
Abstract
Background
The circadian rhythm of about 24 hours is a fundamental physiological function observed in almost all organisms from prokaryotes to humans. Identification of clock genes has allowed us to study the molecular bases for circadian behaviors and temporal physiological processes such as hormonal secretion, and has prompted the idea that molecular clocks reside not only in a central pacemaker, the suprachiasmatic nuclei (SCN) of hypothalamus in mammals, but also in peripheral tissues, even in immortalized cells. Furthermore, previous molecular dissection revealed that the mechanism of circadian oscillation at a molecular level is based on transcriptional regulation of clock and clock-controlled genes.
Results
We systematically analyzed the mRNA expression of clock and clock-controlled genes in mouse peripheral tissues. Eight genes (mBmal1, mNpas2, mRev-erbα, mDbp, mRev-erbβ, mPer3, mPer1 and mPer2; given in the temporal order of the rhythm peak) showed robust circadian expressions of mRNAs in all tissues except testis, suggesting that these genes are core molecules of the molecular biological clock. The bioinformatics analysis revealed that these genes have one or a combination of 3 transcriptional elements (RORE, DBPE, and E-box), which are conserved among human, mouse, and rat genome sequences, and indicated that these 3 elements may be responsible for the biological timing of expression of canonical clock genes.
Conclusions
The observation of oscillatory profiles of canonical clock genes is not only useful for physiological and pathological examination of the circadian clock in various organs but also important for systematic understanding of transcriptional regulation on a genome-wide basis. Our finding of the oscillatory expression of canonical clock genes with a temporal order provides us an interesting hypothesis, that cyclic timing of all clock and clock-controlled genes may be dependent on several transcriptional elements including 3 known elements, E-box, RORE, and DBPE.
Similar content being viewed by others

Background
The circadian rhythm of about 24 hours is a fundamental physiological function observed in almost all organisms from prokaryotes to humans. Circadian rhythms have been known to be generated in pacemaker cells, the suprachiasmatic nuclei (SCN) of hypothalamus in mammals, and entrained by environmental cues, such as light, temperature, noise, feeding or social cues, whereas a recent analysis using mPer2luciferase knockin mice has demonstrated that peripheral tissues express self-sustained circadian oscillations [1]. The output of circadian oscillation appears as locomotive activity, hormonal secretion, the sleep-wake cycle, and many other physiological functions. Disruption of the circadian rhythms has been associated with various kinds of diseases, such as cardiovascular diseases, psychiatric diseases and cancer in humans [2–6]. Identification of clock genes has allowed study of the molecular bases for circadian behaviors and temporal physiological processes and has prompted the idea that molecular clocks reside not only in a central pacemaker, but also in peripheral tissues, even in immortalized cells [2, 3, 6]. Furthermore, previous molecular dissection revealed that the mechanism of circadian oscillation at a molecular level is based on transcriptional regulation of clock and clock-controlled genes, which consists of interwoven positive and negative feedback loops [2, 7–10].
There is a distinct connection between genes and behaviors in circadian rhythms, which is conserved from fly or other lower organisms to humans [6, 8]. The Drosophila period mutants, originally identified as a circadian mutant brought us the first clock gene, period [8, 11], while a point mutation of hPer2 was recently shown to cause a familial advanced sleep phase syndrome [12]. As described above, circadian rhythms rely on a negative feedback loop in gene expression that involves a limited number of clock genes. Recent molecular dissection has increased our understanding of the molecular nature of the transcriptional regulation of some clock genes. The circadian phenotypes at the cellular level may be represented as temporal mRNA expression. Global gene expression profiling using microarrays has led to the discovery of many circadian-regulated genes, but there is only a minor overlap of cycling transcripts between tissues [10, 13, 14]. Thus, circadian rhythms are an appropriate study target for systems biology.
In this study, we systematically examined the mRNA expression of common circadian-regulated genes in several mouse peripheral tissues and made oscillatory profiles of canonical clock genes. Moreover, by bioinformatics, we identified 3 clock elements for circadian transcription (E-box, RORE, DBPE). These 3 elements and their combination would suffice to explain the biological timing of expression of these clock and clock-controlled genes.
Results and discussion
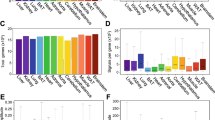
To examine the circadian expression of mouse clock and clock-related genes in peripheral tissues, we performed the quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) method on mRNAs from 7 different mouse peripheral tissues (heart, lung, liver, stomach, spleen, kidney, and testis; Fig. 1). After entrainment of housed mice for 2 weeks under a light-dark (LD) cycle, samples were collected every 4 hr starting at circadian time (CT) 0 (n = 3 at each time point) in the third dark-dark (DD) cycle. Out of 14 mouse genes examined, 8 genes (mBmal1, mNpas2, mRev-erbα, mDbp, mRev-erbβ, mPer3, mPer1 and mPer2; given in the temporal order of the rhythm peak; see also Fig. 2) showed robust circadian of mRNA expression in all tissues except the testis. These genes should thus be considered to be core molecules of the circadian clock. Circadian mRNA expression patterns were similar in each tissue with the exception of testis, where weak or no rhythm was observed. This common pattern of the rhythm throughout many peripheral tissues implies that there may exist a universal mechanism for resetting the peripheral clock. The peak transcript level of each circadian rhythm was as follows: mBmal1 and mNpas2, in subjective night at CT20-CT0; mRev-erbα, in subjective day at CT4-8; mDbp and mRev-erbβ, at CT8; mPer3, at CT8-12; mPer1, at CT12; and mPer2, at CT12-16 (Figs. 1, 2, and 4). In the peripheral tissues mRNA peaks occurred approximately 4 hr later than those in the central pacemaker, SCN [15–20]. In the testis, 4 genes (mBmal1, mNpas2, mRev-erbα, and mDbp) showed weak rhythms of their mRNA expression, while no other genes, including the mPer family, showed any clear oscillation. Surprisingly, the expression of mPer1 transcripts in the testis, which did not show a circadian rhythm, was substantially higher than other tissues, and exceeded RNA expression of other genes studied in the testis. This is consistent with data recently reported [21–23]; although a previous report indicated circadian rhythm of mPer1 in the testis [20]. These findings suggest that mPer1 may play an alternative role in the testis, including developmental regulation during spermatogenesis. The rhythm of mCry1 mRNA expression was obviously circadian (peaking at CT16-20, with a trough at CT4-8) except in the testis, but the peak-trough amplitude was relatively smaller than that of the above genes. mCry2 and mClock RNA levels seemed to be rhythmic except in the testis, but the rhythm was rather weak and not clearly circadian. No circadian rhythms were observed in the remaining 3 genes examined, i.e. mCKI-δ, mCKI-ε, and mTim. Despite a central role in Drosophila for timeless (dTim), the mammalian homologue (mTim) was originally found to show weak or no rhythm in the SCN [24–26]; and its functional role in mammalian clocks remains controversial [27, 28].

Temporal mRNA expression of clock and clock-related genes in mouse peripheral tissues Abscissa presents time at CT (circadian time); and ordinate, mRNA amounts. The relative levels of each RNA were normalized to the corresponding G3-PDH RNA levels. The maximum RNA amount was set to 100. Data are presented as the mean ± SE of triplicate samples.

Circadian mRNA expression of canonical clock genes in mouse peripheral tissues The maximum RNA amount in a 24-h period is indicated by the darkest red (100), while no RNA (0) shown by white. The depth of the color corresponds to the RNA amount.

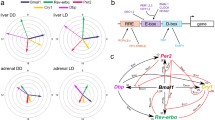
Diagram representing the relationship between 3 transcriptional elements and the peak mRNA expression of canonical clock genes The peak of Per3 mRNA expression is at CT8-12, and its regulatory region includes only conserved DBPEs (yellow) among the 3 elements. The peak of Per2 mRNA is at CT12-16, and its promoter region includes only an E-box (green). The peaks of Bmal1 and Npas2 mRNA are at CT20-0, and their regulatory regions contain only ROREs (blue). The peaks for the other clock genes, which contain 2 or 3 elements, are placed as indicated. The bar at the top represents light (gray) and dark (black) cycles.
As described above, the expression of 8 genes fluctuated in an overt circadian fashion; and so we aligned them in the order of the peak of their oscillatory phase (Fig. 2). Among them, the transcriptional oscillation of 2 representative clock genes, Bmal1 and Per2, was examined by using the real-time luciferase reporter assay. NIH3T3 cells were transfected with the hBmal1-Luc or mPer2-Luc construct and then stimulated with a high concentration of serum. After the serum shock, in the presence of luciferin, light emission was measured and integrated for 1 min at intervals of 15 min. Both promoters fused to luciferase showed circadian rhythms (Fig. 3). The phase of Bmal1 oscillation in cultured cells was almost the opposite of that of Per2, which is consistent with data of mRNA expression in mouse peripheral tissues obtained by real-time RT-PCR (Fig. 2). This result indicates that the promoter regions used in the real-time luciferase reporter assay are sufficient for producing circadian transcriptional oscillation. The promoter analyses of several clock genes have been reported, as described below.

Transcriptional oscillation of Bmal1 and Per2 Transcriptional oscillation of Bmal1 (red) and Per2 (blue) was monitored by using a cell culture-based luminescence reporter assay. NIH3T3 cells were transfected with the hBmal1 -Luc or mPer2 -Luc constructs and then stimulated with a high concentration of serum. After the serum shock, in the presence of luciferin, light emission was measured and integrated for 1 min at intervals of 15 min. Ordinate and abscissa represent relative counts and time after serum shock, respectively. The peak of the count was set to 1. 1440 minutes = 1 day.

Table 1. Three transcriptional elements (RORE, E-box, and DBPE) conserved among the sequences of 3 species (human, mouse, and rat) Numbers in parenthesis indicate the number from the transcriptional start site. The sequences of the binding elements are in bold type, and the sequences matching the consensus sequence are underlined. H: human, M: mouse, R: rat.
A molecular mechanism of canonical clock genes is based on transcriptional regulation via interlocked feedback and/or feed-forward loops [2, 4, 7, 8]. One example of the regulation of a known characterized gene in mammals is that of Per1. The transcription of Per1 is activated by binding of the CLOCK/BMAL1 hetero-complex, both members of which are bHLH-PAS (basic helix-loop-helix-Per-Arnt-Sim) proteins, to the E-boxes in the promoter region of Per1 [29]. The translated PER1 is posttranslationally modified by CKI-ε [30] and, together with other clock proteins such as CRYs [31], is returned to the nucleus to suppress its own transactivation, resulting in closure of the PER1 loop. E-box elements are also known to be essential for transcriptional regulation of many clock-controlled output genes including the vasopression genes [32]. On the other hand, one of the positive elements, BMAL1, whose mRNA expression is cycled antiphase to Per s, as described above, forms another loop [33, 34]. The orphan nuclear receptors RORα and REV-ERBα regulate circadian transcription positively and negatively, respectively, through ROR/REV-ERB elements (ROREs) in the promoter region of Bmal1 [16, 35]. Moreover, an in silico search identified these 2 ROREs in the promoter region of Npas2 (Table 1, see figure 5), the expression pattern of which was very similar to that of Bmal1. These findings gave us the idea that the circadian pattern of RNA expression might be dependent on transcriptional regulation by specific transcription factors.
Using the NCBI database and Celera Database System, we systematically searched for the above 2 elements and another clock element, a DBP-binding element (DBPE), described below. These elements are conserved among human, mouse, and rat genome sequences in the regions 9-kb upstream and 5-kb downstream of the transcription start site (Table 1, see figure 5). Intriguingly, the transcriptional elements corresponded to the cyclic pattern of the clock genes shown in Figure 4. Bmal1 and Npas2, the circadian peaks of which were both in subjective night at CT20-24, included ROREs in their promoters, as described above. In the promoter of Per1 and Per2, the peaks of which were in subjective day at CT12-16, E-boxes were found. In fact, the conserved E-box in Per2 is not a typical E-box of the molecular clock, CACGTG, but an atypical element, CACGTT. Compared with Per1, which contains 5 conserved E-boxes, Per2 may have other unknown factors and elements responsible for its robust transcriptional oscillation. Per1, whose peak was a bit earlier than that of Per2, has another element, a DBPE, in its promoter region, in addition to the 5 E-boxes well studied in vitro [36]. Per3, the peak of which was even earlier at CT8-12, did not have conserved E-boxes but instead contained DBPEs in its gene. Three genes, Rev-erbα, Dbp, and Rev-erbβ, the peaks of which were between those of Bmal1 and Per, had a mixed combination of the elements. The Rev-erbα genome sequence included 1 RORE, 1 DBPE, and 5 E-boxes, whereas Rev-erbβ included all 3 elements only in the mouse genome sequence. Dbp contained 2 ROREs and 2 E-boxes in each genome. Among the elements described above, some of them in (Bmal1, Dbp, and Per1) were experimentally studied and confirmed [16, 29, 35–38].
Transcripts of 8 genes (mBmal1, mNpas2, mRev-erbα, mDbp, mRev-erbβ, mPer3, mPer1, and mPer2) showed a robust circadian rhythm in different peripheral tissues (see Fig. 1). The amount of mRNA in the trough was nearly zero and the peak-trough amplitude of these genes was clearly higher than that of the others examined. Thus, in terms of mRNA expression among the canonical clock genes examined, these 8 genes likely constitute the core molecules of a molecular circadian clock. The expression timing in a 24-h period appears to be conveyed through 3 kinds of sequence elements bound by specific transcription factors (see Fig. 4). Recent genome-wide analyses using microarrays revealed that many genes (about 10 % of the total number of genes studied) oscillated but only several tens of common genes overlapped between two tissues examined [13, 14]. Among the core candidate genes with similar circadian regulation in those 2 tissues, we examined the circadian transcription of 15 candidate genes besides the known clock genes, but could not find genes with oscillatory behavior in different peripheral tissues comparable to that in the 8 genes described above. The 8 genes studied here may approximate the entirety of the core oscillatory genes in the genome. If so, the 3 elements described here may be sufficient for explaining the biological timing of mRNA expression of clock genes. However, our preliminary results showed that an atypical E-box in Per2 promoter may be insufficient for full transcriptional oscillation (Akashi and Takumi, unpublished data). Further detailed studies of each promoter, combined with systematic analyses using microarrays and real-time RT-PCR, will give us a more detailed comprehension of the intertwined positive and negative regulatory loops of molecular biological clocks.
Conclusions
The current study has clarified the detailed circadian expression of mRNAs for clock and clock-related genes in different peripheral tissues of the mouse. The observation of oscillatory profiles of canonical clock genes is not only useful for physiological and pathological examination of the circadian clock in various organs but also important for systematic understanding of transcriptional regulation on a genome-wide basis. Our finding of the oscillatory expression of canonical clock genes in a temporal order provides us an interesting hypothesis, that cyclic timing of all clock and clock-controlled genes may be dependent on several transcriptional elements including 3 known elements, E-box, RORE, and DBPE.
Methods
Animals
Male Balb/c mice purchased 5 weeks postpartum from Japan SLC (Hamamatsu, Japan), were exposed to 2 weeks of light-dark (LD) cycles and then kept in complete darkness as a continuation of the dark phase of the last LD cycle. mRNA expression was examined in the third dark-dark (DD) cycle. All protocols of experiments using animals in this study were approved by the OBI (Osaka Bioscience Institute) Animal Research Committee.
Quantitative RT-PCR
Real-time quantitative RT-PCR was performed by using an ABI PRISM 7000 (Applied Biosystems). The PCR primers were designed with Primer Express software (Applied Biosystems), and the sequences of the forward and reverse primers were as follow: mPer1 FW: CAG GCT AAC CAG GAA TAT TAC CAG C, mPer1 RV: CAC AGC CAC AGA GAA GGT GTC CTG G; mPer2 FW: GGC TTC ACC ATG CCT GTT GT, mPer2 RV: GGA GTT ATT TCG GAG GCA AGT GT; mPer3 FW: CTG CTC CAA CTC AGC TTC CTT T, mPer3 RV: TTA GAC AGC AAG GCT CTG GTT CT; mNpas2 FW: GTA TGC ACA GAG CCA AGT GAT GTT, mNpas2 RV: TGC TCA CTG TGC AGA GAT GTT G; mDbp FW: AAT GAC CTT TGA ACC TGA TCC CGC T, mDbp RV: GCT CCA GTA CTT CTC ATC CTT CTG T; mBmal1 FW: GCA GTG CCA CTG ACT ACC AAG A, mBmal1 RV: TCC TGG ACA TTG CAT TGC AT; mRev-erbα FW: CGT TCG CAT CAA TCG CAA CC, mRev-erbα RV: GAT GTG GAG TAG GTG AGG TC; mRev-erbβ FW: ACG GAT TCC CAG GAA CAT GG, mRev-erbβ RV: CCT CCA GTG TTG CAC AGG TA; G3-PDH FW: ACG GGA AGC TCA CTG GCA TGG CCT T, G3-PDH RV: CAT GAG GTC CAC CAC CCT GTT GCT G; mCry1 FW: CCC AGG CTT TTC AAG GAA TGG AAC A, mCry1 RV: TCT CAT CAT GGT CAT CAG ACA GAG G; mCry2 FW: GGG ACT CTG TCT ATT GGC ATC TG, mCry2 RV: GTC ACT CTA GCC CGC TTG GT; mCKIε FW: GGA TGT GAA GCC CGA CAA CTT, mCKIε RV: TCT CGA CGG CTT TGC TCA AT; mCKIδ FW: CCA GCC TGG AAG ACC TGT TC, mCKIδ RV: TGG CCA GCC CAA AGT CAA; mClock FW: CCT ATC CTA CCT TGG CCA CAC A, mClock RV: TCC CGT GGA GCA ACC TAG AT; mTim FW: ACA TGT GGG CAA TGG CTT, mTim RV: CTG CTC CAC AAA GTG AAA GGT. Specificity of gene amplification was confirmed by measuring the size and purity of the PCR product by gel electrophoresis, and by analyzing the dissociation curve with ABI PRISM 7000 SDS software (Applied Biosystems). For a 25-μl PCR reaction, 50 ng cDNA template was mixed with the forward and reverse primers to a final concentration of 300 nM each and 12.5 μl of 2x SYBR Green PCR Master Mix (Applied Biosystems). The reaction was first incubated at 50°C for 2 min, then at 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Each gene-specific PCR was performed in triplicate. G3-PDH primers were used as the control.
Real-time luciferase reporter assay
NIH3T3 cells were cultured, transfected with hBmal1 -Luc or mPer2 -Luc, and incubated for 24 hours. The medium was then exchanged for serum-rich medium (DMEM, supplemented with 50 % serum). Two hours later this medium was replaced with normal culture medium. In the presence of 0.1 mM luciferin, light emission was measured and integrated for 1 min at intervals of 15 min, with a photomultiplier tube (Hamamatsu Photonics).
In silico search
The sequences were downloaded from the Celera Database System and the NCBI Gene database. Each gene sequence spanning from 9 kb upstream to 4 kb downstream of the transcription start site was examined in each database. Multiple sequence alignments of these sequences for each gene were obtained by Clustalw version 1.83 with default parameters. The binding elements were then searched from these alignments using a pattern finding tool, fuzznuc, with the following consensus sequences allowing for a 1-base mismatch:
DBPE: [GA]T[GT]A[TC]GTAA[TC]
E-Box: CACGTG
RORE: [AT]A[AT]NT[AG]GGTCA
The accession numbers used and the sequence numbers analyzed are as follow: Bmal1; Human, NT_009237.16, 12054318–12069318, Mouse, NT_081129.1, 107781–122781, Rat, NW_047562.1, 13774073–13789073, Npas2; Human, hCG27614, 95632226–65646226, Mouse, mCG8437, 35980102–35994102, Rat, rCT22431, 39204499–39218499, Rev-erbα; Human, hCG93862, 34926094–34912094, Mouse, mCG15360, 105438925–105424925, Rat, rCG33292, 82492796–82478796, Dbp; Human, NT_011109.15, c21417778–21402778, Mouse, NT_078442.1, 59711–74711, Rat, NW_047558.1, 5120734–5135734, Per3; Human, NT_021937.16, 1962822–1977822, Mouse, NT_039268.2, c4331528–4316528, Rat, NW_047727.1, c8016956–8001956, Per1; Human, NT_010718.14, c6905708–6890708, Moues, NT_039515.2, 65661216–65676216, Rat, rCG34390, 52960430–52974430, Per2; Human, NT_005120.14, c5136562–5121562, Mouse, NT_039173.2, c5833757–5818757, Rat, NW_047817.1, c6827703–6812703.
Abbreviations
- SCN:
-
suprachiasmatic nuclei
- RT-PCR:
-
reverse transcription-polymerase chain reaction
- CT:
-
circadian time
- LD:
-
light-dark
- DD:
-
dark-dark
- bHLH-PAS:
-
basic helix-loop-helix-Per-Arnt-Sim
- RORE:
-
ROR/REV-ERB element
- DBPE:
-
DBP-binding element
- NCBI:
-
the National Center for Biotechnology Information.
References
Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M, Takahashi JS: PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004, 101: 5339-5346. 10.1073/pnas.0308709101
Reppert SM, Weaver DR: Coordination of circadian timing in mammals. Nature. 2002, 418: 935-941. 10.1038/nature00965
Schibler U, Sassone-Corsi P: A web of circadian pacemakers. Cell. 2002, 111: 919-922. 10.1016/S0092-8674(02)01225-4
Roenneberg T, Merrow M: The network of time: understanding the molecular circadian system. Curr Biol. 2003, 13: R198-207. 10.1016/S0960-9822(03)00124-6
Fu L, Lee CC: The circadian clock: pacemaker and tumour suppressor. Nat Rev Cancer. 2003, 3: 350-361. 10.1038/nrc1072
Hastings MH, Reddy AB, Maywood ES: A clockwork web: circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci. 2003, 4: 649-661. 10.1038/nrn1177
Dunlap JC: Molecular bases for circadian clocks. Cell. 1999, 96: 271-290. 10.1016/S0092-8674(00)80566-8
Young MW, Kay SA: Time zones: a comparative genetics of circadian clocks. Nat Rev Genet. 2001, 2: 702-715. 10.1038/35088576
Allada R, Emery P, Takahashi JS, Rosbash M: Stopping time: the genetics of fly and mouse circadian clocks. Annu Rev Neurosci. 2001, 24: 1091-1119. 10.1146/annurev.neuro.24.1.1091
Albrecht U, Eichele G: The mammalian circadian clock. Curr Opin Genet Dev. 2003, 13: 271-277. 10.1016/S0959-437X(03)00055-8
Konopka RJ, Benzer S: Clock mutants of Drosophila melanogaster. Proc Natl Acad Sci U S A. 1971, 68: 2112-2116.
Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, Fu YH: An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001, 291: 1040-1043. 10.1126/science.1057499
Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB: Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002, 109: 307-320. 10.1016/S0092-8674(02)00722-5
Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ: Extensive and divergent circadian gene expression in liver and heart. Nature. 2002, 417: 78-83. 10.1038/nature744
Honma S, Ikeda M, Abe H, Tanahashi Y, Namihira M, Honma K, Nomura M: Circadian oscillation of BMAL1, a partner of a mammalian clock gene Clock, in rat suprachiasmatic nucleus. Biochem Biophys Res Commun. 1998, 250: 83-87. 10.1006/bbrc.1998.9275
Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U: The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002, 110: 251-260. 10.1016/S0092-8674(02)00825-5
Takumi T, Matsubara C, Shigeyoshi Y, Taguchi K, Yagita K, Maebayashi Y, Sakakida Y, Okumura K, Takashima N, Okamura H: A new mammalian period gene predominantly expressed in the suprachiasmatic nucleus. Genes Cells. 1998, 3: 167-176. 10.1046/j.1365-2443.1998.00178.x
Takumi T, Taguchi K, Miyake S, Sakakida Y, Takashima N, Matsubara C, Maebayashi Y, Okumura K, Takekida S, Yamamoto S, Yagita K, Yan L, Young MW, Okamura H: A light-independent oscillatory gene mPer3 in mouse SCN and OVLT. Embo J. 1998, 17: 4753-4759. 10.1093/emboj/17.16.4753
Lopez-Molina L, Conquet F, Dubois-Dauphin M, Schibler U: The DBP gene is expressed according to a circadian rhythm in the suprachiasmatic nucleus and influences circadian behavior. Embo J. 1997, 16: 6762-6771. 10.1093/emboj/16.22.6762
Zylka MJ, Shearman LP, Weaver DR, Reppert SM: Three period homologs in mammals: differential light responses in the suprachiasmatic circadian clock and oscillating transcripts outside of brain. Neuron. 1998, 20: 1103-1110. 10.1016/S0896-6273(00)80492-4
Bittman EL, Doherty L, Huang L, Paroskie A: Period gene expression in mouse endocrine tissues. Am J Physiol Regul Integr Comp Physiol. 2003, 285: R561-569.
Alvarez JD, Chen D, Storer E, Sehgal A: Non-cyclic and developmental stage-specific expression of circadian clock proteins during murine spermatogenesis. Biol Reprod. 2003, 69: 81-91. 10.1095/biolreprod.102.011833
Morse D, Cermakian N, Brancorsini S, Parvinen M, Sassone-Corsi P: No circadian rhythms in testis: Period1 expression is clock independent and developmentally regulated in the mouse. Mol Endocrinol. 2003, 17: 141-151. 10.1210/me.2002-0184
Takumi T, Nagamine Y, Miyake S, Matsubara C, Taguchi K, Takekida S, Sakakida Y, Nishikawa K, Kishimoto T, Niwa S, Okumura K, Okamura H: A mammalian ortholog of Drosophila timeless, highly expressed in SCN and retina, forms a complex with mPER1. Genes Cells. 1999, 4: 67-75. 10.1046/j.1365-2443.1999.00238.x
Sangoram AM, Saez L, Antoch MP, Gekakis N, Staknis D, Whiteley A, Fruechte EM, Vitaterna MH, Shimomura K, King DP, Young MW, Weitz CJ, Takahashi JS: Mammalian circadian autoregulatory loop: a timeless ortholog and mPer1 interact and negatively regulate CLOCK-BMAL1-induced transcription. Neuron. 1998, 21: 1101-1113. 10.1016/S0896-6273(00)80627-3
Zylka MJ, Shearman LP, Levine JD, Jin X, Weaver DR, Reppert SM: Molecular analysis of mammalian timeless. Neuron. 1998, 21: 1115-1122. 10.1016/S0896-6273(00)80628-5
Barnes JW, Tischkau SA, Barnes JA, Mitchell JW, Burgoon PW, Hickok JR, Gillette MU: Requirement of mammalian Timeless for circadian rhythmicity. Science. 2003, 302: 439-442. 10.1126/science.1086593
Gotter AL, Manganaro T, Weaver DR, Kolakowski LF, Possidente B, Sriram S, MacLaughlin DT, Reppert SM: A time-less function for mouse timeless. Nat Neurosci. 2000, 3: 755-756. 10.1038/77653
Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ: Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998, 280: 1564-1569. 10.1126/science.280.5369.1564
Vielhaber E, Eide E, Rivers A, Gao ZH, Virshup DM: Nuclear entry of the circadian regulator mPER1 is controlled by mammalian casein kinase I epsilon. Mol Cell Biol. 2000, 20: 4888-4899. 10.1128/MCB.20.13.4888-4899.2000
Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM: mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999, 98: 193-205. 10.1016/S0092-8674(00)81014-4
Jin X, Shearman LP, Weaver DR, Zylka MJ, de Vries GJ, Reppert SM: A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell. 1999, 96: 57-68. 10.1016/S0092-8674(00)80959-9
Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, Kume K, Lee CC, van der Horst GT, Hastings MH, Reppert SM: Interacting molecular loops in the mammalian circadian clock. Science. 2000, 288: 1013-1019. 10.1126/science.288.5468.1013
Yu W, Nomura M, Ikeda M: Interactivating feedback loops within the mammalian clock: BMAL1 is negatively autoregulated and upregulated by CRY1, CRY2, and PER2. Biochem Biophys Res Commun. 2002, 290: 933-941. 10.1006/bbrc.2001.6300
Nakajima Y, Ikeda M, Kimura T, Honma S, Ohmiya Y, Honma K: Bidirectional role of orphan nuclear receptor RORalpha in clock gene transcriptions demonstrated by a novel reporter assay system. FEBS Lett. 2004, 565: 122-126. 10.1016/j.febslet.2004.03.083
Hida A, Koike N, Hirose M, Hattori M, Sakaki Y, Tei H: The human and mouse Period1 genes: five well-conserved E-boxes additively contribute to the enhancement of mPer1 transcription. Genomics. 2000, 65: 224-233. 10.1006/geno.2000.6166
Ripperger JA, Shearman LP, Reppert SM, Schibler U: CLOCK, an essential pacemaker component, controls expression of the circadian transcription factor DBP. Genes Dev. 2000, 14: 679-689.
Ueda HR, Chen W, Adachi A, Wakamatsu H, Hayashi S, Takasugi T, Nagano M, Nakahama K, Suzuki Y, Sugano S, Iino M, Shigeyoshi Y, Hashimoto S: A transcription factor response element for gene expression during circadian night. Nature. 2002, 418: 534-539. 10.1038/nature00906
Acknowledgements
We thank Setsuko Tsuboi, Chiaki Matsubara, Yoko Sakakida, and Dan Trcka for their technical assistance, Paul Burke for his review of the manuscript, and acknowledge Yasuhiro Sakamoto for his administrative assistance. This work was supported in part by a research grant from MEXT. The support of fellowships from the Japan Society for the Promotion of Science (M.A.) is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
TY and YN carried out the molecular biology studies. HS carried out the in silico study. MA carried out the real-time luciferase reporter assay. TM participated in the coordination, and provided financial support. TT conceived of the study, participated in its design and coordination, and drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Yamamoto, T., Nakahata, Y., Soma, H. et al. Transcriptional oscillation of canonical clock genes in mouse peripheral tissues. BMC Molecular Biol 5, 18 (2004). https://doi.org/10.1186/1471-2199-5-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2199-5-18