
Abstract
Background
Retinitis pigmentosa is the most important hereditary retinal degenerative disease, which has a high degree of clinical and genetic heterogeneity. More than half of all cases of retinitis pigmentosa are autosomal recessive (arRP), but the gene(s) causing arRP in most families has yet to be identified. The purpose of this study is to identify the genetic basis of severe arRP in a consanguineous Chinese family.
Methods
Linkage and haplotype analyses were used to define the chromosomal location of the pathogenic gene in the Chinese arRP family. Direct DNA sequence analysis of the entire coding region and exon-intron boundaries of EYS was used to determine the disease-causing mutation, and to demonstrate that the mutation co-segregates with the disease in the family.
Results
A single nucleotide substitution of G to T at nucleotide 5506 of EYS was identified in the Chinese arRP family. This change caused a substitution of a glutamic acid residue at codon 1,836 by a stop codon TAA (p.E1836X), and resulted in a premature truncated EYS protein with 1,835 amino acids. Three affected siblings in the family were homozygous for the p.E1836X mutation, while the other unaffected family members carried one mutant allele and one normal EYS allele. The nonsense mutation p.E1836X was not detected in 200 unrelated normal controls.
Conclusions
The EYS gene is a recently identified disease-causing gene for retinitis pigmentosa, and encodes the orthologue of Drosophila spacemaker. To date, there are only eight mutations in EYS that have been identified to cause arRP. Here we report one novel homozygous nonsense mutation of EYS in a consanguineous Chinese arRP family. Our study represents the first independent confirmation that mutations in EYS cause arRP. Additionally, this is the first EYS mutation identified in the Chinese population.
Similar content being viewed by others

Background
Retinitis pigmentosa (RP; OMIM 268000) is characterized by the constriction of the visual fields, night blindness, changes of the fundi including 'bone corpuscle' lumps of pigment, and the loss of central vision. The worldwide prevalence of RP is about one in 4,000 [1]. RP is inherited most frequently (50-60% of cases) as an autosomal recessive trait, followed by autosomal dominant (30-40%) and then X-linked (5-15%) [1–6]. It is the most common hereditary retinal dystrophy causing irreversible blindness [1–3]. About 30 genes and loci have been implicated in isolated cases of arRP to date [RetNet: http://www.sph.uth.tmc.edu/Retnet/disease.htm]. Mutations in ABCA4, CDHR1, CERKL, CNGA1, CNGB1, CRB1, IDH3B, LRAT, MERTK, NR2E3, NRL, PDE6A, PDE6B, PRCD, PROM1, RBP3, RGR, RHO, RLBP1, RP1, RPE65, SAG, SPATA7, TULP1, USH2A and most recently EYS have been identified to be the cause for arRP [7, 8]. However, the gene(s) causing sporadic arRP in most families has yet to be discovered. The identification of genotypes and phenotypes and the physiological roles of novel RP genes in the retina should provide valuable information for the diagnosis and classification of retinal degeneration. Consequently, genetic studies will result in the scientific basis for the prevention and treatment of RP.
RP25, a genetic locus for arRP, was mapped to a ~ 16 cM region on chromosome 6p12.1-q15 in four Spanish families by Ruiz et al. in 1998 [9]. Then, the pathogenic genes for several arRP families with various ancestral origins, including one Pakistani family and three Chinese families, were also been mapped to this locus [10–12]. By using the 10K genechip array, Barragán I et al. refined the disease interval from the original 16 cM to only a 2.67 cM region between D6S257 and D6S1557 [13]. Most recently, the disease-causing gene at this locus has been identified as the EYS gene. EYS is predicted to be a 3,165 amino acid multi-domain protein, which contains at least 28 epidermal growth factor (EGF)-like domains in its N-terminus, and C-terminal laminin A G-like domains. It is an evolutionarily conserved protein from Drosophila to humans. Finally, eight EYS mutations were detected in eight arRP families from Spanish and Dutch origins at almost the same time in 2008 [7, 8].
In this study, we characterized a consanguineous Chinese family with a severe form of arRP. After linkage analysis with known arRP causative genes and loci, we excluded all other arRP loci except for the RP25 locus. Further direct DNA sequence analysis revealed a homozygous mutation in the EYS. Our data confirmed the conclusion from the two original reports that mutations in EYS can cause arRP, and expanded the mutation spectrum of EYS to the Chinese population.
Methods
The study subjects from a consanguineous Chinese arRP family were recruited from Hubei Province, P. R. China. Informed consent was obtained from the participants in accordance with the study protocols approved by the Ethics Committee of Huazhong University of Science and Technology. Diagnosis of RP was carried out by clinical and ophthalmological examinations. Peripheral blood was collected from the family members and 200 normal Chinese Han controls. Total human genomic DNA was isolated with the DNA Isolation Kit for Mammalian Blood (Roche Diagnostic Co., Indianapolis, IN).
Linkage and haplotype analyses were applied to test the linkage of the family to 22 arRP loci, including ABCA4, CERKL, CNGA1, CNGB1, CRB1, EYS, I8DH3B, LRAT, MERTK, NR2E3, NRL, PDE6A, PDE6B, PRCD, PROM1, RGR, RHO, RLBP1, RPE65, SAG, TULP1, and USH2A. The microsatellite markers flanking these loci were selected from Linkage Mapping Set MD-10 (Applied Biosystems, Inc., Foster City, CA), and genotyped using an ABI 3100 Genetic Analyzer. Genotypes were analyzed by the GeneMapper 2.5 Software program (Applied Biosystems, Inc., Foster City, CA). PCR and genotyping were performed as previously described [14]. When the pathogenic gene was mapped to chromosome 6q12 that harbors the EYS gene, additional SNPs (rs4710437, rs1057530, rs4710457, rs66462731, rs10944813, rs4710292) were employed for the linkage analysis to get unambiguous linked haplotype. The EYS was then subjected to mutational analysis. All coding exons and splice sites of EYS were amplified by PCR and sequenced. The intronic primers and corresponding PCR conditions were as described in the paper of Abd El-Aziz et al. [7].
Results
The RP in the consanguineous Chinese family is inherited in an autosomal recessive mode (figure 1). Parents I1 and I2 are third cousins. The proband II2 and his two sisters had deteriorating vision, while the parents and young brother II4 did not show any RP features. The family history was negative for deteriorating vision or night blindness.

Pedigree structure and haplotype analysis at the RP25 locus. Blackened bars indicate the disease haplotype. Filled squares or filled circles represent male or female individuals affected with RP, respectively. Arrow points to the proband (II2). All patients in the arRP family carry the homozygous haplotype between single nucleotide polymorphisms rs4710292 and rs4710437. The genomic positions of two markers are from Human (Homo sapiens) Genome Browser Gateway, the GRCh37 build version, and six SNPs are from NCBI B37.1 assembly.
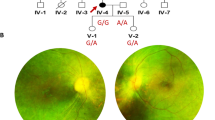
The proband exhibited night blindness at the age of 15 years, and progressively lost his visual acuity to 20/400 at approximately the age of 35-40 years. Funduscopic examinations revealed obvious attenuation of the retinal arteries, waxen color of discs, and bone-spicule pigmentation in the peripheral-mid retina (figure 2A). Electrophysiological examination showed an extinguished or very low amplitude ERG in both eyes (figure 2B).

Fundus photographs and ERG of the proband in the Chinese arRP family. The features of waxy-pale disc, arteriolar attenuation, and bone-spicule pigment deposit in the mid-peripheral retina are shown in A. The full-field ERG in the proband in B, either a-wave or b-wave has obvious reduced amplitude and prolonged response time in both eyes.
The other two affected individuals also had the similar symptoms as the proband. Night blindness appeared late in the second decade of life, low visual acuity between 20/400 and hand movement, and a flat ERG confirmed the diagnosis and severity of the RP disease. No additional symptoms or ocular defects were observed in the affected individuals of the family except for patient II5, who was affected with cataract in both right and left eyes.
After linkage analysis for known arRP causative genes and loci, there was only the RP25 locus that showed a definite linkage with the disease in the family. Homozygosity mapping with haplotype analysis revealed that all three affected siblings (II1, II2, II5) inherited the affected alleles separately from their carrier parents, whereas their unaffected brother II4 inherited two normal chromosomes, and the proband's daughter III1 only one affected allele (Figure 1). These results suggest that EYS may be the pathogenic gene in this family.
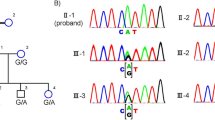
Direct DNA sequence analysis for the entire coding region of EYS (Genbank accession No: FM209056) found a single nucleotide transversion of G to T at nucleotide 5506 of EYS, and it resulted in a substitution of a glutamic acid residue at codon 1836 of EYS by a stop codon TAA (p.E1836X) (Figure 3). The nonsense p.E1836X mutation generates a truncated EYS protein with only 1835 amino acid residues. Three affected siblings are homozygous for the p.E1836X mutation, whereas their mother, and III1, the daughter of proband, carry only one affected allele. The p.E1836X mutation was not detected in 200 unrelated normal controls.

Identification of the homozygous mutation in the consanguineous Chinese arRP family. A and B show the exon 26 of EYS sequences from a normal individual and an affected individual with p.E1836X mutation, respectively. The sequence for individual III1, who is heterozygous for the mutation, is shown in C.
Discussion
A novel EYS nonsense mutation p.E1836X in a consanguineous Chinese arRP family was identified, which is the first EYS mutation discovered in the Chinese population. The p.E1836X mutation is predicted to generate an abnormal EYS protein with only 1,835 of 3,165 amino acid residues. EYS is a multi-domain protein containing 28 epidermal growth factor (EGF)-like domains, and 5 LamG domains at the C-terminus. All 5 LamG domains and 7 EGF-like domains in the C-terminus of EYS were truncated in the arRP family, which may significantly disrupt the normal physiological function of EYS in the retina.
The EYS gene has recently been identified as the disease-causing gene for retinitis pigmentosa, and encodes an orthologue of Drosophila spacemaker. So far, only eight mutations have been reported in arRP families by two groups in 2008. El-Aziz et al identified six mutations, including four homozygous mutations p.D904QfsX17, p.S754AfsX6, p.T657TfsX5, p.W2640X and two compound heterozygous mutations p.E1953X and p.R589RfsX5 [7]. Collin et al. reported a p.Y3156X in two unrelated Dutch families and a p.P2238PfsX16 in an isolated RP patient from Dutch origin [8]. Interestingly, all of these reported mutations, including our finding, resulted in a truncated EYS protein. This suggests that the C-terminus of EYS is essential for its function in the retina, or these EYS mutations (nonsense mutations, deletions) may cause RP by degrading the EYS mRNA through a nonsense-mediated decay mechanism.
RP 25 could be a major locus for arRP because 10-20% of Spanish typical arRP families were found to have mutations in EYS [9, 13]. Consequently, it is interesting to study the relationship between the clinical features of RP and different EYS mutations. Until recently, mutations of EYS have been detected in eight families [7, 8]. Three Chinese families were linked to the RP25 locus, but no mutations were identified in the EYS gene [12]. Similarly, in one Pakistani arRP family, RP was suggested to be caused by an EYS mutation, too [10]. All the patients in these families displayed typical characteristic RP symptoms, including night blindness as the initial symptom, retinal bone-spicule pigmentations, attenuated retinal vessels constriction of visual fields, and absent or flat ERG. However, the age of onset displayed interfamilial differences, ranging from the early second decade to the late forth decade. It is notable that two patients (described by Collin et al.) from two different families with mutation p.Y3156X had combined RP and cataract [8]. One patient II5 in this study was also affected with cataract in both right and left eyes. Further work on deciphering if EYS plays an important role in the cataractogenesis in addition to retinal dystrophy may be useful to understand the function of EYS.
EYS contains 44 exons covering 2.0 Mb, and it is considered to be the largest gene identified and expressed in the human eye as well as the fifth largest overall in the human genome. EYS is highly expressed in the retina, but its biological function remains unknown. In Drosophila, the EYS homological protein spam, and its interactor protein prom, plays a critical role in retina morphogenesis. Drosophila mutant lines for spam and prom can result in a failure of inter-rhabdomeral-space separation [15]. Mutations in human PROM1 were associated with arRP, macular degeneration, and cone-rod dystrophy [16–19]. EYS mutations causing arRP and the data from Drosophila strongly suggest that EYS may have an important function in the photoreceptor morphogenesis. Further investigations of the biochemical function of EYS in retinas will lead to a significant understanding of the pathological mechanism(s) of retina degeneration.
Conclusions
A novel homozygous nonsense mutation in EYS, p.E1836X, was responsible for using arRP in the consanguineous Chinese family. This is the first EYS mutation found in the Chinese population. This research confirms that mutations of EYS can cause arRP. It has also expanded the spectrum of EYS mutations in arRP.
Note Added in Proof
Very recently, three groups of investigators described the identification of EYS novel mutations in patients with retinal dystrophy. Abd El-Aziz et al. [20] identified novel EYS mutations in arRP patients from British and Chinese origins. Bandah-Rozenfeld et al. [21] reported EYS mutations in Israeli and Palestinian families with autosomal recessive retinitis pigmentosa (arRP). Audo I et al. [22] found EYS mutations in French patients with rod-cone dystrophies. These studies supported that EYS is critical for retinal function and confirmed that EYS is a major causative gene for retinal dystrophies in populations of various ethnicities.
References
Hartong DT, Berson EL, Dryja TP: Retinitis pigmentosa. Lancet. 2006, 18; 368 (9549): 1795-809. 10.1016/S0140-6736(06)69740-7.
Boughman JA, Conneally PM, Nance WE: Population genetic studies of retinitis pigmentosa. Am J Hum Genet. 1980, 32 (2): 223-235.
Buch H, Vinding T, La Cour M, Appleyard M, Jensen GB, Nielsen NV: Prevalence and causes of visual impairment and blindness among 9980 Scandinavian adults: the Copenhagen City Eye Study. Ophthalmology. 2004, 111 (1): 53-61. 10.1016/j.ophtha.2003.05.010.
Tous HM, Izquierdo NJ: Retinitis pigmentosa in Puerto Rico. P R Health Sci J. 2006, 25 (4): 315-8.
Wang DY, Chan WM, Tam POS, Baum L, Lam DS, Chong KK, Fan BJ, Pang CP: Gene mutations in retinitis pigmentosa and their clinical implications. Clin Chim Acta. 2005, 351 (1-2): 5-16. 10.1016/j.cccn.2004.08.004.
Bunker CH, Berson EL, Bromley WC, Hayes RP, Roderick TH: Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol. 1984, 97 (3): 357-65.
Abd El-Aziz MM, Barragan I, O'Driscoll CA, Goodstadt L, Prigmore E, Borrego S, Mena M, Pieras JI, El-Ashry MF, Safieh LA, Shah A, Cheetham ME, Carter NP, Chakarova C, Ponting CP, Bhattacharya SS, Antinolo G: EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet. 2008, 40 (11): 1285-7. 10.1038/ng.241.
Collin RW, Littink KW, Klevering BJ, van den Born LI, Koenekoop RK, Zonneveld MN, Blokland EA, Strom TM, Hoyng CB, den Hollander AI, Cremers FP, Khaliq S, Hameed A, Ismail M, Mehdi SQ, Bessant DA, Payne AM, Bhattacharya SS: Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am J Hum Genet. 2008, 83 (5): 594-603. 10.1016/j.ajhg.2008.10.014.
Ruiz A, Borrego S, Marcos I, Antiñolo G: A major locus for autosomal recessive retinitis pigmentosa on 6q, determined by homozygosity mapping of chromosomal regions that contain gamma-aminobutyric acid-receptor clusters. Am J Hum Genet. 1998, 62: 1452-1459. 10.1086/301866.
Khaliq S, Hameed A, Ismail M, et al: Refinement of the locus for autosomal recessive Retinitis pigmentosa (RP25) linked to chromosome 6q in a family of Pakistani origin. Am J Hum Genet. 1999, 65 (2): 571-4. 10.1086/302493.
Barragan I, Marcos I, Borrego S, Antiñolo G: Molecular analysis of RIM1 in autosomal recessive Retinitis pigmentosa. Ophthalmic Res. 2005, 37: 89-93. 10.1159/000084250.
Abd El-Aziz MM, El-Ashry MF, Chan WM, Chong KL, Barragan I, Antiñolo G, Pang CP, Bhattacharya SS: A novel genetic study of Chinese families with autosomal recessive retinitis pigmentosa. Ann Hum Genet. 2007, 71 (Pt 3): 281-94. 10.1111/j.1469-1809.2006.00333.x.
Barragán I, Abd El-Aziz MM, Borrego S, El-Ashry MF, O'Driscoll C, Bhattacharya SS, Antiñolo G: Linkage validation of RP25 Using the 10K genechip array and further refinement of the locus by new linked families. Ann Hum Genet. 2008, 72 (Pt 4): 454-62. 10.1111/j.1469-1809.2008.00448.x.
Liu MG, Ke T, Wang ZX, Yang Q, Chang W, Jiang F, Tang Z, Li H, Ren X, Wang X, Wang T, Li Q, Yang J, Liu J, Wang QK: Identification of a CRYAB mutation associated with autosomal dominant posterior polar cataract in a Chinese family. Invest Ophth Vis Sci. 2006, 47 (8): 3461-3466. 10.1167/iovs.05-1438.
Zelhof AC, Hardy RW, Becker A, Zuker CS: Transforming the architecture of compound eyes. Nature. 2006, 443 (7112): 696-9. 10.1038/nature05128.
Maw MA, Corbeil D, Koch J, Hellwig A, Wilson-Wheeler JC, Bridges RJ, Kumaramanickavel G, John S, Nancarrow D, Röper K, Weigmann A, Huttner WB, Denton MJ: A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum Mol Genet. 2000, 9 (1): 27-34. 10.1093/hmg/9.1.27.
Zhang Q, Zulfiqar F, Xiao X, Riazuddin SA, Ahmad Z, Caruso R, MacDonald I, Sieving P, Riazuddin S, Hejtmancik JF: Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Hum Genet. 2007, 122 (3-4): 293-9. 10.1007/s00439-007-0395-2.
Yang Z, Chen Y, Lillo C, Chien J, Yu Z, Michaelides M, Klein M, Howes KA, Li Y, Kaminoh Y, Chen H, Zhao C, Chen Y, Al-Sheikh YT, Karan G, Corbeil D, Escher P, Kamaya S, Li C, Johnson S, Frederick JM, Zhao Y, Wang C, Cameron DJ, Huttner WB, Schorderet DF, Munier FL, Moore AT, Birch DG, Baehr W, Hunt DM, Williams DS, Zhang K: Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest. 2008, 118 (8): 2908-16.
Pras E, Abu A, Rotenstreich Y, Avni I, Reish O, Morad Y, Reznik-Wolf H, Pras E: Cone-rod dystrophy and a frameshift mutation in the PROM1 gene. Mol Vis. 2009, 15: 1709-16.
Abd El-Aziz MM, O'Driscoll CA, Kaye RS, Barragan I, El-Ashry MF, Borrego S, Antiñolo G, Pang CP, Webster A, Bhattacharya SS: Identification of Novel Mutations in the ortholog of Drosophila eyes shut Gene (EYS) Causing Autosomal Recessive Retinitis Pigmentosa. Invest Ophthalmol Vis Sci. 2010,
Bandah-Rozenfeld D, Littink KW, Ben-Yosef T, Strom TM, Chowers I, Collin RW, den Hollander AI, van den Born I, Zonneveld MN, Merin S, Banin E, Cremers FP, Sharon D: Novel null mutations in the EYS gene are a frequent cause of autosomal recessive retinitis pigmentosa in the Israeli population. Invest Ophthalmol Vis Sci. 2010,
Audo I, Sahel JA, Mohand-Saïd S, Lancelot ME, Antonio A, Moskova-Doumanova V, Nandrot EF, Doumanov J, Barragan I, Antinolo G, Bhattacharya SS, Zeitz C: EYS is a major gene for rod-cone dystrophies in France. Hum Mutat. 2010,
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/11/121/prepub
Acknowledgements
We are grateful to the arRP family for their participation in this study. We appreciate Dr. Stephen R Archacki for his critical reading and revising of the manuscript. This study was supported by grants from Chinese National Natural Science Foundation grant No. 30771199, 30700455, and 30871386.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YH recruited the members of arRP pedigree, performed clinical studies for whole family members, and relationship studies between genotype and phenotype. JZ carried out the extraction of genomic DNA and genotyping assay. CL was responsible for the extraction of genomic DNA and sequencing for mutation detection. GY was involved in linkage analysis and the discussion of the manuscript. ML participated in the original conception of this study, and contributed to the revision of the draft. QKW conceived and designed the experiments and helped the critical revision of the manuscript. ZT designed the experiments, analyzed the data, interpreted the results, and drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Huang, Y., Zhang, J., Li, C. et al. Identification of a novel homozygous nonsense mutation in EYS in a Chinese family with autosomal recessive retinitis pigmentosa. BMC Med Genet 11, 121 (2010). https://doi.org/10.1186/1471-2350-11-121
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-11-121