
Abstract
Background
Individual plants adapt to their immediate environment using a combination of biochemical, morphological and life cycle strategies. Because woody plants are long-lived perennials, they cannot rely on annual life cycle strategies alone to survive abiotic stresses. In this study we used suppression subtractive hybridization to identify genes both up- and down-regulated in roots during water deficit treatment and recovery. In addition we followed the expression of select genes in the roots, leaves, bark and xylem of ‘Royal Gala’ apple subjected to a simulated drought and subsequent recovery.
Results
In agreement with studies from both herbaceous and woody plants, a number of common drought-responsive genes were identified, as well as a few not previously reported. Three genes were selected for more in depth analysis: a high affinity nitrate transporter (MdNRT2.4), a mitochondrial outer membrane translocase (MdTOM7.1), and a gene encoding an NPR1 homolog (MpNPR1-2). Quantitative expression of these genes in apple roots, bark and leaves was consistent with their roles in nutrition and defense.
Conclusions
Additional genes from apple roots responding to drought were identified using suppression subtraction hybridization compared to a previous EST analysis from the same organ. Genes up- and down-regulated during drought recovery in roots were also identified. Elevated levels of a high affinity nitrate transporter were found in roots suggesting that nitrogen uptake shifted from low affinity transport due to the predicted reduction in nitrate concentration in drought-treated roots. Suppression of a NPR1 gene in leaves of drought-treated apple trees may explain in part the increased disease susceptibility of trees subjected to dehydrative conditions.
Similar content being viewed by others


Background
Water is considered to be the most limiting environmental factor with regard to plant growth and maintenance. Furthermore, dehydration is a common component of other abiotic stresses, such as freezing, high temperatures, and salt stress. Loss of water not only affects plants in the short term, but can also weaken them, making them more susceptible to biotic and other abiotic stresses in the long term [1, 2]. Plants have developed a variety of adaptations for ameliorating dehydrative stress. In one strategy, known as drought escape, the plant completes its lifecycle before arrival of the drier summer months. Another strategy, drought tolerance, results in the production of osmoprotectants, i.e. compounds that aid in preventing water loss from the cells or act to relieve the negative impact of the dehydrative stress on cellular components. A third mechanism, drought avoidance, allows plants to circumvent drought periods by morphological changes that allow plants to maintain high water status, for example, by encouraging deeper root penetration in the soil.
Another parameter related to plant water status is WUE which is a function of carbon utilization through photosynthesis and water loss through transpiration. In herbaceous plants there is often a tradeoff between suppression of photosynthesis and prevention of water loss, since WUE is closely tied to stomata function. For some annuals and perennials, increased WUE results in a measure of drought tolerance at the expense of growth and development [3, 4]. On the other hand, some annuals maximize photosynthesis through increased stomatal conductance (defined as the rate of gas exchange through the stomata), thus lowering WUE. Although on the surface this seems counterintuitive, this strategy works because the plants ‘outgrow’ (complete their development) before the onset of seasonal droughts [5].
Regardless of which strategy(ies) a plant uses to survive, adaptation to drought requires complex interactions between anatomy, physiology and biochemistry, all of which are directly or indirectly under genetic control [6–8]. Studies examining genes in herbaceous plants that respond to dehydration have identified a number of common genes potentially related to drought resistance [reviewed in [9]. For example, in Arabidopsis several LEA genes (Xero2, rd22, rab18), metallothionein genes and a ripening-related protein gene have all been shown to respond to drought, as well as to other abiotic stresses [10, 11]. These same genes in other plants (e.g., barley, chick pea, and rice) also show a strong response in transcript level when exposed to drought [12–14].
In woody plants similar studies have identified genes that appear to be significant for drought tolerance [15]. Transcriptional profiling was used to assess gene expression in poplar (Populus euphratica) subjected to a gradual drought treatment [16]. Since these studies were long term in contrast to studies with herbaceous plants, the percentage of genes expressed in response to drought was substantially lower. The expression of these genes may represent gradual adaptation, leading to acclimation to long term, moderate water deficit. In maritime pine (Pinus pinaster Ait.), Dubos and Plomion [17] identified 33 cDNA-AFLP fragments responding to polyethylene drought simulation, most of which were genes of unknown function. Genes which were down-regulated in roots included histone H2B, caffeic acid ortho-methyltransferase and a LEA protein, all of which have been shown to be up-regulated in other systems [11, 14, 18].
Suppression Subtractive Hybridization (SSH) has been used successfully to identify differentially regulated genes in a number of plant and animal systems [19, 20]. Although different methodologies for assessing global gene expression have various strengths and weaknesses, SSH is known for its ability to identify low-abundance transcripts. In the current study we applied this method to identify genes up- and down-regulated in response to a simulated drought and at the end of a one week recovery period. Some of the genes have been previously described in other plants, but several genes crucial to metabolism or defense were unique to this study.
Results and discussion
Genes responding to simulated drought
Genes whose mRNAs respond to drought have been identified and verified in a number of plant systems. We used SSH to identify genes in apple roots that were either up- or down-regulated by a simulated severe drought lasting two weeks (Figure 1). In addition, we identified genes whose expression changed after a week of water deficit recovery. Using bioinformatic tools we were able to design gene-specific primers for select genes from each treatment library to determine whether members of multigene families were identical or different in treatments where they appeared in more than one library (not shown). In this paper we focus on the results of our analysis of roots during water deficit and recovery (genes listed in Additional file 1).

Diagram of water deficit experiment conducted with ‘Royal Gala’. A total of twenty-five trees were selected for the experiment. Five trees were sampled (leaves, bark and roots) at the beginning of the experiment (T = 0), after one week acclimation in the growth chamber. The controls, water deficit and recovery treatments are indicated in the boxes. Black arrows indicate how the experiment was conducted in time. Subtractions are indicated next to the red arrows showing the direction of subtraction, forward or reverse. For example, the forward subtraction between T1E and T1C (AAF) involves T1E cDNA as the tester and ten times concentrated T1C cDNA as the driver. Genes isolated from this subtraction represent those whose levels are upregulated in response to two weeks of water deficit treatment. In the reverse subtraction (AAR), T1C is the tester and T1E (ten times concentrated) is the driver. The reverse subtraction identifies genes downregulated in response to water deficit. The two controls (T1C and T2C) were subtracted to account for differences in gene abundance as a consequence of age (three weeks vs four weeks). This subtraction resulted in only a few sequences.
Tables 1 and 2 contain a list of genes that were up- (AAF library) or down-regulated (AAR library) in response to water deficit treatment compared to well-watered controls run in parallel. Twice as many genes were identified in the library representing genes whose expression was elevated in response to drought compared to those that were down-regulated. Tables 3 and 4 list genes up-regulated after recovery from the drought treatment (BBR library) or elevated during drought treatment (BBF library) relative to recovery. Nearly three times as many genes were identified in the BBF library as in the BBR library. Because samples were taken after two weeks of drought, very early drought-responsive genes, including many transcription factors and signaling components, would not be expected to be identified in our libraries. One exception is the drought-responsive leucine zipper homeobox gene whose transcripts increased in drought-treated roots (manuscript in preparation). Since this gene is a close relative of the Arabiodpsis AtHb7 and AtHb12 genes which are also drought-induced [21], its elevated presence two weeks after the beginning of the drought period may be indicative of a role in maintenance of the drought response.
In three of the SSH libraries copper-binding proteins were identified. The H01BBF sequence (up-regulated in drought) matched (E value: 9e-33) a copper chaperone from Fragaria vesca (ATOX1-like; XP_00408552). Three putative copper binding proteins were also identified in the BBR (down-regulated) subtraction: H11BBR and Contig2BBR aligned with an early nodulin16 precursor (E values: 1e-15 and 4e-17, respectively) from Ricinus communis (XP_002527193), and H04BBR aligned with a predicted copper binding protein from Prunus persica (E value: 1e-18; EMJ04613). In addition, clone D07AAR (down-regulated) aligned with another copper binding protein, mavicyanin, from Ricinus communis (EEF36698). With the exception of H01BBF, all of these genes appear to be down-regulated in apple roots in response to drought. When cellular copper is limiting, copper chaperones are generally required [22, 23]. If Cu+2 uptake is reduced by drought treatment, the up-regulation of H01BBF in apple roots would be consistent with previous studies and may reflect its function as a member of the ATOX copper chaperone family, namely the intracellular delivery of copper to the secretory pathway [23].
Three plasma membrane-intrinsic protein (PIP or aquaporin) ESTs were found in roots (Tables 1, 2 and 3). When the derived amino acids were analyzed with BLASTp against the Arabidopsis genome, the two ESTs from drought up-regulated libraries (Contig4AAF and E10BBF) were found to be most closely matched with Arabidopsis PIP2-7 and PIP2-5, respectively (E values: 2e-69 and 1e-103). The AAR library EST (Contig 9) was more closely related to Arabidopsis PIP2-4 (E value: 2e-27). Comparison of the ESTs with each other indicated that the AAR and AAF sequences both aligned with the BBF sequence, but with no overlap between them. In addition, most of the amino acid differences between AAR and BBF were non-conserved substitutions, as opposed to the conserved differences seen between the AAF and BBF sequences. Taken together the results suggest that these genes in fact represent different family members.
Since abiotic stress affects a number of critical plant processes, the identification of genes representing a variety of cellular functions in response to drought is to be expected. In combining sequences up-regulated in drought, we included genes from both the AAF and BBF libraries. In both cases the total number of up-regulated genes in response to drought exceeded the number of down-regulated genes by nearly two-fold. Many of the identified genes have been previously reported in other plant systems subjected to various types of drought stress [7, 21]. For example, metallothionein and related genes are elevated under water deficit conditions in rice, chick pea, and Arabidopsis[11, 13, 14]; likewise, PIPs and mal d1 have also been commonly associated with dehydration responses.
Analysis of specific genes
In order to confirm treatment differences correlated with SSH and affirm the integration of genes responding to water deficit, we conducted an in-depth analysis of three genes whose role under drought conditions has not been well characterized at the molecular level. These genes included ESTs encoding a high affinity nitrogen transporter (MdHAT2.4), an outermembrane mitochondrial import receptor subunit (MdTom7.1) and a gene (MpNPR1-2) associated with regulons involved in both systemic acquired and basal resistance to biotic stress.
High affinity nitrate transporters
E05BBF is an EST isolated from the root BBF library (up-regulated in drought relative to recovery) and was identified as a high affinity nitrate transporter. In Arabidopsis the high affinity nitrate transporters are represented by seven genes: NRT2.1-2.7. AtNRT2.1 and AtNRT2.2 are the primary genes responsible for transport of nitrate under low nitrogen availability and appear to be inducible, since a mutant lacking both genes fails to achieve nitrate transport levels similar to the wild-type [24]. However, a very high-affinity component is still active in this mutant, and it is thought that this activity corresponds to AtNRT2.4, the most inducible gene under limiting nitrate conditions.
The full length apple gene (MDP0000239537) represented by the E05BBF EST is on chromosome 11. There are three other full length apple genes closely related to E05BBF (Additional file 2). Of these, MDP0000266497 is most like E05BBF and appears to be related to AtNRT2.5 (73% amino acid identity). The remaining two genes are most closely related to AtNRT2.7. MDP0000266497 is located on chromosome 13, whereas MDP0000131368 and MDP0000201530 are located on chromosomes 9 and 17, respectively. All of these predicted proteins are members of the major facilitator superfamily (transport of small solutes across membranes) and possess a nitrogen transporter domain as well [25].
MDP0000239537 (E05BBF; MdNRT2.4) has significant homology to both AtNRT2.1 and AtNRT2.4 (Table 5). In a recent study of Malus hupenensis (Pamp.) Rehder [26], a full length nitrate transporter gene (designated MhNRT2.1) was found to share 98.9% homology with MDP0000239537, whereas a second gene (designated MhNRT2.5) shared 98.6% amino acid identity with MDP0000266497.
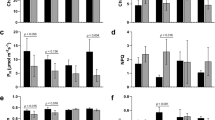
To obtain accurate estimates of the differences in abundance between the drought treatment and recovery, RT-qPCR was performed on all the treatments from roots, as well as samples taken from leaves, bark and xylem subjected to the same conditions (Figure 2). Expression of MdNRT2.4 in leaves was not significantly different in drought-treatment vs. controls (Figure 2C), although the additional week of recovery resulted in an overall decline of transcript abundance for reasons not completely clear. Orsel et al. [25] reported that AtNRT2.4 was substantially inducible in low concentrations of KNO3, whereas Okamoto et al. [27] observed repression of AtNRT2.4 levels in both roots and shoots exposed to 0.5 mM Ca(NO3)2 after nitrogen deprivation. In our study the levels of the MdNRT2.4 transcript from plants under water deficit were 212% of the control in roots and 167% of control in bark; in both organs, recovery resulted in a return to control levels (96% and 115% of controls, respectively). Since nitrogen uptake is by mass flow of water from the soil to the root [28, 29], lower nitrogen levels would be expected in roots of plants under water deficit. In wheat nitrogen-use efficiency was increased by water deficit and diminished in response to increasing concentrations of applied nitrogen [30]. Our observations regarding MdNRT2.4 expression are consistent with these reports and may provide a novel avenue for exploring drought resistance, since links between nitrogen deficiency, water deficit response and ABA/stomatal function have been previously established [31].

Quantitative analysis of MdNRT2.4 expression during water deficit and recovery. Expression was assessed by quantitative RT-PCR as described in Methods. Concentrations were determined by normalization with TEF2, and the results from two independent experiments are shown. Standard error bars are indicated on the columns. A: Root; B: Bark and Xylem; C: Leaf. Con1 and Con2: well watered control plants for water deficit treatment and recovery, respectively, Drt: water deficit treatment and Rec: recovery. Light gray columns = 2005 experiment; dark gray and black (xylem) columns = 2008 experiment.
Analysis of approximately 700 bases upstream of the translation start codon of MdNRT2.4 identified several cis-elements related to stress or hormone response (Additional file 3). Two TATA boxes representing RNA PolII binding sites were found, one approximately 100 bases from the translation start site. There were no consensus G-box elements in either promoter [32, 33]. Elements similar to G-box abscisic acid response elements (ABREs) [34] were present in both the Arabidopsis and apple NRT2.4 promoters. However, NRT2.4 is not likely to respond to drought via ABA induction because the core sequence in both G-box elements does not end in cytosine (C+4) which is essential for ABA induction [35]. On the other hand, a consensus C-repeat binding element linked to both cold and drought response was within a functional distance of the first TATA box in the apple NRT2.4 promoter. The second TATA box is further upstream and is linked to several MYC binding sites, as well as a wound-inducible element [36] and an element associated with hypoosmolarity responsiveness [37]. Both TATA elements could be functional under different regulatory regimes. Mapping transcripts originating from this promoter would determine whether or not both TATA elements are functional. Many of the elements identified in the MdNRT2.4 promoter were absent in the Arabidopsis promoter, and there were considerably fewer MYB binding sites (CANNTG) in the AtNRT2.4 promoter (data not shown).
Mitochondrial import translocase subunit (TOM)
The mitochondrial import complex (TOM for Translocase of the Outer Membrane and TIM for Translocase of the Inner Membrane) is extensively conserved in eukaryotes and typically contains 13 TIM and seven TOM subunits [38]. The TOM subunits consist of two receptors (TOM70 and 40) that interact with the pre-protein and five subunits that compose the translocation channel (TOMs 22, 20, 7, 6, and 5). Only three outer membrane proteins are ubiquitous among eukaryotes: TOM40, TOM22 and TOM7 [39].
An EST (C09AAF) was identified in the drought-treated root subtraction encoding a homolog to the TOM7 subunit. RT-PCR results indicated that C09AAF levels were lower in the drought-treated plants than in the controls. This is not consistent with this sequence having been obtained from the AAF subtraction representing up-regulated sequences. Close inspection of the PCR reaction products indicated marked similarity in band intensity between the treatments and also revealed an unpredicted, higher molecular weight band that was amplified, suggesting that a close relative might be interfering with primer hybridization (data not shown).
BLASTn alignment of C09AAF against the apple genome found three closely related genes on different chromosomes, one of which (MDP0000023053) corresponded perfectly with the coding sequence of C09AAF. The three apple TOM7 genes are found on chromosomes 12, 13 and 16. MDP000023053 (designated as MdTOM7.1) is located on chromosome 13. Several other significant BLASTn hits were also noted, but these sequences maybe pseudo-genes, as the ATG codons are altered. Conservation between the apple derived protein sequences and two Arabidopsis TOM polypeptides is confined mainly to the TOM7 domain characteristic of the superfamily (Additional file 4).
RT-qPCR primers were designed to eliminate any possible contribution from the other two related genes in the RNA populations. Testing of this primer set indicated that only one product was obtained (not shown). This primer set was then used for RT-qPCR analysis and the results are shown in Figure 3. In bark, MdTOM7.1 was moderately elevated under drought conditions compared to its control. On the other hand, MdTOM7.1 in roots and leaves was not appreciably different from the well watered controls and did not decline to control levels during the recovery phase as observed in bark. This is supported by analysis of the MdTOM7 promoters where no ABRE or DRE sequences were found in the first 800 bases upstream of the ATG codon (Additional file 5). In contrast both elements were present just upstream of the TATA box in MdTOM7.2 (MDP0000694615). In apple leaves expression of MdTOM7.1 was similar to the expression of At5g41685 [TOM7-1] and At1g64220 [TOM7-2] (Gene Expression Omnibus, NCBI) in Arabidopsis whole plants treated with salt or exposed to 0 °C, showing little or no change compared to controls. However, both Arabidopsis genes show greater accumulation in the polysomal fraction of plants exposed to dehydration treatment, relative to control polysomes or the non-polysomal fraction of controls and treated plants [40], indicating that regulation of TOM7 may not be solely transcriptional. Based on these observations and the presence of stress-responsive elements in the promoter of MdTOM7.2, it would be of interest to examine expression of this gene in the same RNA populations from the organs of drought-treated apple to determine if it indeed responds to water deficit treatment.

Quantitative analysis of MdTOM7-1 expression during water deficit and recovery. Expression was assessed by quantitative RT-PCR as described in Methods. Concentrations were determined by normalization with TEF2, and the results from two independent experiments were averaged. Standard error bars are indicated on the columns. A: Root; B: Bark and Xylem; C: Leaf. Con1 and Con2: well watered control plants for water deficit treatment and recovery, respectively, Drt: water deficit treatment and Rec: recovery. Light gray columns = 2005 experiment; dark gray and black (xylem) columns = 2008 experiment.
TOM7 binds to TOM40 in the outer mitochondrial membrane where it is thought to modulate pore formation and might be expected to play a role during drought stress. The fact that drought did not appear to alter MdTOM7.1 transcript levels in apple or Arabidopsis suggests that regulation at another level may be more important to plant TOM7.1 or that drought treatment alters another component of the mitochondrial import complex (possibly MdTOM7.2) to allow continual mitochondrial functioning in plants under stress.
Nonexpressor of pathogenesis related genes (MpNPR1-2)
An EST (D05AAF) encoding MpNPR1-2[41] was isolated from the library containing genes up-regulated in response to drought (Table 1). Quantitative assessment of MpNPR1-2 expression in roots, bark and leaves of drought-treated apples indicated that its mRNA was elevated nearly four times in drought-treated roots over control roots in the 2008 experiment, although in an earlier experiment there was essentially no difference (Figure 4A). A smaller increase was noted in drought treated bark and xylem (Figure 4B), but surprisingly drought treatment lowered its expression in leaves from both experiments (Figure 4C).

Quantitative analysis of MpNPR1-2 expression during water deficit and recovery. Expression was assessed by quantitative RT-PCR as described in Methods. Concentrations were determined by normalization with TEF2, and the results from two independent experiments were averaged. Standard error bars are indicated on the columns. A: Root; B: Bark and Xylem; C: Leaf. Con1 and Con2: well watered control plants for water deficit treatment and recovery, respectively, Drt: water deficit treatment and Rec: recovery. Light gray columns = 2005 experiment; dark gray and black (xylem) columns = 2008 experiment.
A previous study of NPR expression in apple identified three NPR1 genes [41]. but only MpNPR1-1 was induced with BTH (Benzo(1,2,3)thiadiazole-7-carbothioic acid) treatment to induce systemic acquired resistance (SAR), suggesting that this may be the ortholog to the Arabidopsis gene, AtNPR1. AtNPR1 encodes a protein with ankyrin repeats that binds to transcription factors of the TGA subfamily of basic leucine zipper proteins [42, 43]. Traditionally, NPR1 has been associated with salicylic acid (SA)-linked systemic acquired resistance affecting a broad spectrum of pathogens, including fungi, bacteria and viruses. A recently described role for cytoplasmic NPR1 in jasmonic acid (JA) suppression has been reported [44], as well as an association with basal defense response [42, 45]. Expression of AtNPR1 is generally constitutive showing only a modest (two time) induction by SA [46].
Cross-talk between signaling pathways has been known for some time. For example, drought-stressed plants are generally more susceptible to pathogen attack than unstressed plants [47, 48]. A study in rice revealed a connection that may be related to the JA suppression role of NPR1 [49]. In this study, constitutive expression of the Arabidopsis AtNPR1 gene in rice leaves conferred resistance to several fungal pathogens and one bacterial pathogen by ‘priming’ the SAR pathway. Interestingly, the same transgenic lines resistant to the pathogens were more sensitive to drought and salt treatments. These results correlated with reduced expression (both in abundance and timing) of some key genes associated with abiotic stress, e.g. Rab21. Our results appear to corroborate suppression of NPR1-2 in leaves under drought stress with studies indicating increased susceptibility of stressed plants to different pathogens. Most interesting is the observation that MpNPR1-2 is significantly elevated in roots at the end of a two-week, continuous water deficit treatment. These results suggest a role for MpNPR1-2 in root-specific protection against soil-borne pathogens during drought stress.
Conclusion
We have identified apple root genes that respond to a two week, moderately severe simulated drought and to a one week period of recovery. Most of the genes identified have been previously reported to be drought- or stress-responsive in other plants. Three genes not previously associated with root response to drought were further characterized. Two genes, MdNRT2.4 and MpNPR1-2 were shown by quantitative RT-PCR analysis to be up-regulated in apple roots subjected to drought. The third gene, MdTOM7.1 was not appreciably expressed in response to drought treatment.
The results from two independent experiments demonstrate that drought treatment increases expression of a high-affinity nitrate transporter (MdNRT2.4) which is consistent with previous research associating drought with nitrogen deficiency. Our results from the 2008 experiment also suggest that MpNPR1-2 may have a defense role in roots, since its up-regulation in this organ in response to abiotic stress has not been previously reported. Finally, a reproducible decrease in MpNPR1 in leaves in association with drought treatment may explain why many plants under abiotic stress are reported to be more susceptible to pathogen attack. Further analysis of these gene families may identify altered functions or expression for individual family members arising as a requirement for adaptation to the varying environmental conditions that most perennial plants face.
Methods
Plant material
Apple plants of a single genotype (Malus × domestica cv. Royal Gala) were initially propagated by asexual in vitro shoot proliferation (clonal replication) culture at the USDA-ARS-NAA-AFRS facility (Kearneysville, WV) as per Norelli et al. [50] and Ko et al. [51], with root induction as per Bolar et al. [52]. After rooting and establishment, the young trees were potted in standard ten-inch nursery pots containing Metromix 310 (composition: horticultural vermiculite, Canadian Sphagnum peat moss, processed bark ash, composted pine bark, and washed sand [Scotts – Sierra Horticultural Products Co., Marysville, OH]) and transferred to a glasshouse. The trees were grown under supplemental lighting to maintain day length at 16 h, and a temperature range of 20–35°C. Trees were watered daily, with weekly application of nutrient solution (MiracleGro) and with supplemental application of Osmocote (Scott’s Miracle-Gro Products; 19-6-12 N-P-K) every two months at the indicated rate of 10 g/pot. Trees were in the glasshouse for a total of 8 months, with final caliper measurements ranging from 0.5 cm to slightly more than 1.0 cm, and heights varying from 1 to 2 m.
Twenty-five trees were placed in a Conviron PGV36 growth chamber (Conviron) at 25°C day (16 h)/18°C night (8 h) with light at 500 μmol photons m−2 s−1 PPFD and acclimated for one week, after which twenty trees were selected for experimentation. Water deficit was imposed essentially as described by Artlip and Wisniewski [53]. The experiment is diagrammed in Figure 1. Water was withheld from ten trees until the pot masses were at 45% of the saturated mass and maintained at this level for two weeks without added fertilizer. Ten control trees were maintained at the saturated mass by daily watering without fertilizer. After two weeks of water limitation, roots, bark and leaves were harvested from five water deficit-treated trees and five well watered controls. The remaining five of the water deficit-treated trees were watered to saturation for one week, along with the five remaining controls. No fertilizer was applied during the experiment. The samples from each individual organ and treatment were pooled, immediately frozen in liquid N2 and stored at -80°C. Harvested leaves were taken from leaf positions 7 through 12 (counting down from the youngest visible leaf at the apex), and typically measured from 6.5 to 9.0 cm in length. These leaves are considered to be at or near full expansion. To avoid complication due to circadian rhythm effects on expression, samples were taken at the same time of day approximately 8 h after ‘lights on’ in the growth chamber.
Suppression subtractive hybridization
Total RNA was isolated from each organ by a method reported by Artlip et al. [54]. The RNA was treated with DNase according to the manufacturer’s (InVitrogen) recommendation and tested for DNA contamination by PCR prior to use in downstream applications. Suppression subtractive hybridization was performed as described previously [20] using the Super SMART method (Clontech, Palo Alto, Calif.) for cDNA synthesis and following the manufacturer’s protocol for subtraction hybridization (Clontech).
PCR analyses
Leaves, roots and bark subjected to the two week drought treatment and one week recovery were collected from two independent experiments conducted in 2005 and 2008. Total RNA was extracted, DNased and quality-assessed on agarose gels. cDNA was synthesized with the Advantage RT kit following the manufacturer’s directions (Clontech).
Primers for RT-qPCR (Additional file 6) were designed with Primer 3 Plus software [55] and tested against genomic DNA for quality assurance. Each primer pair was used to prime RT-qPCR reactions in order to quantify gene expression in different organs. The qPCR reactions were conducted using a kit containing all the reagents (Life Technologies, Applied Biosystems, Grand Island, NY), and the reaction parameters were as follows: 95°C 5 min, followed by 35 cycles of 95°C 1 min, 60-65°C 1 min, 72°C 1 min and a final extension of 72°C for 10 min. Primers for a translation elongation factor (TEF2) were used as an internal control for the qPCR experiments [56]. The relative standard curve method was used to analyze the data.
Bioinformatic analyses
Sequence data and GenBank accession numbers are included in Additional file 1. Remaining vector sequences were manually identified and checked with VecScreen at the National Center for Biotechnology Information (NCBI). Each library was analyzed with the Cap3 Assembly program (Iowa State University) to obtain a unigene file for each forward and reverse subtraction. The unigene files were analyzed with BLASTx, BLASTn and/or tBLASTx (NCBI) to identify specific sequences. Alignments were performed with Cobalt (NCBI) or ClustalW using the BLOSUM matrix. Apple promoter sequences were identified at Genome Database for Rosaceae (http://www.rosaceae.org/) from the whole genome sequence of ‘Golden Delicious’. Promoters were defined as the first one thousand bases upstream of the translation start codon. Cis-elements were identified with PLACE [57], PlantPAN [58] and PlantCare [59, 60].
Availability of supporting data
GeneBank accession numbers are included in Additional file 1.
Authors’ information
Jacob T. Moore and Ryan M. Jenkins are students at Pennsylvania State University; Doug S. Soffe was a student intern from Hagerstown Community College.
Abbreviations
- AAF and AAR:
-
Sequences up- (AAF) or down- (AAR) regulated after two weeks of simulated drought
- ABRE:
-
ABA response element
- BBF and BBR:
-
Sequences up- (BBR) and down- (BBF) regulated after a week of recovery from simulated drought
- CCF and CCR:
-
Sequences up- (CCF) and down- (CCR) regulated between treatment weeks 2 and 3 in trees not exposed to drought
- DRE:
-
Drought response element
- EST:
-
Expressed sequence tag
- LEA:
-
Late embryogenesis abundant
- MDP#:
-
M. × domestica predicted coding region (see http://www.rosaceae.org)
- RT-qPCR:
-
Quantitative reverse transcription-polymerase chain reaction
- SSH:
-
Suppression subtractive hybridization
- WUE:
-
Water use efficiency.
References
Desprez-Loustau M-L, Marçais B, Nageleisen L-M, Piou D, Vannini A: Interactive effects of drought and pathogens in forest trees. Ann For Sci. 2006, 63: 597-612.
Bréda N, Huc R, Granier A, Dreyer E: Temperate forest trees and stands under severe drought: a review of exophysiological responses, adaptation processes and long-term consequences. Ann For Sci. 2006, 63: 625-644.
Querejeta JI, Barea JM, Allen MF, Caravaca F, Roldan A: Differential response of delta C-13 and water-use efficiency to arbuscular mycorrhizal infection in two aridland woody plant species. Oecologia. 2003, 135: 510-515.
Pimentel C, Laffray D, Louguet P: Intrinsic water-use efficiency at the pollination stage as a parameter for drought tolerance selection in Phaseolus vulgaris. Physiol Plant. 1999, 106: 184-189.
McKay JK, Richards JH, Mitchell-Olds T: Genetics of drought adaptation in Arabidopsis thaliana. I. Pleiotropy contributes to genetic correlations among ecological traits. Mol Ecol. 2003, 12: 1137-1151.
Bohnert HJ, Nelson DE, Jensen RG: Adaptations to environmental stresses. Plant Cell. 1995, 7: 1099-1111.
Ingram J, Bartels D: The molecular basis of dehydration tolerance in plants. Annu Rev Plant Physiol Plant Mol Biol. 1996, 47: 377-403.
Street NR, Skogstrom O, Sjodin A, Tucker J, Rodriguez-Acosta M, Nilsson P, Jansson S, Taylor G: The genetics and genomics of the drought response in Populus. Plant J. 2006, 48: 321-341.
Bray EA: Classification of genes differentially expressed during water-deficit stress in Arabidopsis thaliana: an analysis using microarray and differential expression data. Ann Bot. 2002, 89: 03-811.
Reymond P, Weber H, Damond M, Farmer EE: Differential gene expression in response to mechanical wounding and insect feed in Arabidopsis. Plant Cell. 2000, 12: 707-719.
Seki M, Narusaka M, Abe H, Kasuga M, Yamaguchi-Shinozaki K, Carninci P, Hayashizaki Y, Shinozaki K: Monitoring the expression pattern of 1300 Arabidopsis genes under drought and cold stresses by using a full-length cDNA microarray. Plant Cell. 2001, 13: 61-72.
Ozturk AN, Talamé V, Deyholos M, Michalowski CB, Galbraith DW, Gozukimizi N, Tuberosa R, Bohnert HJ: Monitoring large-scale changes in transcript abundance in drought- and salt-stressed barley. Plant Mol Biol. 2002, 48: 551-573.
Boominathan P, Shukla R, Kumar A, Manna D, Negi D, Verma PK, Chattopadhyay D: Long term transcript accumulation during the development of dehydration adaptation in Cicer arietinum. Plant Physiol. 2004, 135: 1608-1620.
Gorantla M, Babu PR, Reddy Lachagari VB, Feltus FA, Paterson AH, Reddy AR: Functional genomics of drought stress response in rice: Transcript mapping of annotated unigenes of an indica rice (Oryza sativa L. cv. Nagina 22). Curr Sci. 2005, 89: 496-514.
Hamanishi ET, Campbell MM: Genome-wide responses to drought in forest trees. Forestry. 2011, 84: 273-283.
Bogeat-Triboulot M-B, Brosché M, Renaut J, Jouve L, Le Thiec D, Fayyaz P, Vinocur B, Witters E, Laukens K, Teichmann T, Altman A, Hausman J-F, Polle A, Kangasjarvi J, Dreyer E: Plant Gradual soil water depletion results in reversible changes of gene expression, protein profiles, ecophysiology, and growth performance in Populus euphratica, a poplar growing in arid regions. Physiol. 2007, 143: 876-892.
Dubose C, Plomion C: Identification of water-deficit responsive genes in maritime pine (Pinus pinaster Ait.) roots. Plant Mol Biol. 2003, 51: 249-262.
Pandey A, Chakraborty S, Datta A, Chakraborty N: Proteomics approach to identify dehydration responsive nuclear proteins from chickpea (Cicer arietinum L.). Mol Cell Proteomics. 2008, 7: 88-107.
Perdiguero P, Collada C, Del Carmen Barbero M, Casado GG, Cervera MT, Soto A: Identification of water stress genes in Pinus pinaster Ait. by controlled progressive stress and suppression-subtractive hybridization. Plant Physiol Biochem. 2012, 50: 44-53.
Bassett CL, Wisniewski ME, Artlip TS, Norelli JL, Renaut J, Farrell RE: Global analysis of genes regulated by low temperature and photoperiod in peach bark. J Amer Soc Hort Sci. 2006, 131: 551-563.
Soderman E, Mattsson J, Engström P: The Arabidopsis homeobox gene ATHB-7 is induced by water deficit and by abscisic acid. Plant J. 1996, 10: 375-381.
Lin SJ, Pufahl R, Dancis A, O’Halloran TV, Culotta VC: A role for the Saccharomyces cerevisiae ATX1 gene in copper trafficking and iron transport. J Biol Chem. 1997, 272: 9215-9220.
Portnoy ME, Rosenzweig AC, Rae T, Huffman DL, O’Halloran TV, Culotta VC: Structure-function analyses of the ATX1 metallochaperone. J Biol Chem. 1999, 274: 15041-15045.
Cerezo M, Tillard P, Gilleur S, Monos S, Daniel-Vedele F, Gojon A: Major alterations of the regulation of root NO3− uptake are associated with the mutation of Nrt2.1 and Nrt2.2 genes in Arabidopsis. Plant Physiol. 2001, 127: 262-271.
Orsel M, Krapp A, Daniel-Vedele F: Analysis of the NRT2 nitrate transporter family in Arabidopsis. Structure and gene expression. Plant Physiol. 2002, 129: 886-896.
Wang XL, Peng FT, Yu W, Li MJ, Wang ZT: Molecular cloning and expression analysis of three cDNAs encoding nitrate transporters in Malus hupenensis Rehd. J Hort Sci Biotech. 2013, 88: 447-456.
Okamoto M, Vidmar JJ, Glass AD: Regulation of NRT1 and NRT2 gene families of Arabidopsis thaliana: responses to nitrate provision. Plant Cell Physiol. 2003, 44: 304-317.
Barber SA, Walker JM, Vaset EH: Mechanisms for the movement of plant nutrients from the soil and fertilizer to the plant root. Crop Food Agriculture. 1963, 11: 204-207.
Gonzalez-Dugo V, Durand J-L, Gastal F: Water deficit and nitrogen nutrition of crops. A review. Agron Sustain Dev. 2010, 30: 529-544.
Fan XL, Li YK: Effect of drought stress and drought tolerance heredity on nitrogen efficiency of winter wheat. Plant Nutrition: Food Security and Sustainability of Agro-Ecosystems. Edited by: Horst WWJ, Schenk MK, Butkert A, Claasen N, Flessa H, Frommer WB, Goldbach HE, Olfs HW, Romheld W, Sattelmacher B, Schmidhalter U, Schubrrt S, Von Wiren H, Wittenmayer L. Madison, USA: American Society of Agronomy, 2001:62-63.
Radin JW, Ackerson RC: Water relations of cotton plants under nitrogen deficiency. III. Stomatal conductance, photosynthesis and abscisic acid accumulation during drought. Plant Physiol. 1981, 67: 115-119.
Williams ME, Foster R, Chua N-H: Sequences flanking the hexameric G-Box Core CACGTG affect the specificity of protein binding. Plant Cell. 1992, 4: 485-496.
Giuliano G, Pichersky E, Malik VS, Timko MP, Scolnik PA, Cashmore AR: An evolutionary conserved protein binding sequence upstream of a plant light-regulated gene. Proc Natl Acad Sci U S A. 1988, 85: 7089-7093.
Shen Q, Ho THD: Functional dissection of an abscisic acid (ABA)-inducible gene reveals two independent ABA-responsive complexes each containing a G-box and novel cis-action element. Plant Cell. 1995, 7: 295-307. Additional Files
Hattori T, Totsuka M, Hobo T, Kagaya Y, Yamamoto-Toyoda A: Experimentally determined sequence requirement of ACGT-containing abscisic acid response element. Plant Cell Physiol. 2002, 43: 136-140.
Pastuglia M, Roby D, Dumas C, Cock JM: Rapid induction by wounding and bacterial infection of an S gene family receptor-like kinase in Brassica oleracea. Plant Cell. 1997, 9: 1-13. Additional Files
Satoh R, Nakashima K, Seki M, Shinozaki K, Yamaguchi Shinozaki K: ACTCAT, a novel cis-acting element for proline- and hypoosmolarity-responsive expression of the ProDH gene encoding proline dehydrogenase in Arabidopsis. Plant Physiol. 2002, 130: 709-719. Additional Files
Dolezal P, Likic V, Tachezy J, Lithgow T: Evolution of the molecular machines for protein import into mitochondria. Science. 2006, 313: 314-318.
Maćasev D, Whelan J, Newbigin E, Silva-Filho MC, Mulhern TD, Lithgow T: Tom22’, an 8-kDa trans-site receptor in plants and protozoans, is a conserved feature of the TOM complex that appeared early in the evolution of eukaryotes. Mol Biol Evol. 2004, 21: 1557-1564.
Kawaguchi R, Bailey-Serres J: mRNA sequence features that contribute to translational regulation in Arabidopsis. Nucleic Acids Res. 2005, 33: 955-965.
Malnoy M, Jin Q, Borejsza-Wysocka EE, He SY, Aldwinckle HS: Overexpression of the apple MpNPR1 gene confers increased disease resistance in Malus × domestica. Molec Plant-Micro Interact. 2007, 20: 1568-1580.
Cao H, Glazebrook J, Clark JD, Volko S, Dong X: The Arabidopsis NPR1 gene that controls systemic acquired resistance encodes a novel protein containing ankyrin repeats. Cell. 1997, 88: 57-63.
Zhang Y, Fan W, Kinkema M, Li X, Dong X: Interaction of NPR1 with basic leucine zipper protein transcription factors that bind sequences required for salicylic acid induction of the PR-1 gene. Proc Natl Acad Sci U S A. 1999, 96: 6523-6528.
Spoel SH, Koornneef A, Claessens SMC, Korzelius JP, Van Pelt JA, Mueller MJ, Buchala AJ, Me´ Traux JP, Brown R, Kazan K, Van Loon LC, Dong X, Pieterse CMJ: NPR1 modulates cross-talk between salicylate- and jasmonate-dependent defense pathways through a novel function in the cytosol. Plant Cell. 2003, 15: 760-770.
Dong X: NPR1, all things considered. Curr Opin Plant Biol. 2004, 7: 547-552.
Cao H, Li X, Dong X: Generation of broad-spectrum disease resistance by overexpression of an essential regulatory gene in systemic acquired resistance. Proc Natl Acad Sci U S A. 1998, 95: 6531-6536.
Schoeneweiss DF: Predisposition, stress and plant disease. Annu Rev Plant Physiol Plant Mol Biol. 1975, 13: 193-211.
Schoeneweiss DF: Role of environmental stress in diseases of woody plants. Plant Dis. 1981, 65: 308-314.
Quilis J, Peñas G, Messeguer J, Brugidou C, San Segundo B: The Arabidopsis AtNPR1 inversely modulated defense responses against fungal, bacterial or viral pathogens while conferring hypersensitivity to abiotic stresses in transgenic rice. Molec Plant-Microbe Interact. 2008, 21: 1215-1231.
Norelli JL, Aldwinckle HS, Beer SV: Virulence of Erwinia amylovora strains to Malus sp. Novole plants grown in vitro and in the greenhouse. Phytopathology. 1988, 78: 1292-1297.
Ko K, Norelli JL, Reynoird J-P, Aldwinckle HS, Brown SK: T4 lysozyme and attacin genes enhance resistance of transgenic ‘Galaxy’ apple against Erwinia amylovora. J Am Soc Hort Sci. 2002, 127: 515-519.
Bolar JP, Hanke V, Norelli JL, Aldwinckle HS: An efficient method for rooting and acclimation of micropropagated apple cultivars. HortSci. 1998, 33: 1251-1252.
Artlip TS, Wisniewski ME: Tissue-specific expression of a dehydrin gene in one-year-old Rio Oso Gem peach trees. J Am Soc Hort Sci. 1997, 122: 784-787.
Artlip TS, Callahan AM, Bassett CL, Wisniewski ME: Seasonal expression of a dehydrin gene in sibling deciduous and evergreen genotypes of peach (Prunus persica [L.] Batsch). Plant Mol Biol. 1997, 33: 61-70.
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM: Primer3Plus, an enhanced web interface to Primer3. Nucl Acids Res. 2007, 35: W71-W74. doi:10.1093/nar/gkm306
Tong Z, Gao Z, Wang F, Zhou J, Zhang Z: Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol. 2009, 10: 71-doi:10.1186/1471-2199-10-71
Higo K, Ugawa Y, Iwamoto M, Korenaga T: Plant cis-acting regulatory DNA elements (PLACE) database:1999. Nucl Acids Res. 1999, 27: 297-300. Additional Files
Chang WC, Lee TY, Huang HD, Huang HY, Pan RL: PlantPAN: Plant Promoter Analysis Navigator, for identifying combinatorial cis-regulatory elements with distance constraint in plant gene group. BMC Genomics. 2008, 9: 561-
Rombauts S, Déhais P, Van Montagu M, Rouzé P: PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999, 27: 295-296.
Lescot M, Dehais P, Thijs G, Marchal K, Moreau Y, Van De Peer Y, Rouze P, Rombauts S: PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucl Acids Res. 2002, 30: 325-327. Additional Files
Beuning LL, Bowen JH, Persson HA, Barraclough D, Bulley S, MacRae EA: Characterization of Mal d 1-related genes in Malus. Plant Molec Biol. 2004, 55: 369-388.
Gao ZS, van de Weg WE, Schaart JG, Schouten HJ, Tran DH, Kodde LP, van der Meer IM, van der Geest AH, Kodde J, Breiteneder H, Hoffmann-Sommergruber K, Bosch D, Gilissen LJWJ: Genomic cloning and linkage mapping of the Mal d 1 (PR-10) gene family in apple (Malus domestica). Theor Appl Genet. 2005, 111: 171-183.
Mohanty B, Krishnan SP, Swarup S, Bajic VB: Detection and preliminary analysis of motifs in promoters of anaerobically induced genes of different plant species. Ann Bot. 2005, 96: 669-681. Additional Files
Maleck K, Levine A, Eulgem T, Morgan A, Schmid J, Lawton KA, Dangl JL, Dietrich RA: The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nat Gen. 2000, 26: 403-410. Additional Files
Lebel E, Heifetz P, Thorne L, Uknes S, Ryals J, Ward E: Functional analysis of regulatory sequences controlling PR-1 gene expression in Arabidopsis. Plant J. 1998, 16: 223-233. Additional Files
Kobayashi T, Nakayama Y, Itai RN, Nakanishi H, Yoshihara T, Mori S, Nishizawa NK: Identification of novel cis-acting elements, IDE1 and IDE2, of the barley ISD2 gene promoter conferring iron-deficiency-inducible, root-specific expression in heterogeneous tobacco plants. Plant J. 2003, 36: 780-793. Additional Files
Abe H, Yamaguchi Shinozaki K, Urao T, Iwasaki T, Hosokawa D, Shinozaki K: Role of Arabidopsis MYC and MYB homologs in drought- and abscisic acid-regulated gene expression. Plant Cell. 1997, 9: 1859-1868. Additional Files
Yamaguchi Shinozaki K, Shinozaki K: Arabidopsis DNA encoding two desiccation-responsive rd29 genes. Plant Physiol. 1993, 101: 1119-1120. Additional Files
Yamaguchi Shinozaki K, Shinozaki K: A novel cis-acting element in an Arabidopsis gene is involved in responsiveness to drought, low-temperature, or high-salt stress. Plant Cell. 1994, 6: 251-264. Additional Files
Baker SS, Wilhelm KS, Thomashow MF: The 5´ region of Arabidopsis thaliana cor15a has cis-acting elements that confer cold-, drought- and ABA-regulated gene expression. Plant Mol Biol. 1994, 24: 701-713. Additional Files
Jansch L, Kruft V, Schmitz UK, Braun HP: Unique composition of the preprotein translocase of the outer mitochondrial membrane from plants. J Biol Chem. 1998, 273: 17251-17257. Additional Files
Cercós M, Gómez Cadenas A, Ho THD: Hormonal regulation of a cysteine proteinase gene, EPB-1, in barley aleurone layers: cis- and trans-acting elements involved in the co-ordinated gene expression regulated by gibberellins and abscisic acid. Plant J. 1999, 19: 107-118. Additional Files
Acknowledgements
The authors would like to thank Jing Ma for her excellent technical assistance and Dr. Michael Glenn for his expert statistical analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
There are no competing interests.
Authors’ contributions
CLB: designed the experiments, conducted RT-qPCR analyses, and performed in-depth bioinformatic analyses of genes AMB: conducted unigene assembly, provided initial bioinformatic analyses of subtraction results. JTM and RMJ: performed quality assurance analyses of DNA and primers and conducted semi-quantitative PCR reactions. DSS: performed semi-quantitative and quantitative PCR reactions. MEW: contributed to the experimental design and interpretation of data. JLN: contributed to the interpretation of data and preparation of manuscript. RJF: conducted SSH experiments and performed quality control experiments on SSH results.
Electronic supplementary material
12870_2014_1595_MOESM1_ESM.xlsx
Additional file 1: Sequences up- (AAF) or down- (AAR) regulated after two weeks of simulated drought; Sequences up- (BBR) and down- (BBF) regulated after a week of recovery from simulated drought; Sequences up- (CCF) and down- (CCR) regulated between treatment weeks 2 and 3 in trees not exposed to drought (recovery control, subtractions T2C/T1C in Figure 1). DNA sequence of unigenes and sequence annotation. Note: sequences not accepted by GenBank dbEST included sequences less than 200 nts, sequences encoding hypothetical or unknown proteins, mitochondrial and chloroplast DNA, and ribosomal DNA. Sequences making up contigs may not be assigned an accession number if one of more of the sequences is less than 200 nts. (XLSX 100 KB)
12870_2014_1595_MOESM2_ESM.docx
Additional file 2: Alignment of full length apple High Affinity Nitrate Transporter genes. MFS: major facilitator superfamily domain and NNP: nitrate/nitrite porter domain defined within alignments. (DOCX 14 KB)
12870_2014_1595_MOESM4_ESM.docx
Additional file 4: Alignment of apple TOM7 predicted polypeptides. Predicted polypeptides for the three apple TOM7 polypeptides and the EST isolated from drought-treated roots are aligned with the two predicted Arabidopsis TOM7 polypeptides [71]. (DOCX 11 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Bassett, C.L., Baldo, A.M., Moore, J.T. et al. Genes responding to water deficit in apple (Malus × domestica Borkh.) roots. BMC Plant Biol 14, 182 (2014). https://doi.org/10.1186/1471-2229-14-182
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-14-182