
Abstract
Smith-Magenis syndrome (SMS) is a complex syndrome involving intellectual disabilities, sleep disturbance, behavioural problems, and a variety of craniofacial, skeletal, and visceral anomalies. While the majority of SMS cases harbor an ~3.5 Mb common deletion on 17p11.2 that encompasses the retinoic acid induced-1 (RAI1) gene, some patients carry small intragenic deletions or point mutations in RAI1. We present data on two cases of Smith-Magenis syndrome with mutation of RAI1. Both cases are phenotypically consistent with SMS and RAI1 mutation but also have other anomalies not previously reported in SMS, including spontaneous pneumothoraces. These cases also illustrate variability in the SMS phenotype not previously shown for RAI1 mutation cases, including hearing loss, absence of self-abusive behaviours, and mild global delays. Sequencing of RAI1 revealed mutation of the same heptameric C-tract (CCCCCCC) in exon 3 in both cases (c.3103delC one case and and c.3103insC in the other), resulting in frameshift mutations. Of the seven reported frameshift mutations occurring in poly C-tracts in RAI1, four cases (~57%) occur at this heptameric C-tract. Collectively, these results indicate that this heptameric C-tract is a preferential hotspot for single nucleotide insertion/deletions (SNindels) and therefore, should be considered a primary target for analysis in patients suspected for mutations in RAI1. We expect that as more patients are sequenced for mutations in RAI1, the incidence of frameshift mutations in this hotspot will become more evident.
Similar content being viewed by others

Background
Smith-Magenis syndrome (SMS, OMIM182290) is a complex syndrome involving intellectual disabilities, sleep disturbance, behavioural problems, and a variety of craniofacial, skeletal, and visceral anomalies (Table 1). The majority of SMS cases harbor an ~3.5 Mb common deletion on 17p11.2 that encompasses the retinoic acid induced-1 (RAI1) gene, which is the causative gene in this disorder [1, 2]. Patients who do not have a cytogenetically detectable deletion of 17p11.2 but whom exhibit phenotypes consistent with SMS are sequenced for mutations in RAI1. To date, fourteen patients with point mutations or small deletions within RAI1 have been reported in the literature (Table 2) [1, 3–6].
Case Presentation
Case 1
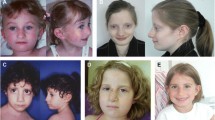
SMS324 is an 18-year-old male who was delivered at 38 weeks gestation following premature labor and antepartum haemorrhage. His birth weight was 2,750 g (10th-50th centile). While his neonatal period was complicated by gastroesophageal reflux and failure to thrive, he was admitted to the hospital at 1 year of age for being considerably overweight, with fat folds on his arms and legs. He spoke his first words at 6 months and walked at 12 months. His medical history includes two episodes of severe asthma, petit mal seizures between the ages of 5-10 y and three spontaneous pneumothoraces (SP). He had a square shaped face, upslanting palpebral fissures, down turned mouth, inverted upper lip, and synophrys (Fig. 1A). Other physical anomalies included brachydactyly, bilateral fifth finger clinodactyly with a small middle phalanx of his fifth fingers, and pes planus. At 16 years of age, height was 163 cm (< 3rd centile), head circumference was 54 cm, and he presented with relative truncal obesity.

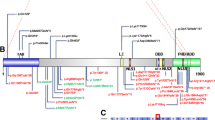
Mutation screening of SMS324 for RAI1 and FLCN. A. Photograph of SMS324 at 18y, exhibiting features characteristic of SMS. B. DNA chromatograph of the normal allele and the c.3103delC change identified, resulting in a frameshift in the mutant allele. This mutation is located within a heptameric C-tract (red C’s in the normal allele). C. Representation of the gene structure of FLCN transcript variant 1(top) and transcript variant 2 (bottom). Non-coding exons are blue while coding exons are red. The FLCN polymorphism identified in this patient is localized with arrows in both transcript variants and is numbered accordingly.
Behaviorally, SMS324 exhibited head-banging and rage attacks starting at age 3. At age 16, he continues to have recurrent episodes of rage attacks, anxiety, and obsessional behavior. He has never slept through the night, with recurrent 3 a.m. wakenings. Apart from finger chewing, he exhibits no self-injurious behaviors, self hugging, onychotillomania or polyembolokomania, which are common behaviors found in SMS patients. He has had a persistent history of sleep disturbance. A formal developmental assessment indicated a mild global developmental delay. He was initially diagnosed with attention deficit disorder; however, a later assessment indicated this diagnosis was incorrect. He was initially mainstreamed in a regular classroom at school but at age 16 now functions in an OI classroom with supervision. The patient was referred for SMS genetic evaluation based upon phenotypic similarities to SMS and/or RAI1 mutation patients (Table 1).
Karyotype analysis and 17p11.2 FISH including RAI1 for this patient was normal, as were 22q11.2 FISH and subtelomeric deletion screening. DNA sequencing of RAI1, however, revealed a deletion of a single cytosine, c.3103delC, in a heptameric C-tract (CCCCCCC) of exon 3 (Fig. 1B). This frameshift mutation predicts the misincorporation of 28 amino acids before a premature stop codon is introduced. The resulting truncated protein is predicted to be non-functional, contributing to the haploinsufficiency of RAI1.
Case 2
SMS335, a 16-year-old female, was delivered full term with a birth weight of 3,398 g (90th percentile). The pregnancy was unremarkable aside from maternal Macrodantin use for urinary tract infection throughout. At the time of evaluation, SMS335 was short in stature (10th -25th percentile), noted to have generalized hypotonia, global developmental delays, significant hearing loss, truncal obesity, and commonly observed behaviours of SMS including sleep disturbance, skin picking, and toenail removal. For SMS335, phenotypes consistent with SMS/mutations in RAI1 are noted in Table 1. Chromosome analyses for fragile-X syndrome, Prader-Willi syndrome and SMS were normal for this patient. DNA sequencing performed by GeneDx (Gaithersburg, MD) identified a c.3103insC, located within the same heptameric C-tract in exon 3 of RAI1, as noted in SMS324.
Conclusions
Single nucleotide insertion/deletions (SNindels) occur at an estimated frequency of 1 for every 11,274 bp of the human genome, with more than half occurring in regions with mononucleotide repeats. SNindels can arise from slipped mispairing in regions with repeating units during DNA synthesis or repair [7]. The c.3103delC and c.3103insC frameshift mutations reported here were previously reported as novel mutations in two other patients [4, 5]. Of the seven reported frameshift mutations occurring in poly C-tracts in RAI1, four cases (~57%) occur at this heptameric C-tract (Table 2). Collectively, these results indicate that this heptameric C-tract is a preferential hotspot for SNindels resulting in a frameshift mutation, and therefore, should be considered a primary target for analysis in patients suspected for mutations in RAI1. We expect that as more patients are sequenced for mutations in RAI1, the incidence of frameshift mutations in this hotspot will become more evident (RAI1 sequencing conditions are available upon request).
Case SMS324 has provided us with an opportunity to evaluate a possible genetic link underlying his history of spontaneous pneumothoraces (SP), a phenotype that has not been documented in any individual with SMS or mutations in RAI1 evaluated to date. Mutations in the folliculin (FLCN) gene have been implicated in Birt-Hogg-Dubé syndrome (BHD, OMIM135150), an autosomal dominant genodermatosis which predisposes for multiple fibrofolliculomas, renal carcinoma, lung cysts and SP [8–11]. Although SMS324 does not have any other features consistent with BHD aside from the three episodes of SP, clinical heterogeneity is known to exist amongst individuals with germline mutations in the FLCN gene. Cases have been reported whereby patients with mutations of FLCN exhibit SP without the involvement of fibrofolliculomas or renal tumours [9, 12–14].
FLCN is located within the SMS region of chromosome 17 and most cases of SMS due to 17p11.2 deletion are heterozygously deleted for this gene. It is alternatively spliced and results in two transcript variants that encode different isoforms. FLCN transcript variant 1 (RefSeq NM_144997) encodes the longer isoform of this protein and is widely expressed in various tissues. Transcript variant 2 (NM_144606) uses an alternative splice site in the 3' coding region to produce a shorter protein product. Direct sequencing of the entire coding region of FLCN, using primers designed based upon the RefSeq, was performed for SMS324. No known causative mutations were identified. However, we detected a single nucleotide alteration of unknown significance within an intronic sequence in the primary transcript, c.871+36G>A, which is considered to be a non-synonymous variant in FLCN (Fig. 1C; dbSNP: rs3744124) [Gunji et al., 2007; Cho et al., 2008; Wei et al., 2009]. Although the coding region of FLCN is highly conserved across vertebrate species, rs3744124 mapped to a region of low conservation amongst 17 vertebral species [Cho et al., 2008].
Alignment of the FLCN polymorphism found in SMS324 against the sequence of transcript variants 1 and 2 reveals a noteworthy observation. This polymorphic change occurs within intron 8 of transcript variant 1 and is expected to have no effect on the production of a fully functional FLCN isoform 1 protein in SMS324. However, when aligned against transcript variant 2 a nucleotide change, c.907G>A, occurs in coding exon 8 for variant 2, resulting in a missense mutation, p.Gly303Arg (Fig. 1C). Thus, while this DNA alteration is considered an intronic SNP for variant 1, it creates a missense mutation in variant 2. Whether or not this discrepancy is significant or has any bearing on the SP observed in SMS324 is unclear at this time. Interestingly, no studies have investigated the frequency and/or expression of transcript variant 2 in normal or neoplastic tissues and moreover, if there is an isoform specific association with certain phenotypes manifested in BHD, such as SP.
These studies add two interesting cases to the growing list of mutations in RAI1 that result in SMS and highlight that key phenotypic differences between patients may exist, even when the underlying etiology is the same. Data presented here highlight a specific poly C tract region within RAI1 that may be more prone to single basepair insertion or deletion, with 57% of C tract RAI1 mutations reported to date. The cases presented also show that even though RAI1 is the primary gene causative of features of SMS, that other genes must be considered in phenotypically complex cases to provide full recurrence and disease risk information for other members of the family.
Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH: Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet. 2003, 33 (4): 466-468. 10.1038/ng1126.
Vlangos CN, Yim DK, Elsea SH: Refinement of the Smith-Magenis syndrome critical region to approximately 950 kb and assessment of 17p11.2 deletions. Are all deletions created equally?. Mol Genet Metab. 2003, 79 (2): 134-141. 10.1016/S1096-7192(03)00048-9.
Girirajan S, Vlangos CN, Szomju BB, Edelman E, Trevors CD, Dupuis L, Nezarati M, Bunyan DJ, Elsea SH: Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet Med. 2006, 8 (7): 417-427. 10.1097/01.gim.0000228215.32110.89.
Bi W, Saifi GM, Girirajan S, Shi X, Szomju B, Firth H, Magenis RE, Potocki L, Elsea SH, Lupski JR: RAI1 point mutations, CAG repeat variation, and SNP analysis in non-deletion Smith-Magenis syndrome. Am J Med Genet A. 2006, 140 (22): 2454-2463.
Bi W, Saifi GM, Shaw CJ, Walz K, Fonseca P, Wilson M, Potocki L, Lupski JR: Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith-Magenis syndrome. Hum Genet. 2004, 115 (6): 515-524. 10.1007/s00439-004-1187-6.
Girirajan S, Elsas LJ, Devriendt K, Elsea SH: RAI1 variations in Smith-Magenis syndrome patients without 17p11.2 deletions. J Med Genet. 2005, 42 (11): 820-828. 10.1136/jmg.2005.031211.
Tan EC, Li H: Characterization of frequencies and distribution of single nucleotide insertions/deletions in the human genome. Gene. 2006, 376 (2): 268-280. 10.1016/j.gene.2006.04.009.
Schmidt LS, Warren MB, Nickerson ML, Weirich G, Matrosova V, Toro JR, Turner ML, Duray P, Merino M, Hewitt S, et al: Birt-hogg-dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001, 69 (4): 876-882. 10.1086/323744.
Toro JR, Pautler SE, Stewart L, Glenn GM, Weinreich M, Toure O, Wei MH, Schmidt LS, Davis L, Zbar B, et al: Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dube syndrome. Am J Respir Crit Care Med. 2007, 175 (10): 1044-1053. 10.1164/rccm.200610-1483OC.
Warren MB, Torres-Cabala CA, Turner ML, Merino MJ, Matrosova VY, Nickerson ML, Ma W, Linehan WM, Zbar B, Schmidt LS: Expression of Birt-Hogg-Dube gene mRNA in normal and neoplastic human tissues. Mod Pathol. 2004, 17 (8): 998-1011. 10.1038/modpathol.3800152.
Zbar B, Alvord WG, Glenn G, Turner M, Pavlovich CP, Schmidt L, Walther M, Choyke P, Weirich G, Hewitt SM, et al: Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dube syndrome. Cancer Epidemiol Biomarkers Prev. 2002, 11 (4): 393-400.
Gunji Y, Akiyoshi T, Sato T, Kurihara M, Tominaga S, Takahashi K, Seyama K: Mutations of the Birt Hogg Dube gene in patients with multiple lung cysts and recurrent pneumothorax. J Med Genet. 2007, 44 (9): 588-593. 10.1136/jmg.2007.049874.
Painter JN, Tapanainen H, Somer M, Tukiainen P, Aittomaki K: A 4-bp deletion in the Birt-Hogg-Dube gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet. 2005, 76 (3): 522-527. 10.1086/428455.
Schmidt LS, Nickerson ML, Warren MB, Glenn GM, Toro JR, Merino MJ, Turner ML, Choyke PL, Sharma N, Peterson J, et al: Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dube syndrome. Am J Hum Genet. 2005, 76 (6): 1023-1033. 10.1086/430842.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/11/142/prepub
Acknowledgements
We sincerely thank SMS324 and SMS335 and their families for participating in our study. We are indebted to Dr. Laura Schmidt for providing FLCN primers for sequencing and discussions on BHD. Patient enrollment and sample collection in this study were in accordance to approved IRB protocols by either Virginia Commonwealth University (VCU3784) or Charles Sturt University (2006/022). This work was funded through resources from Virginia Commonwealth University to S.H.E.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors contributed to the work presented and participated in writing or reviewing the manuscript. HTT performed the experiments and drafted the initial manuscript; TD identified cases and collected clinical data; CLB analysed molecular data and co-supervised the project; and SHE co-supervised the project, analyzed data, and drafted the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Truong, H.T., Dudding, T., Blanchard, C.L. et al. Frameshift mutation hotspot identified in Smith-Magenis syndrome: case report and review of literature. BMC Med Genet 11, 142 (2010). https://doi.org/10.1186/1471-2350-11-142
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-11-142