
Abstract
Background
Autosomal dominant optic atrophy (ADOA, Kjer disease, MIM #165500) is the most common form of hereditary optic neuropathy. Mutations in OPA1 located at chromosome 3q28 are the predominant cause for ADOA explaining between 32 and 89% of cases. Although deletions of OPA1 were recently reported in ADOA, the frequency of OPA1 genomic rearrangements in Denmark, where ADOA has a high prevalence, is unknown. The aim of the study was to identify copy number variations in OPA1 in Danish ADOA patients.
Methods
Forty unrelated ADOA patients, selected from a group of 100 ADOA patients as being negative for OPA1 point mutations, were tested for genomic rearrangements in OPA1 by multiplex ligation probe amplification (MLPA). When only one probe was abnormal results were confirmed by additional manually added probes. Segregation analysis was performed in families with detected mutations when possible.
Results
Ten families had OPA1 deletions, including two with deletions of the entire coding region and eight with intragenic deletions. Segregation analysis was possible in five families, and showed that the deletions segregated with the disease.
Conclusion
Deletions in the OPA1 gene were found in 10 patients presenting with phenotypic autosomal dominant optic neuropathy. Genetic testing for deletions in OPA1 should be offered for patients with clinically diagnosed ADOA and no OPA1 mutations detected by DNA sequencing analysis.
Similar content being viewed by others

Background
Autosomal dominant optic atrophy (ADOA) is the most common hereditary optic neuropathy. The phenotype is characterized by bilateral subnormal visual acuity, colour vision defect, a partial or absolute centrocoecal scotoma, optic nerve pallor, and subnormal retinal nerve fiber layer and ganglion cell layer thickness [1, 2]. The disease has incomplete penetrance and variable expression, ranging from subclinical visual manifestations to legal blindness [3]. The highly variable phenotype, both within and between pedigrees, suggests that genetic and/or environmental cofactors influence the expression of the disease. Kjer's optic atrophy or optic atrophy 1 (MIM #165500) [4], the ADOA originally described by Kjer, is caused by mutations in OPA1 (chromosome 3q28-q29). A specific frameshift mutation in exon 28 is particularly common in Denmark with evidence for a founder effect [5–7]. Other loci for ADOA include OPA4 on chromosome 18q12.2-q12.3 [8] and OPA5 on chromosome 22q12.1-q13.1 [9]. Dominant mutations in OPA3 have been recently reported in ADOA associated with cataract [10]. The gene most commonly involved in ADOA is OPA1 [11, 12] in which 205 unique pathogenic mutations have been identified http://lbbma.univ-angers.fr/eOPA1[13]. Many mutations have only been found in a single family. The prevalence of OPA1 mutations in ADOA patients ranges from 32 to 89%, suggesting the existence of other causative genes or alternative types of genetic defects, including genomic rearrangements [14–18].
Genomic deletions or duplications have been found to account for various genetic disorders [19–21]. Marchbank et al. [22] were first to identify complete deletion of OPA1 as a cause of ADOA. Recently, Fuhrmann et al. have shown that genomic aberrations may explain up to 12.9% of cases of Kjer-type ADOA [23].
Because a large fraction of our ADOA cases lacked a molecular diagnosis, in spite of having typical family histories in most cases, we initiated a study of copy number variation and other genomic rearrangements at the OPA1 locus to supplement the results of direct sequencing. We investigated 40 index patients diagnosed on clinical grounds with ADOA who had previously been found negative for mutations in OPA1 by DNA sequence analysis.
Methods
Patients and control subjects
One hundred unrelated index patients, of Danish origin, with clinically diagnosed ADOA were retrieved from the Danish ophthalmogenetics register and DNA repository of the National Eye Clinic at the Kennedy Center. The study included only cases from families with at least two affected members and an autosomal dominant pattern of inheritance. The diagnosis was based on routine clinical procedures, the standard being refraction and determination of best corrected visual acuity, color vision testing, visual evoked potential recording, fundus photography, Goldmann manual kinetic perimetry, and slit-lamp biomicroscopy of the anterior segment, vitreous, and posterior pole. No other inflammatory, ischemic, toxic causes of optic neuropathies were detected. Genomic DNA was obtained from the index patients and first degree relatives when available. Genomic DNA was extracted from leucocytes using Chemagic Magnetic Separation Module I (Chemagen, Baesweiler, Germany). The patients were screened for mutations in all coding regions and exon-intron boundaries by direct sequencing using BigDye chemistry and analyzed using an ABI 3130 instrument (Applied Biosystems, Foster City, CA, USA) (unpublished data). Of the 100 index patients, 40 were not harboring an identifiable OPA1 mutation. DNA from these 40 index patients was then analyzed for genomic rearrangements in OPA1 using multiplex ligation probe amplification (MLPA). The study was performed in accordance to the Helsinki declaration and was approved by the local ethics committee. Patients and healthy relatives had given their written informed consent.
MLPA Analysis
MLPA analysis was performed using a commercial kit (P229-B1, MRC-Holland, Amsterdam, The Netherlands) following the manufacturer's instructions. The MLPA KIT P229-B1 contains probes for 30 of 31 exons in OPA1. Additional MLPA probes were designed in-house to amplify regions narrowing down the identified deletions and to confirm initial findings. For the reactions we used 150 ng of patient DNA. The reactions were separated and visualized on an ABI 3130 Genetic Analyzer and further analyzed using GeneMarker (SoftGenetics, State College, PA, USA). Patients with all OPA1 exons deleted were further analyzed using both the P264 MLPA kit from MRC-Holland containing additional probes in the 3q29 telomere region (including OPA1, GP5, LSG1, CENTB2, TNK2, UBXD7, PAK2, MFI2, DLG1, BDH, KIAA0226, LMLN) and using manually designed probes with the P200 kit from MRC-Holland (probe sequences are available upon request). The results were considered significant when the peak height ratio of the normalized sample compared to the normalized average of controls was above 1.3 or below 0.65.
Results
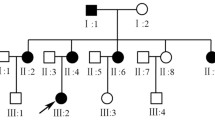
We identified 10 index patients with deletions in OPA1 (Figure 1 and Additional file 1), all patients being heterozygous. Clinical data are presented in table 1. No rearrangements other than deletions were found. Complete deletion of all 31 OPA1 exons was identified in the index patients from families DOA109 and DOA110. Further investigation with MLPA kit P264 showed that the GP5 gene, which is located telomeric to OPA1, was intact in both cases. Segregation analysis of family DOA110 showed that the deletion segregated with the disease (Figure 2B). Eight intragenic deletions spread across the gene were found in OPA1. The most N-terminal deletion was found in index patient from family DOA101 where exons 2-5 were deleted. In the index patient from family DOA102 we found a deletion of exon 10-16. In three independent families (DOA103, DOA104 and DOA105) we identified a deletion of exon 25 and 26. In family DOA103 the deletion was also present in an affected male and his daughter. In family DOA105 the deletion also segregated with the disease as it was present in two affected and absent in two unaffected individuals (Figure 2A).

Deletions and duplications identified in the OPA1 gene. Black boxes illustrate exons and light grey boxes illustrate alternative spliced exons. Arrows above the gene show the functional domains. Arrows below the gene show localization of deletions identified in the present study (blue arrows) and deletions (green arrows) and duplication (red arrow) identified by Fuhrmann et al. (2009) [23].

Pedigrees of three ADOA families A. Family DOA105, B. Family DOA110 C. Family DOA108. Index patients are indicated with an arrow. Filled symbols are individuals affected with ADOA, open symbols are either unaffected or not known. Individuals investigated by deletion analysis are shown with a "+del" (deletion present) or "-del" (no deletion) below symbol.
In family DOA106 we identified a deletion of exons 26 and 27 in the index patient. The deletion was also found in an affected daughter. A deletion of exon 28 was found in index patients from two independent families (DOA107 and DOA108). Only probe 06949-L06529 of P229 was deleted. This probe is located 104 nucleotides downstream of exon 28, and thus a manually designed MLPA probe located in intron 27 was made. As this probe was also deleted we conclude that exon 28 is deleted in these two families. In family DOA107 an affected individual had the deletion while an unaffected brother did not harbor the deletion. Family DOA108 is one of the oldest and largest known families with ADOA registered in Denmark with more than 28 affected and about 50 unaffected or unexamined individuals. Segregation analysis showed that the deletion segregated with the disease (Figure 2C).
Discussion
The main finding of our study is that in a series of 40 unrelated ADOA patients OPA1 deletions were found in 10. The 40 patients were selected from a cohort of 100 unrelated ADAO patients of whom 60 were found to have mutations in OPA1 by sequence analysis (data not shown). Thus assuming that the 60 patients do not have further mutations in OPA1, we find a frequency of 10% of deletions in OPA1 in Danish ADOA patients. Notably, the two patients with complete deletions of OPA1 did not present with any other symptoms than classical ADOA, supporting that haploinsufficiency is the pathogenic mutational mechanism causing classical non-syndromic ADOA phenotype. Additional studies are needed to determine the extent of the deletion by mapping the deletion breakpoints, which is beyond the scope of this report.
Our study is an agreement with the report of Fuhrmann et al. [23] who showed that ADOA can be related to genomic rearrangements of OPA1. We found OPA1 deletions in 10 out of 40 patients selected from a cohort of 100 patients corresponding to a frequency of 10%, which is comparable to 12.9% found by Fuhrmann et al. [23], however, contrary to Fuhrmann et al. [23] we found no duplications. The rearrangements are scattered throughout the gene, affecting single or multiple exons, or even whole gene deletions. Furthermore, the deletions described in this study are different from the rearrangements found by Fuhrmann et al. [23], showing that they arise as separate events, rather than being hotspots for rearrangements. Therefore, we suggest that deletion and duplication analysis of OPA1 should be included in the routine genetic analysis of ADOA patients.
Several studies of various genes have shown that deletions or duplications not detectable with sequencing or screening strategies such as single strand conformational polymorphism (SSCP), contribute to the mutational load, which is confirmed by our study [20, 24]. Thus, genomic rearrangements have to be considered in diseases where a proportion of patients apparently do not harbor mutations in the disease causing gene/genes, since these will be left unrecognized by sequence analysis which is the preferred method for mutation analysis. MLPA is a fast and relatively cheap method to analyze for gene dosage differences among large groups of patients.
Conclusion
OPA1 genomic deletions account for about 10% of ADOA cases in a Danish population, registered at the national center for hereditary eye diseases. Our findings suggest that an analysis of genomic rearrangements is mandatory in the investigation and diagnosis of ADOA.
References
Milea D, Sander B, Wegener M, Jensen H, Kjer B, Jorgensen TM, Lund-Andersen H, Larsen M: Axonal loss occurs early in dominant optic atrophy. Acta Ophthalmol. 2010, 88: 342-346. 10.1111/j.1755-3768.2009.01738.x.
Kerrison JB: Hereditary optic neuropathies. Ophthalmol Clin North Am. 2001, 14: 99-107.
Votruba M, Moore AT, Bhattacharya SS: Clinical features, molecular genetics, and pathophysiology of dominant optic atrophy. J Med Genet. 1998, 35: 793-800. 10.1136/jmg.35.10.793.
Kjer P: Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol Suppl. 1959, 164: 1-147.
Thiselton DL, Alexander C, Morris A, Brooks S, Rosenberg T, Eiberg H, Kjer B, Kjer P, Bhattacharya SS, Votruba M: A frameshift mutation in exon 28 of the OPA1 gene explains the high prevalence of dominant optic atrophy in the Danish population: evidence for a founder effect. Hum Genet. 2001, 109: 498-502. 10.1007/s004390100600.
Eiberg H, Kjer B, Kjer P, Rosenberg T: Dominant optic atrophy (OPA1) mapped to chromosome 3q region. I. Linkage analysis. Hum Mol Genet. 1994, 3: 977-980. 10.1093/hmg/3.6.977.
Kjer B, Eiberg H, Kjer P, Rosenberg T: Dominant optic atrophy mapped to chromosome 3q region. II. Clinical and epidemiological aspects. Acta Ophthalmol Scand. 1996, 74: 3-7. 10.1111/j.1600-0420.1996.tb00672.x.
Kerrison JB, Arnould VJ, Ferraz Sallum JM, Vagefi MR, Barmada MM, Li Y, Zhu D, Maumenee IH: Genetic heterogeneity of dominant optic atrophy, Kjer type: Identification of a second locus on chromosome 18q12.2-12.3. Arch Ophthalmol. 1999, 117: 805-810.
Barbet F, Hakiki S, Orssaud C, Gerber S, Perrault I, Hanein S, Ducroq D, Dufier JL, Munnich A, Kaplan J, et al: A third locus for dominant optic atrophy on chromosome 22q. J Med Genet. 2005, 42: e1-10.1136/jmg.2004.025502.
Reynier P, Amati-Bonneau P, Verny C, Olichon A, Simard G, Guichet A, Bonnemains C, Malecaze F, Malinge MC, Pelletier JB, et al: OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J Med Genet. 2004, 41: e110-10.1136/jmg.2003.016576.
Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, et al: OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000, 26: 211-215. 10.1038/79944.
Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, et al: Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000, 26: 207-210. 10.1038/79936.
Ferre M, Amati-Bonneau P, Tourmen Y, Malthiery Y, Reynier P: eOPA1: an online database for OPA1 mutations. Hum Mutat. 2005, 25: 423-428. 10.1002/humu.20161.
Thiselton DL, Alexander C, Taanman JW, Brooks S, Rosenberg T, Eiberg H, Andreasson S, Van RN, Munier FL, Moore AT, et al: A comprehensive survey of mutations in the OPA1 gene in patients with autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci. 2002, 43: 1715-1724.
Toomes C, Marchbank NJ, Mackey DA, Craig JE, Newbury-Ecob RA, Bennett CP, Vize CJ, Desai SP, Black GC, Patel N, et al: Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum Mol Genet. 2001, 10: 1369-1378. 10.1093/hmg/10.13.1369.
Pesch UE, Leo-Kottler B, Mayer S, Jurklies B, Kellner U, Apfelstedt-Sylla E, Zrenner E, Alexander C, Wissinger B: OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum Mol Genet. 2001, 10: 1359-1368. 10.1093/hmg/10.13.1359.
Baris O, Delettre C, Amati-Bonneau P, Surget MO, Charlin JF, Catier A, Derieux L, Guyomard JL, Dollfus H, Jonveaux P, et al: Fourteen novel OPA1 mutations in autosomal dominant optic atrophy including two de novo mutations in sporadic optic atrophy. Hum Mutat. 2003, 21: 656-10.1002/humu.9152.
Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, Lenaers G, Belenguer P, Hamel CP: Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet. 2001, 109: 584-591. 10.1007/s00439-001-0633-y.
del VJ, Feliubadalo L, Nadal M, Teule A, Miro R, Cuesta R, Tornero E, Menendez M, Darder E, Brunet J, et al: Identification and comprehensive characterization of large genomic rearrangements in the BRCA1 and BRCA2 genes. Breast Cancer Res Treat. 2010, 122: 733-743. 10.1007/s10549-009-0613-9.
Ellard S, Thomas K, Edghill EL, Owens M, Ambye L, Cropper J, Little J, Strachan M, Stride A, Ersoy B, et al: Partial and whole gene deletion mutations of the GCK and HNF1A genes in maturity-onset diabetes of the young. Diabetologia. 2007, 50: 2313-2317. 10.1007/s00125-007-0798-6.
Paracchini V, Seia M, Coviello D, Porcaro L, Costantino L, Capasso P, Degiorgio D, Padoan R, Corbetta C, Claut L, et al: Molecular and clinical features associated with CFTR gene rearrangements in Italian population: identification of a new duplication and recurrent deletions. Clin Genet. 2008, 73: 346-352. 10.1111/j.1399-0004.2007.00957.x.
Marchbank NJ, Craig JE, Leek JP, Toohey M, Churchill AJ, Markham AF, Mackey DA, Toomes C, Inglehearn CF: Deletion of the OPA1 gene in a dominant optic atrophy family: evidence that haploinsufficiency is the cause of disease. J Med Genet. 2002, 39: e47-10.1136/jmg.39.8.e47.
Fuhrmann N, Alavi MV, Bitoun P, Woernle S, Auburger G, Leo-Kottler B, Yu-Wai-Man P, Chinnery P, Wissinger B: Genomic rearrangements in OPA1 are frequent in patients with autosomal dominant optic atrophy. J Med Genet. 2009, 46: 136-144. 10.1136/jmg.2008.062570.
Concolino P, Mello E, Toscano V, Ameglio F, Zuppi C, Capoluongo E: Multiplex ligation-dependent probe amplification (MLPA) assay for the detection of CYP21A2 gene deletions/duplications in congenital adrenal hyperplasia: first technical report. Clin Chim Acta. 2009, 402: 164-170. 10.1016/j.cca.2009.01.008.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/12/49/prepub
Acknowledgements
We wish to thank Nuzha Biari, Kennedy Center, Glostrup, Denmark for technical assistance; Hans Ulrik Møller, Viborg Hospital, Viborg, Denmark and Toke Bek, Aarhus University Hospital, Aarhus, Denmark for clinical assistance. We are grateful to all the patients and their relatives for participating in this study. Very special thanks to Thomas Rosenberg for initiating the work and for help and advice. The work was funded by the Michaelsen Foundation, The Danish Eye Research Foundation and the AP Møller Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors have read, edited and approved the final manuscript.
GJA performed experiments, analyzed data and drafted the manuscript. KG helped designed the study, analyzed data and drafting the manuscript. JE designed the study and experiments. KBN, ML, DM contributed to writing of the paper. ML and DM helped with the ophthalmological clinical information.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Almind, G.J., Grønskov, K., Milea, D. et al. Genomic deletions in OPA1 in Danish patients with autosomal dominant optic atrophy. BMC Med Genet 12, 49 (2011). https://doi.org/10.1186/1471-2350-12-49
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-12-49