
Abstract
Background
Etoposide (epipodophyllotoxin) is a chemical commonly used as an anti-cancer drug which inhibits DNA synthesis by blocking topoisomerase II activity. Previous studies in animal cells have demonstrated that etoposide constitutes a genotoxic stress which may induce genomic instability including mobilization of normally quiescent transposable elements (TEs). However, it remained unknown whether similar genetically mutagenic effects could be imposed by etoposide in plant cells. Also, no information is available with regard to whether the drug may cause a perturbation of epigenetic stability in any organism.
Results
To investigate whether etoposide could generate genetic and/or epigenetic instability in plant cells, we applied etoposide to germinating seeds of six cultivated rice (Oryza sativa L.) genotypes including both subspecies, japonica and indica. Based on the methylation-sensitive gel-blotting results, epigenetic changes in DNA methylation of three TEs (Tos17, Osr23 and Osr36) and two protein-encoding genes (Homeobox and CDPK-related genes) were detected in the etoposide-treated plants (S0 generation) in four of the six studied japonica cultivars, Nipponbare, RZ1, RZ2, and RZ35, but not in the rest japonica cultivar (Matsumae) and the indica cultivar (93-11). DNA methylation changes in the etoposide-treated S0 rice plants were validated by bisulfite sequencing at both of two analyzed loci (Tos17 and Osr36). Transpositional activity was tested for eight TEs endogenous to the rice genome in both the S0 plants and their selfed progenies (S1 and S2) of one of the cultivars, RZ1, which manifested heritable phenotypic variations. Results indicated that no transposition occurred in the etoposide-treated S0 plants for any of the TEs. Nonetheless, a MITE transposon, mPing, showed rampant mobilization in the S1 and S2 progenies descended from the drug-treated S0 plants.
Conclusions
Our results demonstrate that etoposide imposes a similar genotoxic stress on plant cells as it does on animal and human cells, which may induce transgenerational genomic instability by instigating transpositional activation of otherwise dormant TEs. In addition, we show for the first time that etoposide may induce epigenetic instability in the form of altered DNA methylation patterns in eukaryotes. However, penetrance of the genotoxic effects of etoposide on plant cells, as being reflected as genetic and epigenetic instability, appears to be in a strictly genotype- and/or generation-dependent manner.
Similar content being viewed by others


Background
Etoposide (epipodophyllotoxin) is a chemical which has been used widely as an anti-cancer drug as it inhibits DNA synthesis by forming a complex with topoisomerase II [1]. During DNA replication, topoisomerase II can break, unwind and repair both strands of the double-stranded DNA that is supercoiled during the unwinding process, hence releasing the tension built up on the supercoiled DNA and repairing topoisomerase II-associated double-stranded DNA breaks (DSBs) [1]. Accordingly, inhibition of topoisomerase II activity accumulates breaks in DNA, prevents entry into the mitotic phase of cell division, and leads to cell death. Etoposide acts primarily in the G2 and S phases of the cell cycle [2].
Apart from directly generating DSBs, previous studies in animal cells have also shown that etoposide constitutes a genotoxic stress which may induce genomic instability indirectly by instigating mobility of otherwise quiescent transposable elements (TEs), and hence generating insertional mutagenesis [3]. Most plant genomes harbor a large proportion of TEs and some of which are known as inducible to become transcriptionally active and even transpositionally mobile under stress conditions [4]. Therefore, it is interesting to explore whether etoposide treatments may produce similar effects on the activity of TEs in plant genomes, as no information has been available in this aspect.
It becomes increasingly clear that genetic information encoded in the primary DNA sequence is not the only determinant of heritable phenotypes. Epigenetic modifications, collectively known as epigenome, also participate in orchestrating gene expression as well as in maintaining genomic stability [5]. Among the epigenetic modifications, cytosine DNA methylation is the best studied, and plays essential roles as an evolutionarily conserved genome defense device as well as a master regulator of genome-wide temporal and spatial gene expression [6, 7]. More importantly, DNA methylation is sensitive and responsive to environmental cues including genotoxic stress and may generate new and heritable epialleles coping with the particular environmental condition accordingly [8–11]. For instance, it was found in plant cells that various environmental conditions can induce DNA methylation alterations, and those progenies inheriting the altered methylation patterns exhibited enhanced tolerance to the specific stress their progenitors experienced [12–14]. Given the above, it is of apparent interest to test if the genotoxic stress imposed by etoposide may induce epigenetic instability in the form of DNA methylation changes. Surprisingly, however, to our knowledge, there is no report on the epigenetic effects of etoposide treatments in any organism.
The aims of this investigation were to (1) test whether the topoisomerase II inhibitor, etoposide may induce epigenetic instability in the form of DNA methylation changes; (2) explore whether etoposide may also instigate mobilization of transposable elements (TEs) in plant cells. We addressed these two questions in six cultivated genotypes of rice (Oryza sativa L.) representing both subspecies, japonica and indica.
Results
Etoposide-induced epigenetic changes in the form of DNA methylation were manifested in somatic cells of the immediately treated S0 rice plants
For all six studied rice genotypes, the seedling plants treated with etoposide at both concentrations (10 and 20 mg/L, respectively) appeared normal apart from a slight retardation in the overall statue (data not shown), and no difference was observed between the two concentrations. Moreover, after a two-month period of recovering, no discernible difference was observable between the treated and the control plants. This suggests that physiological toxicity to the rice seedlings by the chemical was mild and temporary at the studied concentrations. Nonetheless, it is known from previous studies that some abiotic and genotoxic stresses may impose longer-term or even transgenerational genomic and/or epigenomic effects without expressing immediate phenotypic or physiological effects.
To test whether the etoposide-imposed genotoxic stress may induce changes in cytosine DNA methylation in somatic cells of the immediately treated rice plants (designated as S0) that showed no evidence of phenotypic abnormality, we performed methylation-sensitive Southern blotting with a set of 15 pre-selected probes representing both transposable elements (TEs) and known-function protein-encoding genes, the intrinsic methylation states of which are known to be variable in the wild-type rice genome, i.e., being heavily or moderately methylated or unmethylated, respectively [15–17]. DNA of pooled plants (10–15 individuals) for each genotype was analyzed. Thus, if methylation repatterning would occur due to etoposide treatments, then, both hypo- and hypermethylation could be expected in the gel-blotting patterns by one or more types of these probes on genomic DNAs restricted by the methylation-sensitive endonucleases.
Indeed, the methylation-sensitive Southern blotting patterns indicated that of the six rice genotypes, four showed DNA methylation changes in the etoposide-treated plants vs. their respective controls in at least one of the 15 tested probes (Table 1). Specifically, the following results were obtained: (1) For the two laboratory standard rice genotypes representing respectively the japonica and indica subspecies, Nipponbare and 93-11, only the former showed methylation changes in four (three TEs and one gene) of the 15 probes, and most of the changes belonged to decrease in methylation, i.e., hypomethylation at the CHG sites of the 5’-CCGG tetranucleotide(s) within or immediately flanking the probe sequences (i.e., changes only occurred in MspI digest) (Table 1; Figure 1). (2) For the three RILs (RZ1, RZ2 and RZ35) and their rice parental line (Matsumae), all three RILs showed methylation changes in one to three (two TEs and two genes) of the 15 probes, while Matsumae did not show any change (Table 1; Figure 2). An interesting observation common to the DNA methylation changes in these rice genotypes was that little difference was detected between the two concentrations (10 and 20 mg/L) of the etoposide treatment (Table 1; Figures 1 and 2).

Changes in DNA methylation in the S0 generation of etoposide- treated plants of genotypes Nipponbare and 93-11 relative to their respective wild-type control plants. Each genotype was treated with two concentrations of etoposide, 10 mg/L and 20 mg/L, respectively. “Control” represents plants treated with ddH2O. DNA methylation changes occurred only at the CHG context of the 5’-CCGG sites (in MspI digestion, denoted by rectangles), and the changes occurred only in Nipponbare. Note that the two etoposide concentrations showed the same changes. The Elongation factor gene is intrinsically unmethylated in the rice genome and thus showed a single monomorphic band among all treated plants in both HpaII and MspI digestions of each genotype. This monomorphic hybridizing pattern of Elongation factor also validated complete digestion by the methylation-sensitive isoschizomers in all DNA samples.

Changes in DNA methylation in the S0 generation of etoposide- treated plants of genotypes Matsumae, RZ1, RZ2 and RZ35, relative to their respective control plants. Each genotype was treated by two concentrations of etoposide, 10 mg/L and 20 mg/L, respectively. “Control” represents plants treated with ddH2O. Notably, DNA methylation changes occurred in all three recombinant inbred lines (RZ1, RZ2 and RZ35) but not in their rice parental line Matsumae. Also, the changes occurred in either or both of the enzyme digests, though more abundantly in MspI-digest, and the changes occurred either in the 10 mg/L concentration or in both concentrations. The OsCDPK protein-encoding gene is intrinsically unmethylated and thus showed a monomorphic banding pattern among all treated plants in both HpaII and MspI digestions across the genotypes, indicating complete digestion in all samples.
Further analysis and validation of the etoposide-induced DNA methylation changes in the S0 rice plants by bisulfite sequencing
The methylation-sensitive Southern blotting analysis was capable of only revealing methylation changes at the cytosines that were within or immediately adjacent to the enzyme recognition/restriction site(s). In this case, methylation changes only at the two cytosines (inner and outer) of the 5’-CCGG tetranucleotide(s) were detectable, as which were recognition/restriction sites of the pair of isoschizomers, HpaII/MspI. To further explore whether changes in methylation also occurred at other cytosines, as well as to validate the blotting-detected methylation changes, by an independent approach, we performed bisulfite sequencing for two segments representing the two TEs (Tos17 and Osr36) that showed clear methylation changes in the etoposide-treated S0 plants of Nipponbare (Figure 1). For Tos17, we designed bisulfite sequencing primers that encompassed portion of the 5’-LTR and portion of the internal body-region of the retroelement. The bisulfite sequencing results showed that whereas the LTR portion was intrinsically hypomethylated at all three types of cytosine sites, CG, CHG and CHH, the body-region was moderately methylated at the CG and CHG sites but virtually unmethylated at the CHH sites in the wild-type rice genome; the LTR region did not show discernibly gain of methylation subsequent to the etoposide treatment (10 mg/L), but substantial reduction of CG methylation and minor reduction of CHG methylation occurred in the body-region of the retroelement due to the etoposide treatment, which was accompanied by some de novo hypermethylation at a few CHH sites (Figure 3). The bisulfite sequencing results for this segment were in broad agreement with the methylation-sensitive gel-blotting results (Figure 1). For Osr36, we designed bisulfite sequencing primers within the 5’-LTR region of this retroelement, because this region was known to be heavily methylated according to our previous gel-blotting results in other rice genotypes [18]. The bisulfite sequencing results indeed showed that in the wild-type control plants of Nipponbare, the CG and CHG sites were heavily methylated in the segment whereas the CHH sites were nearly completely (except for one or two positions) devoid of methylation. Only moderate methylation changes were detected in the etoposide-treated plants at the CG sites, but substantial changes were detected at the CHG sites (Figure 4). Surprisingly, in contrast with the changes in this TE detected in the gel-blotting which showed hypomethylation (Figure 1), the collective changes (all cytosines of a given type being considered together) revealed by bisulfite sequencing at both CG and CHG sites represented hypermethylation (Figure 4). Given that the gel-blotting was only capable of detecting methylation changes at the 5-CCGG sites, whereas the bisulfite sequencing results were the collective of all cytosines within the sequenced region, the seemingly discrepant results can be readily reconciled (Figure 4).

Cytosine methylation changes in a fragment of Tos17 (encompassing portions of the 5'-LTR- and body-region) in the S0 generation of the etoposide-treated plants of Nipponbare, determined by bisulfite sequencing. Nine and 10 clones were arbitrarily sequenced for the control and the 10 mg/L etoposide-treated plants, respectively. All three types of cytosines, CG (red circles), CHG (blue circles) and CHH (green circles) were shown in the map. Filled and empty circles denote methylated and unmethylated cytosines, respectively. The red, blue and green columns in the histograms refer to the collective methylation levels (in percentage) respectively of CG, CHG and CHH, of the LTR- and body-regions. The nucleotide sequence of this analyzed fragment was presented in Additional file 1. De novo methylation at six CHH sites as a result of the etoposide-treatment were arrowed.

Cytosine methylation changes in a fragment of Osr36 (portion of the 5'-LTR-region) in the S0 generation of the etoposide-treated plants of Nipponbare, determined by bisulfite sequencing. Ten clones were arbitrarily sequenced for the control and the 10 mg/L etoposide-treated plants, respectively. All three types of cytosines, CG (red circles), CHG (blue circles) and CHH (green circles) were shown in the map. Filled and empty circles denote methylated and unmethylated cytosines, respectively. The red, blue and green columns in the histograms refer to the collective methylation levels (in percentage) respectively of CG, CHG and CHH, of the LTR-region. The seven CHG sites that showed dramatic hypermethylation were arrowed. The nucleotide sequence of this analyzed fragment was presented in Additional file 1.
Transposon mPingwas totally quiescent in the etoposide-treated S0 plants, but showed rampant transgenerational mobilization in their S1 and S2 progenies in one of the genotypes, RZ1
Whole genome sequencing has revealed that a substantial portion of plant genomes is comprised of transposable elements (TEs) and their derivatives, and a small fraction of the TEs still possesses the ability to become transpositionally active (mobile) under specific stress conditions, and which are often accompanied by epigenetic remodeling [19–21]. The most labile TEs to become mobile in the rice genome are the MITE mPing, its autonomous TPase donors Ping and Pong, and a set of low-copy, copia-like, LTR retrotransposons including Tos17, Osr23, Osr35, Osr36 and Osr42 [21, 22]. Given that the etoposide-treated S0 rice plants showed changes in DNA methylation, it was interesting to test whether some of these potentially mobile TEs might become active in these plants. We therefore conducted gel-blotting for these eight TEs (mPing, Ping, Pong, Tos17, Osr23, Osr35, Osr36 and Osr42). We used randomly chosen individual plants of three consecutive generations, S0 (n = 8), S1 (n = 19) and S2 (n = 15), of genotype RZ1 to study this issue because these plants showed heritable phenotypic variations (detailed below). We found that all these TEs showed a monomorphic pattern among the eight etoposide-treated S0 plants of RZ1, and which was identical with the untreated control (Figure 5a), indicating no immediate transpositional activity within the detecting resolution of gel-blotting for any of the tested TEs in the somatic leaf cells of the etoposide-treated S0 plants. Nevertheless, because the activity of many TEs is developmentally regulated [23, 24], it was deemed possible that progenies of these S0 plants might show transposition of the TEs on condition the activity occurred in the germinal cells or progenitor cells thereof in the treated S0 plants. Indeed, we found that at least 10 of the 19 S1 plants showed rampant transpositional events for one of the eight studied TEs, i.e., mPing, as polymorphic patterns including both loss and gain of bands were detected in the gel-blotting probed by this element (Figure 5b, upper panel), consistent with the “cut-and-paste” model of transposition of this element [25]. In contrast, all the rest TEs showed only monomorphic blotting patterns (e.g., Figure 5b, lower panel for probe Osr35), denoting stability of these TEs even in progenies of the etoposide-treated plants. Similarly, at least 14 of the 15 S2 individuals derived from a single S1 plant (S1-7) showed further transpositions of mPing (Figure 5c, upper panel), although the particular S1 plant itself did not show mPing transposition. Taken together, our results indicated that subsequent to the etoposide treatment, activity of mPing was altered in such a way that they became transgenerationally mobilizing.

Stability or mobilization of mPing in different selfed generations of the etoposide-treated plants of RZ1, revealed by DNA gel-blotting. (a) Complete stability of mPing was observed in eight randomly chosen etoposide-treated S0 plants. Also, no evidence for mobility was detected for a set of low-copy LTR retrotransposons (e.g., Osr35) in these S0 plants. (b) Mobilization of mPing was detected in at least 10 of the 19 randomly chosen individual plants of the S1 generation derived from a single S0 plant (S0-1). The empty and solid circles denote excision and reinsertion events, respectively. No evidence for mobility was detected for a set of low-copy LTR retrotransposons (e.g., Osr35) in any of these S1 plants. (c) Additional transposition of mPing was detected in 14 of the 15 S2 plants derived from a single S1 individual (S1-7) in which no transposition of this TE was detected. Only excisions were detected in these plants (marked by empty circles). Again, no evidence for mobility was detected for a set of low-copy LTR retrotransposons (e.g., Osr35) in any of these S2 plants. The DNAs in these blots were digested by XbaI.
To further verify transposition of mPing, we performed mPing-specific transposon display (TD) analysis [19] for a larger number of etoposide-treated S0 (30 individuals), S1 (40 individuals) and S2 (30 individuals) plants of RZ1. Although no polymorphism in the mPing-specific TD profiles was observed in any of the 30 studied S0 plants, we detected a large number of loss and gain of bands in the S1 and S2 plants, denoting transpositional activity of mPing. We then isolated and sequenced a subset of these variant bands and used them as queries to blast against the reference rice genome of Nipponbare (http://rgp.dna.affrc.go.jp). We next designed locus-specific PCR primers (see Additional file 2) based on the matched Nipponbare sequence, which putatively encompassed the mPing copies representing either excision or reinsertion events in the S1 or S2 plants derived from the etoposide-treated S0 plants of RZ1. The results of PCR amplification using DNA of the untreated control plants of RZ1 as a template indicated that all five loci produced PCR amplicons of sizes consistent with harboring an internal mPing copy (430 bp in length), while all 34 loci that presumably represented reinsertion events in the S1 or S2 plants derived from the etoposide-treated S0 plants, and therefore should not contain a mPing copy in the untreated control, indeed had smaller-sized bands consistent with lacking of a mPing copy (data not shown).
With these locus-specific, mPing-containing (n = 5) or -devoiding (n = 34) primer pairs, we first analyzed the etoposide-treated S0 plants (n = 30) of RZ1, and we found only monomorphic patterns identical with those of the control plants (data not shown), confirming that no transpositional events occurred immediately in the leaf somatic cells of the etoposide-treated S0 plants, consistent with the gel-blotting results. We next analyzed the 32 S1 plants (the same as used for TD analysis, described above) derived from the etoposide-treated S0 plants of RZ1, and found that all the five primer pairs harboring mPing showed evidence of excisions (Table 2) in portions of the S1 plants, as concomitant loss of the larger-sized band harboring a mPing and gain of a smaller-sized band lacking a mPing were detected in these plants, with different plants manifesting the changes at different loci (Figure 6a). Similarly, 20 of the 34 primer pairs (58.8%) detected de novo insertions in a large proportion of the 32 S1 plants relative to the RZ1 control plants (Table 3), as gain of a larger-sized band consistent with gaining a mPing and loss of a smaller-sized band originally devoiding of a mPing were detected in each of these cases (e.g., Figure 6b). Further analysis by the locus-specific PCR amplification on a set of 19 S2 plants derived from the studied S1 plants indicated that at least 14 additional new insertions were detected (Table 4), indicating mobility of the activated mPing was retained transgenerationally.

Validation of excision and reinsertion events of mPing by locus-specific PCR amplification in 32 random S1 individuals derived from multiple etoposide-treated S0 plants of RZ1 (in which no evidence for either excision or reinsertion events of mPing was detected) and confirmation of mPing stability in two generations (S0 and S1) of the wild-type control plants of RZ1. (a) The five mPing-containing loci in untreated control of RZ1 showing excisions in the various S1 individuals (depending on locus). Representatives of both the upper- and lower-bands were sequenced, which verified that length difference between the two bands for a given locus was exactly 430, i.e., the full length of mPing, as indicated on the right side of the figure. (b) Five of the 20 characterized mPing-empty loci in untreated control of RZ1 showing reinsertions in the various S1 individuals (depending on locus). Representatives of both the upper- and lower-bands were sequenced, which verified that the length difference between the two bands for a given locus was exactly 430, i.e., the full length of mPing, as indicated on the right side of the figure. (c) and (d) are amplification results by the same five mPing-containing loci and five mPing-devoiding loci as in (a) and (b), respectively, on 40 individual plants of the wild-type control plants of RZ1 at two generations (S0 and S1). Labeling of the band sizes is the same as in (a) and (b).
To test the remote possibility that mPing at these loci in the specific genotype (RZ1) might be intrinsically unstable irrespective to the drug treatment, we further tested 40 individual plants (20 and 20 from the S0 and S1 generations, respectively) of the wild-type RZ1 with the same 10 loci described above. We detected only monomorphic patterns across all tested individual plants (Figure 6, c and d), indicating lack of any excision or insertion event at these loci in these plants, thus lending further support to the conclusion that mPing mobility was causally linked to etoposide treatment.
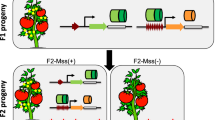
S1 and S2 plants descended from the etoposide-treated S0 plants of RZ1 showed heritable phenotypic variations
We examined the phenotypic stability in the etoposide-treated S0 plants and their S1 and S2 progenies for all six genotypes. We found that only some of the S1 and S2 plants descended from the etoposide-treated S0 plants of RZ1 showed clear phenotypic variations in several traits, with reduced seed-setting (compromised fertility) and kernel shape (elongated kernel length but reduced width) being the most conspicuous (Figures 7 and 8; see Additional file 3). Moreover, the altered phenotypic traits were stably inherited from S1 to S2 (Figures 7 and 8), suggesting that they were likely to have a genetic and/or epigenetic basis rather than being caused by physiological perturbations by the drug treatments applied in the S0 generation.

Illustrations of heritable alteration in fertility (upper panel) and kernel-shape (lower-panel) in progenies (S1 and S2) of the etoposide-treated S0 plants of RZ1. Reduced fertility was observed in the S1 and S2 progenies of the etoposide-treated S0 plants of RZ1, the kernel-shape of which also became elongated. Both altered traits were inheritable (at least from S1 to S2).

Quantification of heritable alteration in fertility (a) and kernel-shape (b) in progenies (S1 and S2) of the etoposide-treated S0 plants of RZ1. Thirty random samples (panicles for fertility and kernels for shape) were measured and quantified. Statistical test was conducted by the Independent-Sample T Test using SPSS 17.0 software (see Additional file 3).
Discussion
As a topoisomerase II inhibitor, the major biological function of etoposide is to inhibit DNA synthesis, and thereby, resulting in accumulation of double-stranded DNA breaks (DSBs) [1, 2]. Consequently, as documented by previous studies in animal and human cells, etoposide constitutes a genotoxic stress which may also induce genomic instability indirectly by causing transpositional activation of otherwise quiescent transposable elements (TEs), and hence generating insertional mutagenesis [3]. Most plant genomes harbor potentially active TEs which can be induced to become transpositionally active under specific conditions [4, 5]. Nonetheless, no information is available regarding whether the chemical etoposide may produce similar effects in plant cells as it does in animals [3].
We have shown in this study that etoposide may indeed cause rampant transposition of a MITE transposon, mPing, endogenous to the rice genome [25]. However, the activation of mPing occurred in a strictly genotype-dependent manner, as only one of the six tested genotypes showed this phenomenon. In addition, the transpositional events did not occur in somatic cells of the immediately treated plants (S0); rather, excision and reinsertion events were detected only in selfed progenies (S1 and S2) of the etoposide-treated S0 plants. This is consistent with developmental regulation of most plant TEs [26]. Thus, for example, if the transpositions of mPing occurred at the gametogenesis stage of the etoposide-treated S0 plants, then the events can not be detected in leaf tissue of treated S0 plants but should be detectable in any sporophyte tissues of the S1 plants.
Authenticity of transposition rather than genomic rearrangements was verified by multiple independent assays including gel-blotting, mPing-specific TD, sequencing and locus-specific PCR amplification. The fact that only one of the eight analyzed TEs with transpositional potentiality was actually activated is in accord with the recent finding that the controlling mechanisms of TEs in plant genomes are highly individualized by diverse repressive epigenetic modifications [4, 27]. The mobilization of only mPing without entailing concomitant transpositional activation of its TPase donors Ping or Pong is also congruent with previous findings on this element [21]. This is understandable as only transcriptional activation of its TPase donor should be sufficient for mPing transposition. Sequencing of the newly inserted mPing copies indicated that, on the one hand, the insertions were randomly distributed with regard to the 12 chromosomes of the rice genome, and on the other hand, the great majority of the insertion sites were landed within low-copy, genic regions. Both characteristics are in line with known propensity of newly induced mPing insertions by various inductive conditions [19, 21, 28].
A hallmark of epigenetic modifications lies in its metastability in response to internal or environmental perturbations. Importantly, alterations of at least some of the epigenetic modifications, e.g., cytosine DNA methylation, are known to produce transgenerational biological effects which could be relevant to coping with the particular stress condition [8–11]. This is a particularly pertinent issue in plants, as altered DNA methylation patterns are more readily transmissible to organismal generations through meiosis in plants than in animals, and thereby, the biological effects they dictate [14]. Therefore, it is apparently interesting to test whether the genotoxic effects of etoposide may also instigate epigenetic instabilities in addition to its genetic mutagenesis. Surprisingly, however, to our knowledge, no such information is available in any organism.
Thus, we have shown in this study for the first time in any organism that the topoisomerase II inhibitor etoposide is also epigenetically mutagenic in the sense that the chemical generated immediate changes in DNA methylation patterns in the leaf somatic cells of the treated S0 rice plants. However, this effect, at least in rice, is of variable penetrance with regard to genetic context, as only four out of the six studied genotypes showed evidence of methylation changes. Interestingly, nearly all the detected methylation changes by methylation-sensitive gel-blotting were found to occur only at the CHG sites, underscoring differential stability of CG vs. CHG methylation modifications in plants. It has been well-established that in plant genomes the three types of cytosine methylation patterns, CG, CHG and CHH, are maintained by distinct yet, to an extent, also overlapping DNA methyltransferases, with CG methylation being predominantly maintained by DNA Methyltransferase 1 (MET1), CHG methylation by Chromomethylase 3 (CMT3, a plant-specific DNA methyltransferase), and CHH methylation by Domains Rearranged Methyltransferase 2 (DRM2, a de novo DNA methyltransferases which is also partly responsible for de novo methylation of all cytosines) [29]. Therefore, it can be envisioned that the etoposide treatments may differentially affect activity or titration of these DNA methyltransferases and result in the observed predominant CHG methylation changes. Nonetheless, based on the higher resolution analytical approach, i.e., bisulfite sequencing, which was capable of revealing methylation change of any single cytosine within the analyzed region, we have found that, for both analyzed loci, all three types of cytosine methylation patterns, CG, CHG and CHH, underwent changes due to the etoposide treatment. But taking both loci together, it still holds that CHG methylation appeared more prone to changing as a result of the etoposide treatment than CG methylation, which is consistent with the situation of other environmental stress-induced cytosine methylation changes in plants [18].
The detected methylation changes included both decrease (hypo) and increase (hyper) in methylation, but with the former seemingly more predominant than the latter. This is consistent with the perturbation of otherwise fine-tuned activity of the various DNA methyltransferases that are responsible for faithfully maintaining the inheritable methylation patterns. However, it has been shown that loss of methylation of certain type (e.g., CG) may activate alternative back-up cellular systems, which in turn may catalyze hypermethylation of other types of methylation (e.g., CHH) and produce aberrant methylation patterns [30]. This scenario may explain the detected hypermethylation in the bisulfite sequencing results.
It seems surprising that little difference was detected between the two concentrations (10 mg/L vs. 20 mg/L) of the etoposide treatment with regard to the induced DNA methylation changes. It has been established in human cells that each molecule of etoposide stabilizes only one single-stranded DNA break [1]. Therefore, it can be deduced that if 10 mg/L has already reached the saturated concentration for the germinating rice seeds, then no difference should be expected with elevated concentrations.
Phenotypic examination under paddy-field condition of the etoposide-treated S0 rice plants and their selfed S1 and S2 generations showed that of the six genotypes, only S1 and S2 plants of RZ1 showed clear and heritable (at least between S1 and S2) variations in several phenotypic traits like fertility and kernel-shape. Because mPing transpositional activity was concomitantly detected in these plants, it suggests that the co-occurrence of the two phenomena may be more than coincidental. Additional investigations are needed to explore the correlative or causal relationships between the two phenomena.
Conclusions
Results of this study have demonstrated that the eukaryotic topoisomerase II-blocking drug, etoposide, imposes a similar genotoxic stress on plant cells as it does on animal and human cells. In rice, rampant mobilization of an endogenous MITE transposon, mPing, was detected in selfed progenies of the etoposide-treated plants in one of the studied cultivars, pointing to transgenerational genomic instability by the drug treatment. In addition, epigenetic instability in the form of altered DNA methylation patterns was even more generally observed in somatic cells of rice plants shortly after the etoposide treatment, indicating immediate epigenetic effects of the drug. However, it should be noted that the mutagenic effects of etoposide at both the genetic and epigenetic levels are genotype- as well as organismal generation-dependent.
Materials and Methods
Plant materials
Seeds of six cultivated rice (Oryza sativa L.) genotypes, including the standard laboratory cultivars for each of the two subspecies, japonica (Nipponbare) and indica (93-11), were used. Four additional genotypes (Matsumae, RZ1, RZ2 and RZ35) all belonging to the japonica subspecies are akin to each other, as the three recombinant inbred lines (RILs), RZ1, RZ2 and RZ35, are derived from the same rice genotype (Matsumae) and a wild rice species, Zizania Latifolia Griseb [31]. All the genotypes were maintained in our hands by strict selfing.
Etoposide treatment
Uniform seeds of each of the six rice genotypes were germinated in three different solutions, double-distilled water (used as the control), 10 mg/L etoposide solution and 20 mg/L etoposide solution, respectively. These two concentrations of etoposide were chosen on the basis of both previous studies in animal cells and our preliminary experiments in rice (unpublished data). The solutions were changed every day, for seven days. The germinating seeds were allowed to develop into seedlings in petri dishes and then be transplanted to regular paddy-fields. Randomly selected individuals (designated as S0) were tagged and later analyzed. Seeds were collected from each individual plant and designated as S1 for selfed progenies of both the controls and etoposide-treated plants. Similarly, we designated selfed progenies of the S1 generation as S2. We extracted genomic DNAs from leaves of all these plant materials for molecular analyses.
Conventional and methylation-sensitive gel-blotting analyses
Conventional and methylation-sensitive gel-blotting analyses followed similar protocols, the only difference lied in using different restriction enzymes for DNA digestion. The former employed XbaI as restriction enzyme, while the latter employed a pair of methylation-sensitive isoschizomers, HpaII and MspI instead. All restriction enzymes were purchased from New England Biolabs Inc. (Beverly, Massachusetts). Specifically, genomic DNA was isolated from expanded leaves of individual plants by a modified CTAB method [32] and purified by phenol extraction. The DNA was digested by restriction enzyme(s). To ensure complete digestion, an excess of enzyme (10 units enzyme per μg DNA) was used and the incubation time was extended to 48 h. Digested DNA was run through 1% agarose gel and transferred onto Hybond N+ nylon membranes (Amersham Pharmacia Biotech, Piscataway, New Jersey) by the alkaline transfer method recommended by the supplier. For probes, we used eight transposable elements (TEs) for conventional gel-blotting, and eight transposable elements (TEs) and seven known-function, protein-encoding genes for methylation-sensitive gel-blotting. Specific primers (see Additional file 4) for amplifying each of these probes were designed based on sequences deposited at GenBank, and the fragments were amplified by PCR at an annealing temperature of 58-60°C. The probe fragments were verified by sequencing, and then gel-purified and labeled with fluorescein-11-dUTP by the Gene Images Random Prime-labeling Module (Amersham Pharmacia Biotech). Hybridization signals were detected by the Gene Images CDP-Star detection module (Amersham Pharmacia Biotech) after washing at a stringency of 0.2× SSC, 0.1% SDS for 2× 50 min. Finally, the filters were exposed to X-ray films.
Bisulfite sequencing analysis
Bisulfite sequencing was carried out as described [33], which involved bisulfite treatment of single stranded DNA by the EZ DNA Methylation-GoldTM Kit (ZYMO Research Corporation). Bisulfite treatment converts unmethylated cytosines into uracils while methylated cytosines remain unconverted. After treatment, the region of interest was PCR amplified by the designed primers for each of the two studied TEs, Tos17 and Osr36 (see Additional file 5), and the PCR products were cloned and sequenced. The PCR amplification of the converted cytosine (to uracil) would result in the replacement of uracil with thymine. Analyses of the bisulfite-sequencing results were conducted at the Kismeth website (http://katahdin.mssm.edu/kismeth).
Transposon display
Transposon display (TD) [34] was performed by combining the mPing sub-terminal-specific primers [25] with MseI-adaptor-specific primers (see Additional file 6) and the amplicons were visualized on 5% polyacrylamide gels after silver-staining. Compared with the control plants, novel bands and lost bands in TD gels of etoposide treated plants (S0, S1 and S2) were considered as putative mPing de novo insertions and excisions, respectively. A subset of the variable bands were eluted, cloned and sequenced. The sequences were then blasted against the whole genome sequence of rice cv. Nipponbare (http://rgp.dna.affrc.go.jp). Based on this analysis, a set of locus-specific primers were designed, as detailed below.
PCR-based locus assay on mPingtransposition
To validate the putative transpositions of mPing in the S0, S1 and S2 generations of the etoposide-treated rice plants, revealed in the TD analysis, a set of 39 pairs of locus-specific primers each containing an intact mPing (see Additional file 2) were used for PCR amplification at annealing temperatures ranging from 56°C to 60°C depending on the primers. The amplicons were visualized by ethidium bromide staining after electrophoresis through 1% agarose gels. The identified excision and insertion sites of mPing were isolated, cloned and sequenced for confirmation.
References
Bromberg KD, Burgin AB, Osheroff N: A two-drug model for etoposide action against human topoisomerase IIα. J Biol Chem. 2003, 278 (9): 7406-7412. 10.1074/jbc.M212056200.
Montecucco A, Biamonti G: Cellular response to etoposide treatment. Cancer Lett. 2007, 252 (1): 9-18. 10.1016/j.canlet.2006.11.005.
Hagan CR, Sheffield RF, Rudin CM: Human Alu element retrotransposition induced by genotoxic stress. Nat Genet. 2003, 35 (3): 219-220. 10.1038/ng1259.
Lisch D: Epigenetic regulation of transposable elements in plants. Annu Rev Plant Biol. 2009, 60: 43-66. 10.1146/annurev.arplant.59.032607.092744.
Feng S, Jacobsen SE: Epigenetic modifications in plants: An evolutionary perspective. Curr Opin Plant Biol. 2011, 14 (2): 179-186. 10.1016/j.pbi.2010.12.002.
Slotkin RK, Vaughn M, Borges F, Tanurdžić M, Becker JD, Feijó JA, Martienssen RA: Epigenetic Reprogramming and Small RNA Silencing of Transposable Elements in Pollen. Cell. 2009, 136 (3): 461-472. 10.1016/j.cell.2008.12.038.
Pai AA, Bell JT, Marioni JC, Pritchard JK, Gilad Y: A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genetics. 2011, 7 (2): e1001316-10.1371/journal.pgen.1001316.
Bossdorf O, Richards CL, Pigliucci M: Epigenetics for ecologists. Ecol Lett. 2008, 11 (2): 106-115.
Bird A: DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16 (1): 6-21. 10.1101/gad.947102.
Boyko A, Kathiria P, Zemp FJ, Yao Y, Pogribny I, Kovalchuk I: Transgenerational changes in the genome stability and methylation in pathogen-infected plants: (Virus-induced plant genome instability). Nucleic Acids Res. 2007, 35 (5): 1714-1725. 10.1093/nar/gkm029.
Bond DM, Finnegan EJ: Passing the message on: inheritance of epigenetic traits. Trends Plant Sci. 2007, 12 (5): 211-216. 10.1016/j.tplants.2007.03.010.
Boyko A, Kovalchuk I: Epigenetic control of plant stress response. Environ Mol Mutagen. 2008, 49 (1): 61-72. 10.1002/em.20347.
Wang WS, Pan YJ, Zhao XQ, Dwivedi D, Zhu LH, Ali J, Fu BY, Li ZK: Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J Exp Bot. 2011, 62 (6): 1951-1960. 10.1093/jxb/erq391.
Kou HP, Li Y, Song XX, Ou XF, Xing SC, Ma J, Von Wettstein D, Liu B: Heritable alteration in DNA methylation induced by nitrogen-deficiency stress accompanies enhanced tolerance by progenies to the stress in rice (Oryza sativa L.). J Plant Physiol. 2011, 168 (14): 1685-1693. 10.1016/j.jplph.2011.03.017.
Takata M, Kiyohara A, Takasu A, Kishima Y, Ohtsubo H, Sano Y: Rice transposable elements are characterized by various methylation environments in the genome. BMC Genomics. 2007, 8 (1): 469-10.1186/1471-2164-8-469.
Rabinowicz PD, Citek R, Budiman MA, Nunberg A, Bedell JA, Lakey N, O'Shaughnessy AL, Nascimento LU, McCombie WR, Martienssen RA: Differential methylation of genes and repeats in land plants. Genome Res. 2005, 15 (10): 1431-1440. 10.1101/gr.4100405.
Rabinowicz PD, Palmer LE, May BP, Hemann MT, Lowe SW, McCombie WR, Martienssen RA: Genes and transposons are differentially methylated in plants, but not in mammals. Genome Res. 2003, 13 (12): 2658-2664. 10.1101/gr.1784803.
Ou X, Long L, Zhang Y, Xue Y, Liu J, Lin X, Liu B: Spaceflight induces both transient and heritable alterations in DNA methylation and gene expression in rice (Oryza sativa L.). Mutat Res Fund Mol Mech Mutagen. 2009, 662 (1–2): 44-53.
Ngezahayo F, Xu C, Wang H, Jiang L, Pang J, Liu B: Tissue culture-induced transpositional activity of mPing is correlated with cytosine methylation in rice. BMC Plant Biology. 2009, 9 (1): 91-10.1186/1471-2229-9-91.
Wang N, Wang H, Zhang D, Wu Y, Ou X, Liu S, Dong Z, Liu B: Transpositional reactivation of the Dart transposon family in rice lines derived from introgressive hybridization with Zizania latifolia. BMC Plant Biol. 2010, 10: 190-10.1186/1471-2229-10-190.
Lin X, Long L, Shan X, Zhang S, Shen S, Liu B: In planta mobilization of mPing and its putative autonomous element Pong in rice by hydrostatic pressurization. J Exp Bot. 2006, 57 (10): 2313-2323. 10.1093/jxb/erj203.
Shen Y, Lin XY, Shan XH, Lin CJ, Han FP, Pang JS, Liu B: Genomic rearrangement in endogenous long terminal repeat retrotransposons of rice lines introgressed by wild rice (Zizania latifolia Griseb.). J Integr Plant Biol. 2005, 47 (8): 998-1008. 10.1111/j.1744-7909.2005.00103.x.
Vicient CM: Transcriptional activity of transposable elements in maize. BMC Genomics. 2010, 11 (1): 601-10.1186/1471-2164-11-601.
Blumenstiel JP: Evolutionary dynamics of transposable elements in a small RNA world. Trends Genet. 2011, 27 (1): 23-31. 10.1016/j.tig.2010.10.003.
Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, Wessler SR: An active DNA transposon family in rice. Nature. 2003, 421 (6919): 163-167. 10.1038/nature01214.
Raizada MN, Benito MI, Walbot V: The MuDR transposon terminal inverted repeat contains a complex plant promoter directing distinct somatic and germinal programs. Plant J. 2001, 25 (1): 79-91. 10.1046/j.1365-313x.2001.00939.x.
Cantu D, Vanzetti LS, Sumner A, Dubcovsky M, Matvienko M, Distelfeld A, Michelmore RW, Dubcovsky J: Small RNAs, DNA methylation and transposable elements in wheat. BMC Genomics. 2010, 11 (1): 408-10.1186/1471-2164-11-408.
Naito K, Cho E, Yang G, Campbell MA, Yano K, Okumoto Y, Tanisaka T, Wessler SR: Dramatic amplification of a rice transposable element during recent domestication. Proc Natl Acad Sci U S A. 2006, 103 (47): 17620-17625. 10.1073/pnas.0605421103.
Cao X, Jacobsen SE: Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol. 2002, 12 (13): 1138-1144. 10.1016/S0960-9822(02)00925-9.
Tränkenschuh W, Puls F, Christgen M, Albat C, Heim A, Poczkaj J, Fleming P, Kreipe H, Lehmann U: Frequent and distinct aberrations of DNA methylation patterns in fibrolamellar carcinoma of the liver. PLoS ONE. 2010, 5 (10): e13688-10.1371/journal.pone.0013688.
Wang YM, Dong ZY, Zhang ZJ, Lin XY, Shen Y, Zhou D, Liu B: Extensive de novo genomic variation in rice induced by introgression from wild rice (Zizania latifolia Griseb.). Genetics. 2005, 170 (4): 1945-1956. 10.1534/genetics.105.040964.
Allen GC, Flores-Vergara MA, Krasynanski S, Kumar S, Thompson WF: A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat Protoc. 2006, 1 (5): 2320-2325. 10.1038/nprot.2006.384.
Hayatsu H, Shiraishi M, Negishi K: Bisulfite modification for analysis of DNA methylation. Current Protocols in Nucleic Acid Chemistry. 2008, 6 (Suppl 33): 6.10.11-16.10.15.
Casa AM, Brouwer C, Nagel A, Wang L, Zhang Q, Kresovich S, Wessler SR: The MITE family Heartbreaker (Hbr): Molecular markers in maize. Proc Natl Acad Sci U S A. 2000, 97 (18): 10083-10089. 10.1073/pnas.97.18.10083.
Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China (No. 30990243), the Programme for Introducing Talents to Universities (B07017) and a Young Faculty Incubation Project of NENU (No. 09QNJJ21).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
XY and YY carried out major parts of the experiments, analyzed the data and drafted the manuscript. XL, LJ, CZ and XO participated in all the experiments. KO edited the manuscript. BL designed the work and finalized the manuscript. All authors read and approved the final manuscript.
Xuejiao Yang, Yingjie Yu contributed equally to this work.
Electronic supplementary material
12870_2011_1090_MOESM3_ESM.doc
Additional file 3: Statistical analysis of kernel shape and fertility between etoposide-treated plants and their controls in each generation. (DOC 28 KB)
12870_2011_1090_MOESM4_ESM.doc
Additional file 4: The studied probe sequences and primers for probe fragment amplification in gel-blotting analysis. (DOC 38 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yang, X., Yu, Y., Jiang, L. et al. Changes in DNA methylation and transgenerational mobilization of a transposable element (mPing) by the Topoisomerase II inhibitor, Etoposide, in rice. BMC Plant Biol 12, 48 (2012). https://doi.org/10.1186/1471-2229-12-48
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-12-48