
Abstract
Background
Pancreatic cholesterol esterase has three proposed functions in the intestine: 1) to control the bioavailability of cholesterol from dietary cholesterol esters; 2) to contribute to incorporation of cholesterol into mixed micelles; and 3) to aid in transport of free cholesterol to the enterocyte. Inhibitors of cholesterol esterase are anticipated to limit the absorption of dietary cholesterol.
Results
The selective and potent cholesterol esterase inhibitor 6-chloro-3-(1-ethyl-2-cyclohexyl)-2-pyrone (figure 1, structure 1) was administered to hamsters fed a high cholesterol diet supplemented with radiolabeled cholesterol ester. Hamsters were gavage fed 3H-labeled cholesteryl oleate along with inhibitor 1, 0–200 micromoles. Twenty-four hours later, hepatic and serum radioactive cholesterol levels were determined. The ED50 of inhibitor 1 for prevention of the uptake of labeled cholesterol derived from hydrolysis of labeled cholesteryl oleate was 100 micromoles. The toxicity of inhibitor 1 was investigated in a 30 day feeding trial. Inhibitor 1, 100 micromoles or 200 micromoles per day, was added to chow supplemented with 1% cholesterol and 0.5% cholic acid. Clinical chemistry urinalysis and tissue histopathology were obtained. No toxicity differences were noted between control and inhibitor supplemented groups.
Conclusions
Inhibitors of cholesterol esterase may be useful therapeutics for limiting cholesterol absorption.
Similar content being viewed by others
Background
Primary hypercholesterolemia is an established risk factor of atherosclerosis and coronary heart disease (CHD) [1]. Epidemiological data indicate a positive relationship between serum LDL-cholesterol and CHD, which is the leading cause of death in both men and women in the United States. Clinical trials have shown that cholesterol-lowering regimens are beneficial for prevention of CHD morbidity and mortality. A variety of regimens are being used to lower serum cholesterol including diet restriction, nicotinic acid, bile acid sequestering agents, and HMGCoA reductase inhibitors. Reductase inhibitors have become widely used [2]. Although generally well tolerated and effective, side effects have been reported in significant numbers of participants in controlled trials, including increases in serum levels of hepatic transaminases, increases in creatine kinase, muscle weakness, GI disturbances, headache, and sleep disorders. With prolonged use, other side effects have been noted including depression [3], sensorimotor neuropathy [4], and eczema [5]. Alternative therapies are needed, especially for populations that cannot tolerate reductase inhibitors
Dietary cholesterol is comprised of free and esterified cholesterol, the ratio depending upon dietary source. In diets rich in meats, a significant percentage of cholesterol is esterified. Hydrolysis of cholesterol ester in the lumen of the small intestine is catalyzed by cholesterol esterase (CEase), EC3.1.1.13, which liberates free cholesterol. Free cholesterol mixes with cholesterol contained in bile secretions to form the pool of absorbable cholesterol. Due to the low solubility of cholesterol, solubilization of cholesterol by bile salts and lecithin into micelles is essential. In addition, transport proteins are required to deliver cholesterol from micelles to the enterocytes for absorption. CEase provides the hydrolytic activity for hydrolysis of cholesterol ester and may provide the transport function for delivery of cholesterol from micelles to enterocytes [6], although this has not been clearly established [7]. Inhibitors of CEase may provide a method to limit the bioavailability of dietary cholesterol derived from cholesterol esters and may also limit the absorption of free cholesterol.
Recently, the feasibility of limiting the bioavailability of cholesterol derived from cholesterol esters by inhibiting CEase was reported by Bailey, Gallo and coworkers. Intragastric administration of a single dose of 3-benzyl-6-chloro-2-pyrone, 2 (figure 1) to rats simultaneous with feeding of cholesterol ester resulted in a 60% drop in cholesterol absorption, which resulted from a 63% inactivation of lumenal CEase activity [8]. Compound 2 is a prototype haloenol lactone developed by Katzenellenbogen and coworkers as an inhibitor of chymotrypsin, although it is not highly selective [9]. It effectively inhibits or inactivates numerous serine hydrolases, including CEase. However, despite its lack of selectivity, compound 2 did provide the opportunity to demonstrate in an animal study that inhibition of CEase is a new approach to the treatment of hypercholesterolemia

The selective and potent cholesterol esterase inhibitor 6-chloro-3-(1-ethyl-2-cyclohexyl)-2 pyrone (1) and the prototype haloenol lactone, 3-benzyl-6-chloro-2-pyrone (2)
To circumvent the lack of selectivity of 2, we developed 6-chloro-3-(1-ethyl-2-cyclohexyl)-2-pyrone, 1 (figure 1) as a selective and reversible inhibitor of CEase [1]. This involved replacing the aromatic 3-benzyl group in 2 with an aliphatic ring tethered to the 3-position. Inhibition of CEase is very sensitive to the length of the tether. Compound 1 is a potent inhibitor of CEase (Kd = 25 nanomolar) and is highly selective for CEase compared to other serine hydrolases such as chymotrypsin [10]. Therefore, compound 1 can now be considered the prototype for development of selective inhibitors of CEase. In the present study, we carried out an animal model study of the effects of 1 on the absorption of cholesterol derived from cholesteryl oleate to test whether 1 is active in vivo.
Results
Appearance in the serum of free, labeled cholesterol derived from intestinal hydrolysis of 100 micromoles 3H-cholesteryl oleate was followed over a 24 hour period. Absorption occurred in a time-dependent fashion, as shown in figure 2. The inhibition of uptake of labeled cholesterol by 100 micromoles of inhibitor 1 was significant within 6 hours (p < 0.001). Inhibition of uptake was maximum by 12 hours, and this inhibition was maintained throughout the 24 hour observation period, during which time absorption continued in the control animals. Hepatic levels of labeled cholesterol were also determined. Hepatic uptake of labeled cholesterol derived from hydrolysis of 3H-cholesteryl oleate was apparent within 6 hours, as shown in figure 3, and was inhibited by administration of 100 micromoles of inhibitor 1 (p < 0.002). Thus, inhibitor 1 is effective at preventing the hydrolysis of cholesteryl oleate, thereby limiting the bioavailability of cholesterol derived from cholesterol esters.

Inhibition of the absorption of cholesterol derived from intestinal hydrolysis of 3H-cholesteryl oleate was significant within 6 hours (p < 0.001) in serum in hamsters treated with 100 umole of inhibitor 1. The inhibition continued throughout the 24 hour observation period (p < 0.0002 at 24 hours)

Inhibition of hepatic uptake of cholesterol derived from 3H-cholesteryl-oleate in hamsters was also significant within 6 hours (p < 0.002).
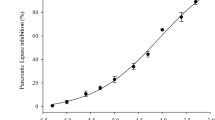
The dose-dependence of inhibitor 1 on the uptake of labeled cholesterol from hydrolysis of 3H-cholesteryl oleate was determined. Appearance of cholesterol in serum and liver was prevented by inhibitor 1 in a dose-dependent manner, as shown in figures 4a and 4b. Inhibitor 1 administered at dosages between 20–200 micromoles resulted in an approximate ED50 of 100 micromoles when assessed as a reduction in cholesterol appearance in both liver and serum.

Dose response curves of the effects of inhibitor 1 on hepatic and serum absorption of cholesterol derived from hydrolysis of cholesteryl oleate indicated an approximate ED50 = 100 umoles.
To investigate the toxic effects of 1, the inhibitor was administered to animals for 30 days. Animals treated with inhibitor 1 at 100 and 200 micromoles per day were compared to controls through clinical chemistries, necropsy and histopathology. There were no differences between treated and control animals based upon clinical chemistries (liver enzymes and proteins, renal function indices, and electrolytes). No lesions were noted on necropsy. In addition, there were no noted differences between treated and control animals in the histopathology exam (heart, lung, liver, pancreas, GI tract, thyroid, lymph nodes).
Discussion
There may be additional physiological roles of pancreatic cholesterol esterase
CEase is also synthesized in lactating mammary glands and is secreted into milk in a number of species including human. Pancreatic and milk CEase are derived from the same gene [11]. It appears that the same enzyme is present in serum [12] and is synthesized by human macrophages [13], eosinophils [14], endothelial cells [15], and liver [16]. The physiological roles of CEase that is present in these various locations are not clear, nor is it known whether inhibitors designed to block hydrolysis of dietary cholesterol esters through inhibition or inactivation of CEase in the intestine will have additional biological effects. It is noteworthy that the lipid-lowering effect of WAY-121,898, a novel carbamate inhibitor of CEase, was observed both by oral and by parenteral administration of the drug [17]. This apparent broad distribution of CEase expression, however, has been questioned in a recent report of tissue levels of CEase mRNA in which only pancreatic and intestinal tissues were positive [18].
Cholesterol esterase has a major physiological role in cholesterol ester hydrolysis
The major physiological role of CEase in determining the bioavailability of dietary cholesterol esters is supported by in vivo studies of CEase inhibitors [8, 17, 19] and by studies with CEase gene knockout mice where impaired absorption of cholesterol ester was observed [7]. Gene knockout mice, however, did not exhibit impaired absorption of unesterified cholesterol which suggests that CEase does not have a direct cholesterol transport function. This conclusion differs from the results of a number of studies with rats in which unesterified cholesterol absorption was decreased in animals made CEase deficient by removing pancreatic juice [20] or by treating pancreatic juice with antibody to CEase [21]. CEase increased the absorption of free cholesterol into cultured Caco-2 cells [22] and into gut sacs [23], further suggesting a transport role for CEase. Other studies, however, did not support a role for CEase in transport of unesterified cholesterol to enterocytes; uptake of cholesterol by Caco-2 cells has been reported to be independent of added CEase or of transfection with CEase cDNA [24, 25]. These conflicting reports regarding a transport function for CEase may reflect structural differences in the various CEase used in these studies. The C-termini of CEase possess a consensus proline-rich eleven-amino acid repeat sequence (PVPPTGDSGAP)n, which differs among species in the number of repeats, followed by an extreme C-terminus conserved nine-amino acid consensus sequence (AQMPVVIGF). The number of repeats varies from 2–4 in most animals to 16 in human. These proline-rich repeats allow CEase to bind to membrane-associated heparin, and it has been suggested that this may facilitate cholesterol transport [26]. However, a recent clinical trial with CVT-1 (sodium cellulose sulfate), a potent and specific inhibitor of human CEase that binds to the heparin binding site on CEase, did not block absorption of unesterified cholesterol [27], which suggests that CEase does not have a role in transport of free cholesterol in humans.
Cholesterol esterase appears to have an indirect but essential role in absorption of free cholesterol
Recently, a study was reported [28] that may resolve these conflicting suggestions of a transport role for CEase. It is known that CEase can also hydrolyze phospholipids [28], a role normally assigned to pancreatic phospholipase A2 (PLA2) [29]. Biliary secretions include hepatic micelles consisting of bile salts, cholesterol and lecithin (phosphatidyl choline). This bile mixes with dietary lipid in the proximal part of the small intestine where pancreatic enzymes including CEase participate. The digestion of dietary lipid and the formation of intestinal mixed micelles containing free cholesterol, biles salts, lecithin and fatty acids are highly dependent on the emulsifying and detergent properties of the hepatocyte-derived lecithin and bile salts. Several studies indicate that the lecithin-rich micelles formed in the intestine are not efficient transporters of cholesterol to the enterocyte unless the lecithin is hydrolyzed to lysolecithin, a reaction presumed to be catalyzed mainly by PLA2. The recent study that addressed the conflicting reports concerning a transport role for CEase indicated that CEase also has a major role in the hydrolysis of lecithin [28]. Lymphatic recovery of free cholesterol from cannulated rat duodenum as a lecithin-containing micelle was enhanced by co-administration of CEase in rats drained of bile and pancreatic juice. In addition, cholesterol uptake into Caco-2 cells (an enterocyte model) from these same micelles was accelerated by CEase in a dose-dependent manner that depended upon the catalytic activity of CEase. The acceleration disappeared when CEase was inactivated, and the acceleration in cholesterol uptake by these cells in response to CEase occurred only when the micelles contained lecithin. If the lecithin in the micelles was replaced with lysolecithin, cholesterol uptake was efficient and did not depend upon CEase or PLA2. This suggests a new model for cholesterol absorption (figure 5) with a dual role for CEase. In this model, CEase does not have a direct transport role in the absorption of free cholesterol but rather has an indirect role by virtue of its participation with PLA2 in the formation of lysolecithin-containing micelles. In this model, therefore, inhibitors of the catalytic activity of CEase, such as inhibitor 1, would be expected to limit the availability of free cholesterol derived from hydrolysis of dietary cholesterol esters and limit the uptake of free cholesterol by inhibiting the CEase-catalyzed hydrolysis of lecithin, thereby limiting the formation of lysolecithin-containing micelles that are the vehicles for efficient delivery of cholesterol to the enterocytes.

Inhibitors of pancreatic cholesterol esterase have the potential to modify cholesterol metabolism at multiple sites. In this model, cholesterol esterase is proposed to contribute to the availability of free cholesterol by catalyzing the hydrolysis of cholesterol esters and contribute to the formation of lysolecithin-containing micelles, which are the vehicles for efficient absorption of cholesterol, by catalyzing the hydrolysis of lecithin.
A number of different types of inhibitors of cholesterol esterase have been developed
Numerous types of chemicals have been studied as either mechanism-based or reversible inhibitors of CEase, including boronic and borinic acids, arylhaloketones, aryl phosphates and phosphonates, and aryl and cholesteryl carbamates [30, 31]. Application of carbamate chemistry has been especially useful for development of structure-activity relationships (SAR). Carbamates are transient inhibitors (pseudosubstrate inhibitors) of CEase in which the acylation (carbamoylation) step is followed by a slower deacylation step. SAR can be developed for the individual steps of binding, acylation and deacylation. It has been suggested from studies of carbamates that bile salts increase the hydrophobicity of both the fatty acid and cholesterol binding sites of CEase [32]. 6-Chloro-2-pyrones, which were developed as inhibitors of serine proteases, have the potential (figure 6) to function as 1) substrates; 2) pseudosubstrate inhibitors; 3) suicide inhibitors; 4) reversible inhibitors; or 5) inactivators. Initial studies of the inhibition of chymotrypsin by 3-benzyl-6-chloro-2-pyrone were interpreted as demonstrating suicide inhibition [33]. Subsequent studies indicated that this pyrone is a pseudosubstrate inhibitor in which acylation is much faster than deacylation such that the acylenzyme accumulates and rearranges to the acid chloride, which hydrolyzes while tethered to the active site serine, followed by slow hydrolysis and release of product [34]. This interpretation was supported by crystallographic analysis of the acylenzyme [35]. By comparison, treatment of chymotrypsin with 5-benzyl-6-chloro-2-pyrone led to inactivation, in which the active site serine was covalently attached to the 6-position of an intact pyrone, with loss of chloride. Other related studies of inhibition of serine proteases by haloenol lactones such as 3-chloroisocoumarins and halomethylene furanones have demonstrated mechanism-based (suicide) inactivation [36, 37]. Inhibitor 1 was shown to be a reversible inhibitor of CEase [10].

Substituted 6-chloro-2-pyrones can function as (1) pseudosubstrates of serine hydrolases in which the acylenzyme intermeditate B accumulates; (2) suicide inhibitors in which formation of B is followed by attack of an active site nucleophile to form C and deacyclation to form D; (3) simple substrates in which the reactive acylchloride B hydrolyzes to acylenzyme E followed by deacylation to regenerate active enzyme F with liberation of the substituted glutaconic acid product. Alternatively, substituted 6-chloro-2-pyrones can be simple reversible inhibitors or irreversible active site inhibitors where an active site nucleophile attacks the 6-postion of the pyrone with release of chloride.
Conclusions
In summary, inhibitor 1 was demonstrated to limit the bioavailability of cholesterol derived from hydrolysis of cholesterol ester. Inhibitor 1 can be considered a prototype of selective inhibitors of CEase that appear to have low toxicity in vivo. It remains to be demonstrated whether inhibition of CEase may also limit the absorption of free cholesterol.
Methods
Animals and diets
All animals were housed and treated in compliance with the NIH's Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act. Outbred male Golden Syrian hamsters, 100–125 gm (Charles River Labs.), were housed individually. Animals were acclimated to a light cycle of 12 hours light, 12 hours dark, for one week prior to the beginning of any experiment and randomly assigned to treatment and control groups. Unless indicated, all animals in the study were fed a custom prepared rodent diet, AIN76 (ICN Biologicals, Aurora, Ohio) and water ad libitum. Treatment protocols consisted of feeding a custom rodent diet containing unlabeled free cholesterol and cholic acid to which was added cholesteryl oleate and varying amounts of inhibitor 1. Diets were provided daily in predetermined amounts calculated to maintain normal growth. The basal composition of the semipurified hamster diet contained: casein, 20%; DL-methionine, 0.3%; sucrose, 50%; corn starch, 15%; Alphacel, 5%; corn oil, 5%; choline bitartrate, 0.2%; mineral mix (AIN), 3.5%; vitamin mix (AIN) 1%.
Acute cholesterol absorption measurements
Cholesterol absorption was assessed using a modification of the method reported by Harwood et al. [38]. Male Golden Syrian hamsters were fed the custom diet containing 1% cholesterol and 0.5% cholic acid for three days and then fasted overnight. The following morning hamsters were gavage fed inhibitor 1 at various doses of 0, 20, 40, 80, 120, 160 and 200 micromoles (corresponding to 0 to 48 mg of inhibitor 1), respectively, suspended in a 10% aqueous ethanol solution containing 0.25% methylcellulose and 0.6% Tween-80 as suspending agents. Dosing was 0.5 ml/100 g of body weight. Immediately thereafter, hamsters received an oral bolus, 1.5 ml/hamster, of vegetable oil containing 3H-labeled cholesteryl oleate (1 μCi), and unlabeled cholesteryl oleate (15 mg). The degree of cholesterol absorption was assessed by the serum and hepatic levels of 3H-cholesterol measured over the next 24 hours. Hamsters were anesthetized with isoflourane and blood was obtained by cardiac puncture for determination of serum cholesterol. Animals were euthanized, while anesthetized, by exsanguination. At necropsy, organs including liver, gall bladder, spleen, kidneys, heart, lungs, stomach and intestines, testicles, and the carcass were weighed. Livers were excised and saponified in 5 ml of 2.5 M KOH for 2 hours at 70°C. Aliquots (0.2 mL) of the saponified livers were decolorized with H2O2 (0.2 mL of 30%), neutralized with 3 N HCl (0.2 mL), and then counted in a liquid scintillation counter. Total hepatic radioactivity was calculated from the radioactivity in each aliquot after adjusting for the total volume of the saponified liver. The percent inhibition of cholesterol absorption was determined by comparing the hepatic radioactivity in hamsters receiving inhibitor and hamsters receiving only the vehicle.
Thirty day toxicity study of inhibitor 1
Toxicity data were obtained by monitoring blood chemistry values and urine over a 30 day period, using a modified method reported by DeNinno et al [39]. Male Golden Syrian hamsters were acclimated for 1 week to eating powdered custom diet. Weights of the animals and food consumption were monitored daily. Three groups of hamsters were fed the powdered custom diet containing 1% cholesterol and 0.5% cholic acid. The control group (n = 6) ate only custom diet for 30 days. One group of 6 hamsters was given the optimum dose of inhibitor (100 micromoles) added to the powdered diet daily for 30 days. The third group received the powdered diet with 200 micromoles of inhibitor for 30 days. Blood and urine samples were collected every 5 days by chamber induction of Isoflurane anesthesia, cardiac puncture, and cystocentesis. At 30 days the hamsters were anesthetized by chamber induction and euthanized by exsanguination. At necropsy, organs including liver, gall bladder, spleen, kidneys, heart, lungs, stomach and were weighed. A complete necropsy with histopathology of all organs was carried out. Saponification of liver tissue was carried out as described as above. Tubes were mixed and placed at -20°C for 15 min to enhance precipitation of protein before addition of 4 ml of hexane. After vigorous vortexing, samples were centrifuged at 1500 g for 5 min to separate phases; 3 ml of the hexane phase was evaporated to dryness under N2 before being redissolved in 0.3 mL of 1% (v/v) Triton X-100 in ethanol. Cholesterol levels were analyzed using a commercial assay kit. Triplicate 15 uL aliquots were analyzed in 96-well microtiter plates; 0.2 mL of reagent was added and the plates incubated at room temperature for 1 hr. Absorbance was read at 490 nm using a microtiter spectrophotometer and compared to a standard curve to calculate cholesterol mass.
References
Gordon T, Kannel WB, Castelli WP, Dawber TR: Lipoproteins, cardiovascular disease, and death: the Framingham Study. Arch Intern Med. 1981, 141: 1128-1131. 10.1001/archinte.141.9.1128.
Hsu I, Spinler SA, Johnson NE: Comparative evaluation of the safety and efficacy of HMG-CoA reductase inhibitor monotherapy in the treatment of primary hypercholesterolemia. Ann Pharmacotherapy. 1995, 29: 743-759.
Boumendil E, Tubert-Bitter P: Depression-induced absenteeism in relation to antihyperlipidemic treatment: a study using GAZEL cohort data. Epidemiology. 1995, 6: 322-325.
Phan T, McLeod JG, Pollard JD, Peiris O, Rohan A, Halpern JP: Peripheral neuropathy associated with simvastatin. J Neurology Neurosurgery & Psychiatry. 1995, 58: 625-628.
Proksch E: Antilipemic drug-induced skin manifestations. Hautartz. 1995, 46: 76-80. 10.1007/s001050050213.
Myers-Payne SC, Hui DY, Brockman HL, Schroeder F: Cholesterol esterase: a cholesterol transfer protein. Biochem. 1995, 34: 3942-3947.
Howles PN, Carter CP, Hui DY: Dietary free and esterified cholesterol absorption in cholesterol esterase (bile salt-stimulated lipase) gene-targeted mice. J Biol Chem. 1996, 271: 7196-7202. 10.1074/jbc.271.12.7196.
Bailey JM, Gallo L, Gillespie J: Inhibition of dietary cholesterol ester absorption by 3-BCP, a suicide inhibitor of cholesterol-esterase. Biochem Soc Trans. 1995, 23: 408S-
Daniels SB, Cooney E, Sofia MJ, Chakravarty PK, Katzenellenbogen JA: Haloenol lactones: potent enzyme-activated irreversible inhibitors for a-chymotrypsin. J Biol Chem. 1983, 250:
Deck LM, Baca ML, Salas SL, Hunsaker LA, Vander Jagt DL: 3-Alkyl-6-chloro-2-pyrones: Selective inhibitors of pancreatic cholesterol esterase. J Med Chem. 1999, 42: 4250-4256. 10.1021/jm990309x.
Nilsson J, Blackberg L, Carlsson P, Enerback S, Hernell O, Bjursell G: cDNA cloning of human-milk bile-salt-stimulated lipase and evidence for its identity to pancreatic carboxylic ester hydrolase. Eur J Biochem. 1990, 192: 543-550.
Brodt-Eppley J, White P, Jenkins S, Hui DY: Plasma cholesterol esterase level is a determinant for an atherogenic lipoprotein profile in normolipidemic human subjects. Biochim Biophys Acta. 1995, 1272: 69-72. 10.1016/0925-4439(95)00083-G.
Li F, Hui DY: Modified low density lipoprotein enhances the secretion of bile salt-stimulated cholesterol esterase by human monocyte-macrophages: species-specific differences in macrophage cholesteryl ester hydrolase. J Biol Chem. 1997, 272: 28666-28671. 10.1074/jbc.272.45.28666.
Holtsberg FW, Ozgur LE, Garsetti DE, Myers J, Egan RW, Clark MA: Presence in human eosinophils of a lysophospholipase similar to that found in the pancreas. Biochem J. 1995, 309: 141-144.
Li F, Hui DY: Synthesis and secretion of the pancreatic-type carboxyl ester lipase by human endothelial cells. Biochem J. 1998, 329: 675-679.
Winkler KE, Harrison EH, Marsh JB, Glick JM, Ross AC: Characterization of a bile salt-dependent cholesteryl ester hydrolase activity secreted from HepG2 cells. Biochim Biophys Acta. 1992, 1126: 151-158.
Krause BR, Sliskovic DR, Anderson M, Homan R: Lipid-lowering effects of WAY-121,898, an inhibitor of pancreatic cholesteryl ester hydrolase. Lipids. 1998, 33: 489-498.
Lindquist S, Blackberg L, Hernell O: Distribution of the bile-salt-stimulated lipase in human tissues. FASEB. 2001, 15: A193-
Jeon SM, Kim HS, Lee TG, Ryu SH, Suh PG, Byun SJ, Park YB, Choi MS: Lower absorption of cholesteryl oleate in rats supplemented with areca catechu L. extract. Ann Nutr Metab. 2000, 44: 170-176. 10.1159/000012841.
Borja CR, Vahouny GV, Treadwell CR: Role of bile and pancreatic juice in cholesterol absorption and esterification. Am J Physiol. 1964, 206: 223-228.
Gallo LL, Clark SB, Myers S, Vahouny GV: Cholesterol absorption in rat intestine: Role of cholesterol esterase and acyl coenzyme A:cholesterol acyltransferase. J Lipid Res. 1984, 25: 604-612.
Lopez-Candales A, Bosner MS, Spilburg CA, Lange LG: Cholesterol transport function of pancreatic cholesterol esterase: Directed sterol uptake and esterification in enterocytes. Biochemistry. 1993, 32: 12085-12089.
Bhat SG, Brockman HL: The role of cholesteryl ester hydrolysis and synthesis in cholesterol transport across rat intestinal mucosal membrane. A new concept. Biochem Biophys Res Comm. 1982, 109: 486-492.
Huang Y, Hui DY: Metabolic fate of pancreas-derived cholesterol esterase in intestine: An in vivo study using Caco-2 cells. J Lipid Re. 1990, 31: 2029-2037.
Shamir R, Johnson WJ, Zolfaghari R, Lee HS, Fisher EA: Role of bile salt-dependent cholesteryl ester hydrolase in the uptake of micellar cholesterol by intestinal cells. Biochemistry. 1995, 34: 6351-6358.
Spilburg CA, Cox DG, Wang X, Bernat BA, Bosner MS, Lange LG: Identification of a species specific regulatory site in human pancreatic cholesterol esterase. Biochemistry. 1995, 34: 3942-3947.
Bosner MS, Wolff AA, Ostlund RE: Lack of effect of cholesterol esterase inhibitor CVT-1 on cholesterol absorption and LDL cholesterol in humans. Cardiovasc Drugs Ther. 1999, 13: 449-454. 10.1023/A:1007864208932.
Ikeda I, Matsuoka R, Hamada T, et al: Cholesterol esterase accelerates intestinal cholesterol absorption. Biochim Biophys Acta. 2002, 1571: 34-44. 10.1016/S0304-4165(02)00204-0.
Homan R, Hamelehle KL: Phospholipase A2 relieves phosphatidylcholine inhibition of micellar cholesterol absorption and transportby human intestinal cell line Caco-2. J Lipid Res. 1998, 39: 1197-1209.
Feaster SR, Quinn DM: Mechanism-based inhibitors of mammalian cholesterol esterase. Methods Enzymol. 1997, 286: 231-252. 10.1016/S0076-6879(97)86013-2.
Feaster SR, Lee K, Baker N, Hui DY, Quinn DM: Molecular recognition by cholesterol esterase of active site ligands: structure-activity effects for inhibition by aryl carbamates and subsequent carbamylenzyme turnover. Biochemistry. 1996, 35: 16723-16734. 10.1021/bi961677v.
Lin G, Shieh C-T, Ho H-C, Chouhwang J-Y, Lin W-Y, Lu C-P: Structure-reactivity relationships for the inhibition mechanism at the second alkyl-chain-binding site of cholesterol esterase and lipase. Biochemistry. 1999, 38: 9971-9981. 10.1021/bi982775e.
Gelb MH, Abeles RH: Mechanism of inactivation of chymotrypsin by 3-benzyl-6-chloro-2-pyrone. Biochemistry. 1984, 23: 6596-6604.
Ringe D, Mottonen JM, Gelb MH, Abeles RH: X-ray diffraction analysis of the inactivation of chymotrypsin by 3-benzyl-6-chloro-2-pyrone. Biochemistry. 1986, 25: 5633-5638.
Ringe D, Seaton BA, Gelb MH, Abeles RH: Inactivation of chymotrypsin by 5-benzyl-6-chloro-2-pyrones: 13C NMR and X-ray diffraction analysis of the inactivator-enzyme complex. Biochemistry. 1985, 24: 64-68.
Powers JC, Kam CM: Isocoumarin inhibitors of serine peptidases. Methods Enzymol. 1994, 244: 442-457. 10.1016/0076-6879(94)44033-6.
Daniels SB, Katzenellenbogen JA: Haloenol-lactones: studies on the mechanism of inactivation of alpha-chymotrypsin. Biochemistry. 1986, 25: 1436-1444.
Harwood HJ, Chandler CE, Pellarin LD, Bangerter FW, Wilkins RW, Long CA, Cosgrove PC, Malinow MR, Marzetta CA, Pettini JL, Savoy YE, Mayne JT: Pharmocological consequencess of cholesterol absorption inhibition: alteration in cholesterol metabolism and reduction in plasma cholesterol concentration induced by the synthetic saponin B-tigogenin cellbioside (CP-88,818 tiqueside). J Lipid Res. 1993, 34: 377-395.
DeNinno MP, McCarthy PA, Duplantier KC, Eller C, Etienne JB, Zawistoski MP, Bangerter FW, Chandler CE, Morehouse LA, Sugarman ED, Wilkins RW, Woody HA, Zaccaro LM: Steroidal Glycoside Cholesterol Absorption Inhibitors. J Med Chem. 1997, 40: 2547-2555. 10.1021/jm9702600.
Acknowledgements
Supported by SBIR grant DK58440 and NIH grant HL68598
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
JEH and LMC carried out the animal studies. LAH conducted the laboratory measurements. LMD provided the inhibitor and helped conceive the study. DLVJ conceived the study and wrote the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Heidrich, J.E., Contos, L.M., Hunsaker, L.A. et al. Inhibition of pancreatic cholesterol esterase reduces cholesterol absorption in the hamster. BMC Pharmacol 4, 5 (2004). https://doi.org/10.1186/1471-2210-4-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2210-4-5