nothing to declare. PH is an employee of QIAGEN Pty Ltd.
Authors’ contributions
Authors obtaining funding included RLP and JRL; Authors participated in conception and design were JRL, TJM, PH, LJL, JW and RLP; Authors carried out data analysis and interpretation included JRL, TJM, PH, JZW, LJL and RLP; Authors who did drafting of the manuscript and critical revision were JRL, TJM, PH, JT, JZW, LJL and RLP. All authors have read and approved the final manuscript.
Joshua R Lewis, Tegan J McNab contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lewis, J.R., McNab, T.J., Liew, L.J. et al. DNA methylation within the I.4 promoter region correlates with CYPl19A1 gene expression in human ex vivo mature omental and subcutaneous adipocytes. BMC Med Genet 14, 87 (2013). https://doi.org/10.1186/1471-2350-14-87
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-14-87