
Abstract
The peptides angiotensin IV and LVV-hemorphin 7 were found to enhance memory in a number of memory tasks and reverse the performance deficits in animals with experimentally induced memory loss. These peptides bound specifically to the enzyme insulin-regulated aminopeptidase (IRAP), which is proposed to be the site in the brain that mediates the memory effects of these peptides. However, the mechanism of action is still unknown but may involve inhibition of the aminopeptidase activity of IRAP, since both angiotensin IV and LVV-hemorphin 7 are competitive inhibitors of the enzyme. IRAP also has another functional domain that is thought to regulate the trafficking of the insulin-responsive glucose transporter GLUT4, thereby influencing glucose uptake into cells. Although the exact mechanism by which the peptides enhance memory is yet to be elucidated, IRAP still represents a promising target for the development of a new class of cognitive enhancing agents.
Similar content being viewed by others
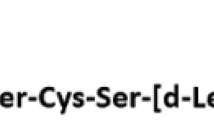


Background
The incidence of age-related neurological diseases is escalating due primarily to the increased life expectancy in the general population of many developed nations. One of the more prevalent and debilitating neurological disorders is Alzheimer's disease (AD). The widely accepted 'amyloid hypothesis' of AD has resulted in drug development strategies focused on the modulation of the amyloid-β (Aβ) processing enzymes (β and γ secretases) and prevention of Aβ aggregation or oligomerization (vaccine for Aβ) [1]. The role of hyperphosphorylated tau protein has recently gained prominence and some new intervention therapies have focussed on the 'responsible' kinases, including glycogen synthase kinase 3β and cyclin-dependent kinase [1]. The quandary with these disease-modifying approaches is that the etiology of AD is still not well understood. Although the disease is characterized by the deposition of amyloid plaques, and neurofibrillary tangles, it is not known if these pathological hallmarks play causative, in addition to indicative, roles.
Currently, all drugs approved by the Food and Drug Administration (FDA) for AD address its symptoms. Most belong to the class of cholinesterase inhibitors, are of limited efficacy [2], and are indicated for the treatment of mild-to-moderate forms of the disease [3]. In spite of this, many drugs currently being developed to treat cognitive decline in AD are still targeting central cholinergic systems http://www.alzforum.org/drg/drc. These include the new generation cholinesterase inhibitors, cholinergic receptor agonists, and drugs that facilitate cholinergic transmission. The exception is the NMDA receptor antagonist memantine, which works to prevent excitotoxicity and cell death, and is the only medication that has been approved in the European Union, Australia, and by the FDA, for the treatment of moderate-to-severe AD. It is currently not approved for the treatment of early AD, as its efficacy has not as yet been substantiated for mild-to-moderate AD [4]. More innovative approaches are required in the development of symptomatic treatments. This has recently been realised in the form of ampakines and modulators of the CREB pathway [5]. Development of memory-enhancing drugs is gaining momentum because of their increasingly widespread application in the treatment of other forms of memory disorders, including mild cognitive impairment, as well as that resulting from brain trauma and ischemic damage.
Rationale for proposing that insulin-regulated aminopeptidase is a novel target for the development of cognitive enhancers
Our discovery that peptide inhibitors of insulin-regulated aminopeptidase (IRAP) elicit significant effects on memory acquisition and retrieval provides the basis for the proposition that IRAP is a novel target for the discovery of cognitive enhancers. Central administration of the two peptides angiotensin (Ang) IV (Ang IV) or LVV-hemorphin 7 (LVV-H7) results in facilitation of memory, as demonstrated in the conditioned and passive avoidance paradigm [6–8], and enhanced performance in the spatial memory tasks, as in the swim and Barnes mazes [9, 10]. More importantly, these peptides reverse performance deficits induced by global ischemia [11], bilateral perforant pathway lesion [9], perturbations of central cholinergic systems [12–15], and chronic alcohol exposure [16].
At the cellular level, Ang IV has been shown to facilitate long-term potentiation in the dentate gyrus of rats in vivo [17] and in the CA1 region of the hippocampus in vitro [18]. In view of the fact that long-term potentiation is considered to be a cellular marker for memory formation, these findings provide further evidence that Ang IV does indeed play a role in memory processing.
In 2001, the specific target for Ang IV and LVV-H7 was identified – these peptides bind specifically, and with high affinity, to the transmembrane aminopeptidase IRAP [19]. Furthermore, Ang IV and LVV-H7 were found to be competitive inhibitors of IRAP, inhibiting its aminopeptidase activity by binding to the catalytic site [20, 21]. We therefore propose that these peptides mediate their effects on memory by binding to IRAP in specific brain nuclei. In support of this hypothesis, IRAP is found in high concentrations in areas of the brain that are important for memory processing, including the hippocampus, amygdala, and prefrontal cortex [22–24].
Target validation – characterization of the IRAP knockout mouse
Studies on the global IRAP knockout mouse revealed an important proof-of-concept: that IRAP is the specific binding site in the brain for the peptide IRAP inhibitors, Ang IV and LVV-H7. In the absence of IRAP expression, there is a complete loss of the Ang IV binding site in the brain. Preliminary studies demonstrated that the IRAP knockout mice performed better than their wild-type littermates in the swim maze [25]. However improvements in performances were not detected with other memory paradigms including the novel object recognition and the spontaneous alternation T maze. In fact, an age-related deficit was detected in the 6-month-old IRAP knockout mice in the Y maze paradigm (Albiston, personal communication).
Although the mechanism via which Ang IV and LVV-H7, by binding to IRAP, facilitate memory acquisition and retrieval is not fully understood, it is irrefutable that these peptides enhance memory and are specific, high affinity inhibitors of IRAP. There is now sufficient evidence to support the use of IRAP as a target for the identification of a new class of cognitive enhancers.
Intracellular 'trafficking' domain of IRAP
IRAP was first identified as a protein that accompanies the insulin-responsive glucose transporter GLUT4 within specialized vesicular compartments of fat and muscle cells [26]. The movement of these GLUT4/IRAP-containing vesicles to the plasma membrane is under the tight control of insulin, which facilitates an up to ten-fold increase in the uptake of glucose into the cell [27]. IRAP is the only transmembrane enzyme of the M1 family of aminopeptidases that contains a large intracellular domain. In the specialized GLUT4 vesicles, this 109 amino acid amino-terminal tail of IRAP projects into the cytoplasm (Figure 1). This intracellular domain contains two dileucine motifs preceded by acidic regions, which are thought to play important roles in vesicular trafficking. Injection of this IRAP domain into fat cells results in the translocation of GLUT4 vesicles to the plasma membrane [28]. Moreover, this domain has been shown to interact with several cytoplasmic proteins, including AS160 [29, 30], tankyrase [31], acyl-coenzyme A dehydrogenase [32], FHOS, the 'so called' formin homolog overexpressed in spleen [33], and p115 [29, 34]; some of these proteins are associated with intracellular protein transport machinery. It is therefore possible that the amino-terminal cytoplasmic tail of IRAP plays a significant role in the retention or trafficking of GLUT4-containing vesicles in insulin-responsive cells.

Predicted domain structure of insulin-regulated aminopeptidase with key features highlighted.
Extracellular catalytic domain of IRAP
IRAP is a type II membrane protein of 160 kDa and belongs to the gluzincin family of aminopeptidases [26] that includes aminopeptidase A [35], aminopeptidase N [36], and leukotriene A4 hydrolase [37]. The large carboxy-terminal domain of IRAP contains the proteolytic active site characteristic of the M1 aminopeptidase family consisting of the Zn2+ binding motif, HEXXH-(18X)-E, and an exopeptidase motif, GAMEN (Figure 1). Mutational analysis of the active site of IRAP revealed classic characteristics of an aminopeptidase with single residue mutations of the Zn2+ binding motif resulting in complete loss of activity [38]. Rudberg et al. [39] proposed in 2002 that the GAMEN sequence is a consensus motif for an amino-terminal recognition site in zinc aminopeptidases with the glutamate (E) residue acting as an anionic binding site for substrates. Mutational studies reveal that the G428, A429 and N432 residues in IRAP are important for binding of both peptide substrates and inhibitors [40].
In many cell types and under basal conditions, IRAP occurs predominantly in vesicles resembling large, dense core vesicles, where the intraluminal location of its catalytic domain can facilitate the processing of precursor peptide sequences. Upon stimulation by insulin in fat and muscle cells [41], IRAP translocates to the plasma membrane, presenting its catalytic domain to the extracellular surface.
IRAP had also been cloned from a human placental cDNA library (as oxytocinase), based on the capacity of IRAP to readily cleave oxytocin in vitro [42]. The plasma level of IRAP is increased during the later stages of pregnancy and it is thought that this enzyme is involved in the regulation of circulating oxytocin to prevent the onset of premature labour [43]. Interestingly, IRAP cleaves the first three residues from the amino terminus of a structurally related peptide hormone, arginine-vasopressin, more efficiently than oxytocin. There is current debate as to which peptide is the more relevant, endogenous substrate of IRAP. Findings from the IRAP knockout mice favour vasopressin [44], whereas in the female reproductive system, oxytocin appears more physiologically relevant [42]. Other in vitro peptide substrates include somatostatin, lys-bradykinin, met-enkephalin, dynorphin A, neurokinin A, neuromedin B, and cholecystokinin 1–8 [21, 45, 46].
Development of inhibitors of IRAP as cognitive enhancers
Although IRAP was cloned more than a decade ago, the first and only specific, high affinity inhibitors of IRAP that are used to investigate the physiological roles of the enzyme are the peptides Ang IV and LVV-H7. However, these peptides are not the ideal pharmacological tools, in particular Ang IV, which is unstable in the circulation, having a half-life of only seconds. In spite of their robust memory-enhancing properties in rodents, these peptides are unlikely to be useful for human therapy unless the problems of oral bioavailability, stability and blood brain barrier permeation can be overcome. Some attempts have been made to design small molecule peptidomimetic compounds based predominantly on the structure of Ang IV (see below).
Peptide inhibitors of IRAP
The two archetypal competitive inhibitors of IRAP are the peptides Ang IV (VYIHPF) and LVV-H7 (LVVYPWTQRF), with Ki values of 113 nM and 845 nM, respectively [21]. The majority of structure-activity data has focussed on sequence truncation or residue replacement of those two peptides. The literature surrounding the activity of Ang IV analogues, with respect to building structure-activity relationships, is somewhat confused by the historic controversy over the macromolecular target. There are separate but overlapping collections of data that assess either competition binding to membrane fractions, or inhibition of IRAP catalytic activity; there is clear evidence that these two measures do not correlate. The significant factor appears to be the presence (or absence) of zinc at the catalytic site in the different assays, which contributes to substrate and inhibitor affinity. The presence of metal ions in tissue preparations can also confound binding data due to the degradation of the test peptides by metalloproteases. That said, there does appear to be some overlap in the general descriptors of binding and enzyme inhibition that might be utilized in future design. Suffice it to say, compounds unable to bind IRAP fail to inhibit its activity.
Design of inhibitors of IRAP based on the known peptide inhibitors
The available data relating to inhibition of IRAP (summarized in Table 1) point to a primary pharmacophore centred on the VYI/VYP motifs of the parent peptides. Modulation of affinity is achieved by additional substituents, particularly on the carboxy-terminal side of the tripeptide [47–49] (Figure 2). Several groups have looked at structural modification by variation of the peptide amide backbone. Divalinal-Ang IV was initially described as an AT4-receptor antagonist, but displays an IRAP inhibitory property, albeit with lower affinity to Ang IV. A group of non-peptide tyrosine derivatives (Figure 2) display structural similarities to the Ang IV tripeptide, and have been described in the patent literature as potent AT4 receptor agonists [50]. Unfortunately, no data are available describing IRAP inhibition of these compounds. One potential outcome of these structural changes is stabilization against peptidase activity, an important criterion in developing effective therapeutics. Introduction of modified amino acids can also achieve this goal, and replacement of the amino-terminal valine with β2-homovaline, reportedly stabilizes the analogue relative to the parent while maintaining inhibitory potency [51].

Chemical structures of angiotensin IV and reported peptidomimetic analogues.
Little has been attempted to date with respect to learning the conformational requirements for enzyme inhibitory activity of either Ang IV or LVV-H7. Fledgling attempts have been described that utilize peptide cyclization to stabilize specific conformational populations of Ang IV, and the results would appear quite encouraging [49]. For example, the use of cystine or homocystine disulfide linkages as replacements for pairs of residues at both the amino- and carboxy-terminal regions of Ang IV, have yielded analogues that compete in binding, and in the latter case, inhibit enzyme activity. The use of conformational constraint, particularly around the key tripeptide motif, would seem a potentially fruitful avenue to improving potency, reducing susceptibility to other peptidases, and possibly facilitating central nervous system penetration. The tyrosine residue has been resistant to any structural perturbation to date however. The successful incorporation of conformational constraints, and subsequent conformational analyses, would allow the development of a three-dimensional pharmacophore, expediting the drug design process.
In summary, a body of data is emerging that offers the promise of turning the peptide lead Ang IV into a small molecule peptidomimetic, and developing a refined understanding of the ligand/enzyme interactions that must be maintained for inhibitory potency.
In silico-derived IRAP inhibitors
Alternative approaches include a large scale screening of compound libraries using IRAP as the target to identify novel, small molecule inhibitors of the enzyme. The fluorometric assay to monitor the catalytic activity of IRAP is readily adaptable for high throughput. The enzyme kinetics and mutational studies have provided some, albeit limited, understanding of the enzyme-inhibitor and enzyme-substrate interactions [21, 38, 40], which is required to enable the more cost- and time-effective computational screening approach to drug discovery. In this method, the structures of millions of commercially available compounds are docked into a three-dimensional atomic model of the active site of IRAP, one at a time, using powerful computing clusters. A binding score is calculated and the compounds are ranked in the order in which they are predicted to perform. The compounds are then assayed for inhibitory activity.
The crystal structure of IRAP has not yet been solved. However, the structure of a close family member, leukotriene A4 hydrolase, was published a few years ago [52] and has been used to generate a homology model of the catalytic domain of IRAP (residues L140 to S533; Figure 3). Both IRAP and leukotriene A4 hydrolase belong to the M1 aminopeptidase family, and while the overall identity between the sequences is low, the region immediately surrounding the active site residues, including the region of HEXXH and GAMEN motifs, is highly conserved (41% identity). The central catalytic domain is structurally conserved across a wide-range of zinc-dependent peptidases. For example, although Thermoplasma acidophilum Tricorn Interacting Factor F3 has only 16% identity with leukotriene A4 hydrolase at the amino acid level, the catalytic domains are structurally similar based on a comparison of their crystal structures with a root-mean-square deviation on 395 alpha-carbon atoms of 2.1 Å [52].

Ribbon representation of a homology model of the complete extracellular region of insulin-regulated aminopeptidase. The amino-terminal catalytic domain is highlighted in red with the catalytic zinc ion shown as a grey sphere.
This molecular model of IRAP has been used to screen a compound database containing 1.5 million compounds assembled from commercially available libraries with filters to exclude compounds on the basis of poor pharmacokinetic properties and presence of highly reactive moieties. Iterative screens based on the shape or structure of the most potent 'hits' led to the identification of a family of small molecule IRAP inhibitors [53, 54]. These compounds act as competitive inhibitors and do have memory enhancing properties in rodents.
Conclusion
The fundamental discovery of peptide inhibitors of IRAP and their ability to enhance memory underpins this research. The utilization of a range of strategies will enable the successful identification and development of new IRAP inhibitors with potentially therapeutic applications for the treatment of memory loss in AD.
Abbreviations
- Aβ:
-
amyloid-β
- AD:
-
Alzheimer's disease
- Ang:
-
angiotensin
- FDA:
-
Food and Drug Administration
- IRAP:
-
insulin-regulated aminopeptidase
- LVV-H7:
-
LVV-hemorphin 7.
References
Wolfe MS: Therapeutic strategies for Alzheimer's disease. Nat Rev Drug Discov. 2002, 1: 859-866. 10.1038/nrd938.
Birks J: Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Systematic Reviews. 2006, CD005593-
Kaduszkiewicz H, Zimmermann T, Beck-Bornholdt H, Bussche van den H: Cholinesterase inhibitors for patients with Alzheimer's disease: systematic review of randomised clinical trials. BMJ. 2005, 331: 321-327. 10.1136/bmj.331.7512.321.
Cosman KM, Boyle LL, Porsteinsson AP: Memantine in the treatment of mild-to-moderate Alzheimer's disease. Expert Opin Pharmacother. 2007, 8: 203-214. 10.1517/14656566.8.2.203.
Marshall E: A star-studded search for memory-enhancing drugs. Science. 2004, 304: 36-38. 10.1126/science.304.5667.36.
Braszko JJ, Kupryszewski G, Witczuk B, Wisniewski K: Angiotensin II-(3–8)-hexapeptide affects motor activity, performance of passive avoidance and a conditioned avoidance response in rats. Neuroscience. 1988, 27: 777-783. 10.1016/0306-4522(88)90182-0.
Wright JW, Miller-Wing AV, Shaffer MJ, Higginson C, Wright DE, Hanesworth JM, Harding JW: Angiotensin II(3–8) (ANG IV) hippocampal binding: potential role in the facilitation of memory. Brain Res Bull. 1993, 32: 497-502. 10.1016/0361-9230(93)90297-O.
Braszko JJ: Involvement of D1 dopamine receptors in the cognitive effects of angiotensin IV and des-Phe6 angiotensin IV. Peptides. 2004, 25: 1195-1203. 10.1016/j.peptides.2004.04.014.
Wright JW, Stubley L, Pederson ES, Kramar EA, Hanesworth JM, Harding JW: Contributions of the brain angiotensin IV-AT4 receptor subtype system to spatial learning. J Neurosci. 1999, 19: 3952-3961.
Lee J, Albiston AL, Allen AM, Mendelsohn FAO, Ping SE, Barrett GL, Murphy M, Morris MJ, McDowall SG, Chai SY: Effect of intracerebroventricular injection of AT4 receptor ligands, Nle1-angiotensin IV and LVv-hemorphin 7, on spatial learning in rats. Neuroscience. 2004, 124: 341-349. 10.1016/j.neuroscience.2003.12.006.
Wright JW, Clemens JA, Panetta JA, Smalstig EB, Weatherly LA, Kramar EA, Pederson ES, Mungall BH, Harding JW: Effects of LY231617 and angiotensin IV on ischemia-induced deficits in circular water maze and passive avoidance performance in rats. Brain Res. 1996, 717: 1-11. 10.1016/0006-8993(95)01454-3.
Olson ML, Olson EA, Qualls JH, Stratton JJ, Harding JW, Wright JW: Norleucine1-angiotensin IV alleviates mecamylamine-induced spatial memory deficits. Peptides. 2004, 25: 233-241. 10.1016/j.peptides.2003.12.005.
Pederson ES, Harding JW, Wright JW: Attenuation of scopolamine-induced spatial learning impairments by an angiotensin IV analog. Regul Pept. 1998, 74: 97-103. 10.1016/S0167-0115(98)00028-7.
Pederson ES, Krishnan R, Harding JW, Wright JW: A role for the angiotensin AT4 receptor subtype in overcoming scopolamine-induced spatial memory deficits. Regul Pept. 2001, 102: 147-156. 10.1016/S0167-0115(01)00312-3.
Albiston AL, Pederson ES, Burns P, Purcell B, Wright JW, Harding JW, Mendelsohn FA, Weisinger RS, Chai SY: Reversal of scopolamine-induced memory deficits by LVV-hemorphin 7 in rats in the passive avoidance and Morris water maze paradigms. Behav Brain Res. 2004, 154: 239-243.
Wisniewski K, Borawska M, Car H: The effect of angiotensin II and its fragments on post-alcohol impairment of learning and memory. Pol J Pharmacol. 1993, 45: 23-29.
Wayner MJ, Armstrong DL, Phelix CF, Wright JW, Harding JW: Angiotensin IV enhances LTP in rat dentate gyrus in vivo. Peptides. 2001, 22: 1403-1414. 10.1016/S0196-9781(01)00475-2.
Kramar EA, Armstrong DL, Ikeda S, Wayner MJ, Harding JW, Wright JW: The effects of angiotensin IV analogs on long-term potentiation within the CA1 region of the hippocampus in vitro. Brain Res. 2001, 897: 114-121. 10.1016/S0006-8993(01)02100-X.
Albiston AL, McDowall SG, Matsacos D, Sim P, Clune E, Mustafa T, Lee J, Mendelsohn FA, Simpson RJ, Connolly LM, et al: Evidence that the angiotensin IV (AT4) receptor is the enzyme insulin regulated aminopeptidase. J Biol Chem. 2001, 276: 48263-48266. 10.1074/jbc.C100512200.
Albiston AL, Mustafa T, McDowall SG, Mendelsohn FA, Lee J, Chai SY: AT(4) receptor is insulin-regulated membrane aminopeptidase: potential mechanisms of memory enhancement. Trends Endocrinol Metab. 2003, 14: 72-77. 10.1016/S1043-2760(02)00037-1.
Lew RA, Mustafa T, Ye S, McDowall SG, Chai SY, Albiston AL: Angiotensin AT4 ligands are potent, competitive inhibitors of insulin regulated aminopeptidase (IRAP). J Neurochem. 2003, 86: 344-350. 10.1046/j.1471-4159.2003.01852.x.
Moeller I, Paxinos G, Mendelsohn FA, Aldred GP, Casley D, Chai SY: Distribution of AT4 receptors in the Macaca fascicularis brain. Brain Res. 1996, 712: 307-324. 10.1016/0006-8993(95)01482-9.
Chai SY, Bastias MA, Clune EF, Matsacos DJ, Mustafa T, Lee JH, McDowall SG, Mendelsohn FA, Paxinos G, Albiston AL: Distribution of angiotensin IV binding sites (AT(4) receptor) in the human forebrain, midbrain and pons as visualised by in vitro receptor autoradiography. J Chem Neuroanat. 2000, 20: 339-348. 10.1016/S0891-0618(00)00112-5.
Fernando R, Larm J, Albiston AL, Chai SY: Distribution and cellular localization of the insulin regulated aminopeptidase (IRAP) in the rat central nervous system. J Comp Neurol. 2005, 487: 372-390. 10.1002/cne.20585.
Albiston AL, Burns P, Daswani D, Fernando RN, Yeatman HR, Chai SY: Characterisation of the behavioural phenotype of the IRAP knockout mouse. 7th IBRO World Congress of Neuroscience; Melbourne, Australia, 12–17. 2007, Abstract 225., July.
Keller SR, Scott HM, Mastick CC, Aebersold R, Lienhard GE: Cloning and characterization of a novel insulin-regulated membrane aminopeptidase from Glut4 vesicles. J Biol Chem. 1995, 270: 23612-23618. 10.1074/jbc.270.35.20497.
James DE, Brown R, Navarro J, Pilch PF: Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature. 1988, 333: 183-185. 10.1038/333183a0.
Waters SB, D'Auria M, Martin SS, Nguyen C, Kozma LM, Luskey KL: The amino terminus of insulin-responsive aminopeptidase causes Glut4 translocation in 3T3-L1 adipocytes. J Biol Chem. 1997, 272: 23323-23327. 10.1074/jbc.272.37.23323.
Peck GR, Ye S, Pham V, Fernando RN, Macaulay SL, Chai SY, Albiston AL: Interaction of the Akt substrate, AS160, with the glucose transporter 4 vesicle marker protein, insulin-regulated aminopeptidase. Mol Endocrinol. 2006, 20: 2576-2583. 10.1210/me.2005-0476.
Larance M, Ramm G, Stockli J, van Dam EM, Winata S, Wasinger V, Simpson F, Graham M, Junutula JR, Guilhaus M, et al: Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005, 280: 37803-37813. 10.1074/jbc.M503897200.
Chi NW, Lodish HF: Tankyrase is a golgi-associated mitogen-activated protein kinase substrate that interacts with IRAP in GLUT4 vesicles. J Biol Chem. 2000, 275: 38437-38444. 10.1074/jbc.M007635200.
Katagiri H, Asano T, Yamada T, Aoyama T, Fukushima Y, Kikuchi M, Kodama T, Oka Y: Acyl-coenzyme A dehydrogenases are localized on GLUT4-containing Vesicles via association with insulin-regulated aminopeptidase in a manner dependent on its dileucine motif. Mol Endocrinol. 2002, 16: 1049-1059. 10.1210/me.16.5.1049.
Tojo H, Kaieda I, Hattori H, Katayama N, Yoshimura K, Kakimoto S, Fujisawa Y, Presman E, Brooks CC, Pilch PF: The Formin family protein, formin homolog overexpressed in spleen, interacts with the insulin-responsive aminopeptidase and profilin IIa. Mol Endocrinol. 2003, 17: 1216-1229. 10.1210/me.2003-0056.
Hosaka T, Brooks CC, Presman E, Kim SK, Zhang Z, Breen M, Gross DN, Sztul E, Pilch PF: p115 Interacts with the GLUT4 vesicle protein, IRAP, and plays a critical role in insulin-stimulated GLUT4 translocation. Mol Biol Cell. 2005, 16: 2882-2890. 10.1091/mbc.E05-01-0072.
Nanus DM, Engelstein D, Gastl GA, Gluck L, Vidal MJ, Morrison M, Finstad CL, Bander NH, Albino AP: Molecular cloning of the human kidney differentiation antigen gp160: human aminopeptidase A. Proc Natl Acad Sci USA. 1993, 90: 7069-7073. 10.1073/pnas.90.15.7069.
Olsen J, Cowell GM, Konigshofer E, Danielsen EM, Moller J, Laustsen L, Hansen OC, Welinder KG, Engberg J, Hunziker W, et al: Complete amino acid sequence of human intestinal aminopeptidase N as deduced from cloned cDNA. FEBS Lett. 1988, 238: 307-314. 10.1016/0014-5793(88)80502-7.
Funk CD, Radmark O, Fu JY, Matsumoto T, Jornvall H, Shimizu T, Samuelsson B: Molecular cloning and amino acid sequence of leukotriene A4 hydrolase. Proc Natl Acad Sci USA. 1987, 84: 6677-6681. 10.1073/pnas.84.19.6677.
Laustsen PG, Vang S, Kristensen T: Mutational analysis of the active site of human insulin-regulated aminopeptidase. Eur J Biochem. 2001, 268: 98-104. 10.1046/j.1432-1327.2001.01848.x.
Rudberg PC, Tholander F, Thunnissen MM, Haeggstrom JZ: Leukotriene A4 hydrolase/aminopeptidase. Glutamate 271 is a catalytic residue with specific roles in two distinct enzyme mechanisms. J Biol Chem. 2002, 277: 1398-1404. 10.1074/jbc.M106577200.
Ye S, Chai SY, Lew RA, Albiston AL: Insulin-regulated aminopeptidase: analysis of peptide substrate and inhibitor binding to the catalytic domain. Biol Chem. 2007, 388: 399-403. 10.1515/BC.2007.044.
Ross SA, Herbst JJ, Keller SR, Lienhard GE: Trafficking kinetics of the insulin-regulated membrane aminopeptidase in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 1997, 239: 247-251. 10.1006/bbrc.1997.7459.
Rogi T, Tsujimoto M, Nakazato H, Mizutani S, Tomoda Y: Human placental leucine aminopeptidase/oxytocinase. A new member of type II membrane-spanning zinc metallopeptidase family. J Biol Chem. 1996, 271: 56-61. 10.1074/jbc.271.1.56.
Yamahara N, Nomura S, Suzuki T, Itakura A, Ito M, Okamoto T, Tsujimoto M, Nakazato H, Mizutani S: Placental leucine aminopeptidase/oxytocinase in maternal serum and placenta during normal pregnancy. Life Sciences. 2000, 66: 1401-1410. 10.1016/S0024-3205(00)00451-3.
Wallis MG, Lankford MF, Keller SR: Vasopressin is a physiological substrate for the insulin-regulated aminopeptidase IRAP. Am J Physiol Endocrinol Metab. 2007, 293: E1092-1102. 10.1152/ajpendo.00440.2007.
Herbst JJ, Ross SA, Scott HM, Bobin SA, Morris NJ, Lienhard GE, Keller SR: Insulin stimulates cell surface aminopeptidase activity toward vasopressin in adipocytes. Am J Physiol. 1997, 272: E600-606.
Matsumoto H, Nagasaka T, Hattori A, Rogi T, Tsuruoka N, Mizutani S, Tsujimoto M: Expression of placental leucine aminopeptidase/oxytocinase in neuronal cells and its action on neuronal peptides. Eur J Biochem. 2001, 268: 3259-3266. 10.1046/j.1432-1327.2001.02221.x.
Krishnan R, Hanesworth JM, Wright JW, Harding JW: Structure-binding studies of the adrenal AT4 receptor: analysis of position two- and three-modified angiotensin IV analogs. Peptides. 1999, 20: 915-920. 10.1016/S0196-9781(99)00081-9.
Lee JH, Mustafa T, McDowall SG, Mendelsohn FAO, Brennan MB, Lew R, Albiston AL, Chai SY: Structure-activity study of LVV-hemorphin-7: angiotensin AT4 receptor ligand and inhibitor of insulin-regulated aminopeptidase (IRAP). J Pharmacol Exp Ther. 2003, 305: 205-211. 10.1124/jpet.102.045492.
Axen A, Lindeberg G, Demaegdt H, Vauquelin G, Karlen A, Hallberg M: Cyclic insulin-regulated aminopeptidase (IRAP)/AT4 receptor ligands. J Pept Sci. 2006, 12: 705-713. 10.1002/psc.782.
Kobori T, Goda K, Sugimoto K, Ota T, Tomisawa K: Amino compounds and angiotensin IV receptor agonists. US Patent. 1998
Lukaszuk A, Demaegdt H, Szemenyei E, Toth G, Tymecka D, Micicka A, Vanderheyden P, Vauquelin G, Tourwé D: β-Homo-amino acid scan of Angiotensin IV. 29th European Peptide Symposium; Gdansk, Poland: 3–8. 2006, Abstract 282., September.
Thunnissen MM, Nordlund P, Haeggstrom JZ: Crystal structure of human leukotriene A(4) hydrolase, a bifunctional enzyme in inflammation. Nat Struct Biol. 2001, 8: 131-135. 10.1038/84117.
Chai SY, Parker MW, Albiston AL, Morton CJ, Ng HL, Ye S, Mendelsohn FAO: Enzyme inhibitors and uses thereof. US Patent. 2006
Albiston AL, Morton CJ, Ng HL, Pham V, Yeatman HR, Ye S, Fernando RN, De Bundel D, Ascher DB, Mendelsohn FAO, Parker MW, Chai SY: Identification and characterization of a new class of cognitive enhancers based on inhibition of insulin-regulated aminopeptidase. FASEB J. 2008,.
Acknowledgements
DBA is an Australian Postgraduate Award Scholar and a recipient of a St Vincent's Institute Foundation Scholarship sponsored by Colin North and Major Engineering. MWP is an Australian Research Council Federation Fellow and a National Health and Medical Research Council (NHMRC) Honorary Fellow. SYC is an NHMRC Senior Research Fellow. HRY is supported by a Sylvia and Charles Viertel Alzheimer's Postgraduate Scholarship. This work is supported by the Alzheimer's Drug Discovery Foundation/Institute for the Study of Aging, Neurosciences Victoria, NHMRC Development grants (IDs: 454714 & 520695) and the CASS Foundation of Australia.
This article has been published as part of BMC Neuroscience Volume 9 Supplement 2: 2008 Proceedings of the 8th International Conference on Alzheimer's Disease Drug Discovery The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2202/9?issue=S2.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chai, S.Y., Yeatman, H.R., Parker, M.W. et al. Development of cognitive enhancers based on inhibition of insulin-regulated aminopeptidase. BMC Neurosci 9 (Suppl 2), S14 (2008). https://doi.org/10.1186/1471-2202-9-S2-S14
Published:
DOI: https://doi.org/10.1186/1471-2202-9-S2-S14