
Abstract
Background
Transferrin binding protein B (tbpB), an outer membrane lipoprotein, is required for the acquisition of iron from human transferrin. Two tbpB families have been documented in Neisseria meningitidis: an isotype I tbpB gene of 1.8 kb and an isotype II tbpB gene of 2.1 kb, the former expressed by meningococci in the disease-associated ST-11 clonal complex and the latter found among meningococci belonging to the hyper-invasive clonal complexes including ST-8, ST-18, ST-32, ST-41/44 as well as N. gonorrhoeae isolates. The origin of the isotype I tbpB gene is unknown, however several features in common with non-pathogenic Neisseria and the ST-11 clonal complex N. meningitidis isolate FAM18 have been documented leading to the hypothesis that the isotype I tbpB gene may also be shared between non-pathogenic Neisseria and ST-11 meningococci. As a result, the diversity of the tbpB gene was investigated in a defined collection of Neisseria species.
Results
Two families of isotype I tbpB were identified: family A containing conserved genes belonging to ST-11 meningococci, N. polysaccharea and N. lactamica isolates and family B including more diverse isotype I tbpB genes from N. sicca, N. mucosa, N. flava, N. subflava as well as N. cinerea, N. flavescens and N. polysaccharea isolates. Three isotype II tbpB families were identified with: family C containing diverse tbpB genes belonging to N. polysaccharea, N. lactamica, N. gonorrhoeae and N. meningitidis isolates, family D including another subset of isotype II tbpB genes from N. lactamica isolates and family E solely composed of N. gonorrhoeae tbpB genes.
Conclusion
This study reveals another instance of similarity between meningococci of the ST-11 clonal complex and non-pathogenic Neisseria with the origin of the isotype I tbpB gene resulting from a horizontal genetic transfer event occurring between these two populations.
Similar content being viewed by others

Background
The genus Neisseria contains 12 species and biovars colonising humans most of which are non-pathogenic colonisers of the upper respiratory tract [1, 2]. Only two species, Neisseria gonorrhoeae, the etiological agent of gonorrhoea and Neisseria meningitidis, a major cause of meningitis and septicaemia worldwide, regularly cause disease in humans [3, 4].
A major determinant in the survival of Neisseria within the human host is the ability to acquire iron, the majority of which is not circulating freely in the human body but is stored in ferritin and haemoglobin or is complexed with the glycoproteins transferrin and lactoferrin [5]. Neisseria have devised ways to counteract this iron limitation through the evolution of several high-affinity receptor systems including the lactoferrin binding proteins A and B, the transferrin binding proteins A and B, and the haptoglobin-haemoglobin receptor HpuAB, each composed of an accessory lipoprotein subunit and a TonB-dependent gated porin [6–11]. In addition, Neisseria can obtain iron through the expression of the surface-exposed haemoglobin receptor HmbR [12, 13].
Based on antigenic and genomic features of TbpB and tbpB, N. meningitidis isolates can be classified into two major families: isotype I (tbpB gene of 1.8 kb and TbpB protein with a mass of approximately 68 kDa) and isotype II (tbpB gene of 2.1 kb and TbpB protein with a mass of approximately 80 to 90 kDa) [14]. Isotype II tbpB genes have been documented in several N. meningitidis clonal complexes including ST-8, ST-32, ST-18 and ST-41/44 as well as non-pathogenic Neisseria [14–17]. Isotype I tbpB genes, on the other hand, have solely been identified among N. meningitidis isolates belonging to the ST-11 clonal complex and have not been detected among other Neisseria species. Four tbpB families were recently described based on partial nucleotide sequences from serogroup A clonal complex ST-4 N. meningitidis and N. lactamica isolates collected in The Gambia [16, 17]. Families one and four contained diverse isotype II tbpB alleles from N. meningitidis and N. lactamica isolates and families two and three included isotype I tbpB alleles. Importantly, among the 138 isolates analysed (98 serogroup A ST-4 meningococci, 12 unrelated meningococci, 22 N. lactamica isolates, and 6 unidentified Neisseria spp.) only three isotype I tbpB alleles were found, all of which belong to meningococci [16, 17].
Meningococci from the ST-11 clonal complex have been a major cause of meningococcal disease worldwide throughout the last century and despite low carriage rates continue to be associated with disease [18]. In addition to the isotype I tbpB gene, ST-11 meningococci can be distinguished from other hyper-virulent clonal complexes by the absence of an opcA gene and the possession of a class 2 porB gene [19–21]. Furthermore, similarities between the ST-11 clonal complex isolate FAM18 and non-pathogenic Neisseria spp. have been reported including the clustering of pilE sequences [22] and the comparable genetic organisation of the opcA negative locus in two N. polysaccharea isolates [23]. Taken together, these observations indicate the occurrence of specific horizontal genetic exchange events between commensal Neisseria and ST-11 meningococci which may have contributed to the described genetic isolation of this clonal complex [24]. The origin of the isotype I tbpB gene is unknown. Consequently, the distribution of the gene in a defined collection of Neisseria spp. was investigated with the hypothesis that the isotype I tbpB gene was present in other Neisseria spp.
Results
Identification of the tbpBgene
Isotype I tbpB genes were isolated from the non-pathogenic Neisseria spp. Two families of the gene became apparent. The first contained sequences closely related to meningococcal ST-11 tbpB genes belonging to three N. polysaccharea and two N. lactamica isolates. The second included more divergent isotype I tbpB genes from the non-pathogenic Neisseria spp. N. sicca, N. mucosa, N. flava, N. subflava, N. cinerea, N. flavescens as well as from another three N. polysaccharea isolates (Table 1). Isotype II tbpB genes were obtained from the remaining six N. lactamica and two N. polysaccharea isolates, while in agreement with previous studies, N. gonorrhoeae isolates contained isotype II tbpB genes (Table 1) [25]. A further 23 non-pathogenic Neisseria isolates were analysed and found to be negative for the tbpB gene. Among these were N. polysaccharea, N. perflava, N. sicca, N. subflava, N. flava and N. mucosa isolates. These isolates may contain divergent or truncated tbpB genes unable to be amplified with the primers used, however the remainder of this study will focus on the tbpB genes that were sequenced.
Functional assessment of the protein was beyond the scope of the present study. Nevertheless, previously documented putative transferrin binding sites were observed based on predicted translations of the nucleotide sequences [26, 27]. In particular, the highly conserved domains N3, N4 and C3, critical for efficient iron uptake and located in both the N- and C- terminal segments among isolates N. gonorrhoeae FA19, N. meningitidis M982, Moraxella catarrhalis 4223 and Acinetobacter pleuropneumoniae serotype 7, were also identified [27]. Domain N3 (residues 377 to 388 in N. gonorrhoeae FA19) displayed 100% sequence identity in both isotype I TbpB families, whereas six non-synonymous changes were observed among isotype II TbpB. Domain N4 (residues 409 to 422 in N. gonorrhoeae FA19) was also highly conserved among isotype I TbpB with the occurrence of three non-synonymous substitutions. Five variable sites were present among isotype II TbpB. Domain C3 (residues 703 to 713 in N. gonorrhoeae FA19) showed the most diversity with the occurrence of four non-synonymous substitutions among both TbpB isotypes.
Phylogenetic relationships inferred from novel Neisseria tbpBsequences
All of the sequences were aligned manually with sequences starting from and ending at the same amino acid residue in each isolate. Published isotype I and II tbpB sequences from isolates B16B6, M982, 8680, 8726, 2713, 2717 and FA19, used in previous analyses [14, 16, 17, 27], were also included in the alignment as well as those from the sequenced genomes of N. meningitidis isolates FAM18, Z2491, MC58 and N. gonorrhoeae FA1090.
Phylogenetic analysis was undertaken using the software package ClonalFrame version 1.1, which is a statistical model-based method initially described for inferring bacterial clonal relationships using multilocus sequence data [28]. Inference is performed in a Bayesian framework and a neutral coalescent model is assumed based on the hypothesis that the bacteria in the sample come from a constant-sized population in which each bacterium is equally likely to reproduce, irrespective of its previous history. The key assumption of ClonalFrame is that recombination events introduce a constant rate of substitutions to a contiguous region of sequence with the end result that a clonal frame can be inferred. In the present study, over 50,000 iterations were performed with every hundredth tree sampled after which a 95% majority-rule consensus tree was derived. Annotation was then undertaken by importing the tree into the Molecular Evolutionary Genetics Analysis software package (MEGA version 4.0) [29].
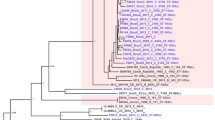
The two major isotype families were evident with each family containing a distinct cluster of genes (Fig. 1). The shortness of the branches for isotype I tbpB genes indicated that these were a closely related group of sequences compared with the depth of the branches seen for isotype II tbpB genes where greater diversity is known to occur [30]. Closer inspection of the tree revealed the two families of isotype I tbpB genes observed by sequence analysis as well as three clusters of isotype II tbpB genes. For ease of interpretation, the two isotype I tbpB families have been named tbpB A and tbpB B with the isotype II clusters named tbpB C through to tbpB E (Fig. 1 and Table 1). This nomenclature is proposed according to published guidelines in bacterial genetics [31] and is recommended in light of the emergence of the new families revealed in this study. Hitherto, studies in tbpB genetic diversity have focussed on a specific Neisseria spp. or meningococcal clonal complex and have not encompassed all of the Neisseria species included in the present work. This inclusion has provided a more detailed analysis of tbpB diversity and will allow a more flexible nomenclature for tbpB genes.

Phylogenetic tree inferred from aligned tbpB genes. Over 500 trees were generated using Clonalframe from which a 95% majority-rule consensus tree was derived and imported into MEGA version 4.0 for further annotation. Meningococcal reference tbpB genes (accession numbers in brackets) belonging to isolates B16B6, M982, 2713, 2717, 8710, 8680, FA19, FA1090, FAM18, Z2491 and MC58 [Genbank: Z15129, Z15130, AJ223044, AJ279554, Y09618, Y09977, U05205, U65219, AM421808, AL157959 and AE002098, respectively] were also included in the phylogenetic analysis. The proposed nomenclature for each tbpB family is indicated by large encircled letters. The nodes 1, 2 and 3 depicted with a diamond correspond to the recombination events presented in Figure 2. Open squares denote N. gonorrhoeae tbpB sequences, open circles N. lactamica, open triangles all of the other Neisseria spp. excluding N. polysaccharea, which are depicted with black circles and N. meningitidis which are represented by black squares.
Family tbpB A was comprised of genes most closely related to those of clonal complex ST-11 meningococci with four of these belonging to N. lactamica and N. polysaccharea isolates. Family tbpB B included a divergent cluster of isotype I genes (75% identity) belonging to a variety of Neisseria spp. as well as containing a subset of N. polysaccharea isolates (Fig. 1 and Tables 1 &2).
Three distinct isotype II tbpB families were apparent (Fig 1 and Tables 1 &2). Several gonococcal genes have clustered together and can be found in family tbpB E with families tbpB C and tbpB D containing genes belonging to N. lactamica, N. polysaccharea, N. gonorrhoeae and N. meningitidis isolates. Throughout the tree isolates have clustered by Neisseria species indicative of within species conservation of tbpB genes. The Genbank accession numbers for new tbpB genes sequenced in this study are listed in Table 2 as well as those belonging to previously submitted tbpB sequences.
Genetic diversity of the tbpBgenes
Genes belonging to family tbpB A were the least diverse (mean p-distance ranging from 0.001 to 0.040) with 85 polymorphic sites, the majority of which occur among N. polysaccharea and N. lactamica isolates. Overall, six fixed differences were observed between the genes of ST-11 meningococci and those of N. lactamica and N. polysaccharea with no shared polymorphisms between the two populations. Family B tbpB genes were more diverse (mean p-distance value 0.117) with 415 polymorphic sites. In a comparison of both gene families, there were 210 fixed differences and 54 shared mutations.
As expected, families tbpB C , D and E were more diverse (mean p-distances = 0.187, 0.140 and 0.112 respectively). Genes belonging to the N. polysaccharea isolates shared 99% identity with 16 segregating sites, seven of which encoded non-synonymous changes. N. lactamica tbpB genes were more diverse with 696 polymorphic sites while 829 polymorphisms were observed among N. gonorrhoeae tbpB genes. A total of 445 shared mutations were observed between N. lactamica and N. meningitidis tbpB C genes indicative of recombination, while only six were apparent between N. polysaccharea and N. meningitidis.
Very little recombination was noticeable among tbpB A genes or between these and family tbpB B , however a mosaic gene structure was present among the latter indicating that recombination occurred frequently among tbpB B genes from N. sicca, N. flava, N. subflava, N. mucosa, N. flavescens, N. cinerea and N. polysaccharea (Fig. 2a). As expected, tbpB genes from families tbpBC, Dand E recombined often (Fig. 2b &2c) with most of this occurring from bases 200 to 800.

ClonalFrame representation of tbpB recombination events. The nucleotide sequence of tbpB genes is represented on the x axis with the red line indicating at each locus the probability for an import on a scale from 0 (bottom of the y axis) to 1 (top of the y axis). Each inferred substitution in the graph is represented by a cross, the intensity of which indicating the posterior probability for that substitution. In panel A, recombination events occurring at node 1 in the phylogenetic tree (fig. 1) are represented. A mosaic gene structure is evident with fragments present between bases 150 and 300, 800 and 1000 and 1400 and 1800. In panel B, horizontal genetic exchange at node 2 are depicted occurring from bases 200 to 800 and, in panel C node 3 is represented.
Discussion
The aim of this study was to identify the origin of the isotype I tbpB gene. Previous observations have determined that these were confined to meningococci belonging to the ST-11 clonal complex [14, 32]. In contrast, isotype II genes were widely distributed among N. meningitidis clonal complexes and N. lactamica isolates [14, 16, 17]. The results presented here reveal the existence of isotype I tbpB genes among diverse Neisseria spp. Based on phylogenetic analysis these could be divided into two families: tbpB A containing genes homologous to ST-11 meningococci and tbpB B including more distantly related isotype I genes belonging to diverse non-pathogenic Neisseria spp. (Table 1 and Fig. 1). N. lactamica and N. polysaccharea isolates were found with both tbpB isotypes while, in agreement with previous studies, N. gonorrhoeae isolates solely contained isotype II tbpB genes [25]. Phylogenetic analysis demonstrated the presence of three isotype II families, named tbpB C through to tbpB E . Family C contained genes belonging to N. lactamica, N. polysaccharea, N. meningitidis and N. gonorrhoeae isolates, family D included another subset of isotype II tbpB genes belonging to N. lactamica and N. meningitidis isolates and finally, family E comprised N. gonorrhoeae genes (Table 1 and Fig. 1). In light of the tbpB families now present a new nomenclature is proposed according to published guidelines in bacterial genetics [31]. Previously, studies in tbpB genetic diversity focussed on a specific Neisseria spp. or meningococcal clonal complex and did not encompass all of the Neisseria spp. included in the present work [14–17, 25]. This inclusion has provided a more detailed analysis of tbpB diversity with the proposed nomenclature allowing more flexibility for future tbpB genes. Using this scheme genes can be grouped according to the family they belong to followed by an allele number.
A number of features are shared between clonal complex ST-11 N. meningitidis isolates and non-pathogenic Neisseria. Sequences upstream of the pilE gene from the class II pilin-producing N. meningitidis strain FAM18 were identical to the short region characterised upstream from N. polysaccharea pilE [22]. The N. polysaccharea isolate analysed (ATCC 43768) was included in the present study and harboured a tbpB gene similar to that of N. meningitidis isolate FAM18. Furthermore, opcA genes are absent among meningococci belonging the ST-11 clonal complex and were also undetectable among N. polysaccharea isolates 87043 and 90400 [19, 20], which were found in this study with isotype I tbpB genes. The identification of isotype I family A and B genes among Neisseria spp. is another characteristic shared with N. meningitidis isolates belonging to clonal complex ST-11 and is indicative of the occurrence of several horizontal genetic transfer events between non-pathogenic Neisseria, in particular N. polysaccharea and meningococci belonging to this clonal complex.
The evolutionary reasons leading to the existence of two tbpB isotypes among Neisseria are unknown. However, seclusion of isotype I tbpB to ST-11 clonal complex meningococci may be due to clonal expansion or selection for this isotype. Indeed, the isotype I TbpB protein has been shown to play an essential role in iron acquisition from human transferrin with isogenic mutants deficient in TbpB failing to grow on hTf as a sole iron source [33, 34]. Thus, both the TbpA and TbpB parts of the transferrin complex are critical. This was reflected in the lower diversity observed among tbpB genes belonging to families A (mean p-distance ranging from 0.001 to 0.040) and B (mean p-distance 0.117), highlighting the importance of this gene in contributing to the fitness of the organism. There has been selection for isotype I tbpB among meningococci belonging to the ST-11 clonal complex such that it has become restricted to this clonal complex. In contrast, isotype II tbpB genes have been found to provide a purely facilitating role such that TbpB-deficient mutants were only incapacitated with slower growth [34]. This has been confirmed in isogenic mutagenesis studies of both TbpA and TbpB in N. gonorrhoeae, H. influenzae and M. catarrhalis (which all contain isotype II-like tbpB genes) [35–37]. The non-essential role the isotype II tbpB gene has in iron acquisition may contribute to its hyper-variability. Indeed, Zhu et al found that the high rate of import among isotype II tbpB genes, although providing a temporary advantage because of antigenic composition, resulted in reduced fitness of the isolates [16, 17]. The higher recombination patterns observed in the present study among isotype II tbpB genes (Fig. 2b &2C) combined with the deeper phylogeny seen (Fig. 1) support this.
Conclusion
This work investigated the distribution of the two tbpB variants among Neisseria spp. and aimed to discover the origin of the isotype I tbpB gene. Results revealed this gene was found among diverse Neisseria spp. indicating the occurrence of a horizontal genetic transfer event between N. meningitidis and non-pathogenic Neisseria. Three features shared between ST-11 meningococci and non-pathogenic Neisseria have now been described: (i) the presence of isotype I tbpB genes (ii) the identical sequences upstream of the pilE gene and (iii) the analogous genetic organisation of the opcA negative locus.
A revised nomenclature was proposed according to the published guidelines [31]. The scheme now distinguishes isotype I tbpB genes into two new families: tbpB A and tbpB B the former contained tbpB genes closely related to ST-11 clonal complex meningococci, the latter included the more distantly related tbpB genes belonging to many non-pathogenic Neisseria species. The scheme also separates isotype II tbpB genes into three new families: tbpB C comprising tbpB genes from N. meningitidis, N. lactamica, N. polysaccharea and N. gonorrhoeae isolates, tbpB D consisting of tbpB genes from N. lactamica and N. meningitidis isolates and finally, tbpB E containing N. gonorrhoeae tbpB genes.
Methods
Growth of isolates and DNA preparation
The non-pathogenic Neisseria and N. gonorrhoeae isolates used in this study are listed in Table 2. Isolates were cultured overnight on GC agar (Difco) supplemented with 1% isovitalex (Oxoid) and grown at 37°C in the presence of 10% CO2. Boiled cell suspensions were prepared for each isolate. Briefly, a PBS solution of overnight GC grown bacteria was boiled for 5 minutes, centrifuged and the supernatant stored at +4°C before being directly used for PCR.
Nucleotide sequence determination
Amplification and sequencing of tbpB genes were completed using primers listed in Table 3. Degenerate primers were used for some of the sequencing steps. PCR products were PEG purified and either sequenced directly or cloned using the TOPO PCR TA cloning kit for sequencing (Invitrogen). Nucleotide sequence determination was carried out using the Li-Cor Global IR2 system along with the Sequitherm Excel II DNA sequencing kit (ScienceTec, France). Additional sequencing was carried out by cycle sequencing with BigDye Ready Reaction Mix (Applied Biosystems) according to manufacturer's instructions and using an ABI 377 automated DNA sequencer.
Data manipulation and analysis
The tbpB nucleotide sequences were assembled using the Staden sequence analysis package [38] and all sequences aligned manually in the Seqlab alignment program (Genetics Computer Group, Madison, Wis.). Phylogenetic analysis was undertaken using the software package ClonalFrame version 1.1, which is a statistical model-based method initially described for inferring bacterial clonal relationships using multilocus sequence data [28]. In the present study, over 50,000 iterations were performed with every hundredth tree sampled after which a 95% majority-rule consensus tree was derived. Annotation was then undertaken by importing the tree into the Molecular Evolutionary Genetics Analysis software package (MEGA version 4.0) [29].
The level of sequence diversity between tbpB genes was assessed by calculating p-distances within each tbpB family revealing the proportion (p) of nucleotide sites at which sequences differed. These analyses were conducted using MEGA. The number of fixed differences and shared polymorphisms were obtained using the software DnaSP [39]. Old and new accession numbers for tbpB genes are listed in table 2.
References
Knapp JS: Historical perspectives and identification of Neisseria and related species. Clin Microbiol Rev. 1988, 1: 415-431.
Vedros NA: Genus I. Neisseria Trevissan 1885. Bergey's Manual of systematic bacteriology. Edited by: Krieg NR and Holt JG. 1984, Baltimore, The Williams & Wilkins Co, 1: 290-296.
Gerbase AC, Rowley JT, Mertens TE: Global epidemiology of sexually transmitted diseases. Lancet. 1998, 351 : 2-4. 10.1016/S0140-6736(98)90001-0.
Rosenstein NE, Perkins BA, Stephens DS, Popovic T, Hughes JM: Meningococcal disease. N Engl J Med. 2001, 344: 1378-1388. 10.1056/NEJM200105033441807.
Otto BR, Verweij-van Vught AM, MacLaren DM: Transferrins and heme-compounds as iron sources for pathogenic bacteria. Crit Rev Microbiol. 1992, 18: 217-233. 10.3109/10408419209114559.
Prinz T, Meyer M, Pettersson A, Tommassen J: Structural characterization of the lactoferrin receptor from Neisseria meningitidis. J Bacteriol. 1999, 181: 4417-4419.
Schryvers AB, Morris LJ: Identification and characterization of the human lactoferrin-binding protein from Neisseria meningitidis. Infect Immun. 1988, 56: 1144-1149.
Ala'Aldeen DA, Borriello SP: The meningococcal transferrin-binding proteins 1 and 2 are both surface exposed and generate bactericidal antibodies capable of killing homologous and heterologous strains. Vaccine. 1996, 14: 49-53. 10.1016/0264-410X(95)00136-O.
Schryvers AB, Morris LJ: Identification and characterization of the transferrin receptor from Neisseria meningitidis. Mol Microbiol. 1988, 2: 281-288. 10.1111/j.1365-2958.1988.tb00029.x.
Lewis LA, Dyer DW: Identification of an iron-regulated outer membrane protein of Neisseria meningitidis involved in the utilization of hemoglobin complexed to haptoglobin. J Bacteriol. 1995, 177: 1299-1306.
Lewis LA, Gray E, Wang YP, Roe BA, Dyer DW: Molecular characterization of hpuAB, the haemoglobin-haptoglobin-utilization operon of Neisseria meningitidis. Mol Microbiol. 1997, 23: 737-749. 10.1046/j.1365-2958.1997.2501619.x.
Stojiljkovic I, Hwa V, de Saint Martin L, O'Gaora P, Nassif X, Heffron F, So M: The Neisseria meningitidis haemoglobin receptor: its role in iron utilization and virulence. Mol Microbiol. 1995, 15: 531-541. 10.1111/j.1365-2958.1995.tb02266.x.
Stojiljkovic I, Larson J, Hwa V, Anic S, So M: HmbR outer membrane receptors of pathogenic Neisseria spp.: iron-regulated, hemoglobin-binding proteins with a high level of primary structure conservation. J Bacteriol. 1996, 178: 4670-4678.
Rokbi B, Renauld-Mongenie G, Mignon M, Danve B, Poncet D, Chabanel C, Caugant DA, Quentin-Millet MJ: Allelic diversity of the two transferrin binding protein B gene isotypes among a collection of Neisseria meningitidis strains representative of serogroup B disease: implication for the composition of a recombinant TbpB-based vaccine. Infect Immun. 2000, 68: 4938-4947. 10.1128/IAI.68.9.4938-4947.2000.
Rokbi B, Mignon M, Caugant DA, Quentin-Millet MJ: Heterogeneity of tbpB, the transferrin-binding protein B gene, among serogroup B Neisseria meningitidis strains of the ET-5 complex. Clin Diagn Lab Immunol. 1997, 4: 522-529.
Linz B, Schenker M, Zhu P, Achtman M: Frequent interspecific genetic exchange between commensal Neisseriae and Neisseria meningitidis. Mol Microbiol. 2000, 36: 1049-1058. 10.1046/j.1365-2958.2000.01932.x.
Zhu P, van der Ende A, Falush D, Brieske N, Morelli G, Linz B, Popovic T, Schuurman IG, Adegbola RA, Zurth K, Gagneux S, Platonov AE, Riou JY, Caugant DA, Nicolas P, Achtman M: Fit genotypes and escape variants of subgroup III Neisseria meningitidis during three pandemics of epidemic meningitis. Proc Natl Acad Sci U S A. 2001, 98: 5234-5239. 10.1073/pnas.061386098.
Bentley SD, Vernikos GS, Snyder LA, Churcher C, Arrowsmith C, Chillingworth T, Cronin A, Davis PH, Holroyd NE, Jagels K, Maddison M, Moule S, Rabbinowitsch E, Sharp S, Unwin L, Whitehead S, Quail MA, Achtman M, Barrell B, Saunders NJ, Parkhill J: Meningococcal genetic variation mechanisms viewed through comparative analysis of serogroup C strain FAM18. PLoS Genet. 2007, 3: e23-10.1371/journal.pgen.0030023.
Seiler A, Reinhardt R, Sarkari J, Caugant DA, Achtman M: Allelic polymorphism and site-specific recombination in the opc locus of Neisseria meningitidis. Mol Microbiol. 1996, 19: 841-856. 10.1046/j.1365-2958.1996.437970.x.
Zhu P, Morelli G, Achtman M: The opcA and (psi)opcB regions in Neisseria: genes, pseudogenes, deletions, insertion elements and DNA islands. Mol Microbiol. 1999, 33: 635-650. 10.1046/j.1365-2958.1999.01514.x.
Wang JF, Caugant DA, Morelli G, Koumare B, Achtman M: Antigenic and epidemiologic properties of the ET-37 complex of Neisseria meningitidis. J Infect Dis. 1993, 167: 1320-1329.
Aho EL, Urwin R, Batcheller AE, Holmgren AM, Havig K, Kulakoski AM, Vomhof EE, Longfors NS, Erickson CB, Anderson ZK, Dawlaty JM, Mueller JJ: Neisserial pilin genes display extensive interspecies diversity. FEMS Microbiol Lett. 2005, 249: 327-334. 10.1016/j.femsle.2005.06.035.
Zhu P, Klutch MJ, Derrick JP, Prince SM, Tsang RS, Tsai CM: Identification of opcA gene in Neisseria polysaccharea: interspecies diversity of Opc protein family. Gene. 2003, 307: 31-40. 10.1016/S0378-1119(02)01208-8.
Claus H, Stoevesandt J, Frosch M, Vogel U: Genetic isolation of meningococci of the electrophoretic type 37 complex. J Bacteriol. 2001, 183: 2570-2575. 10.1128/JB.183.8.2570-2575.2001.
Cornelissen CN, Anderson JE, Sparling PF: Characterization of the diversity and the transferrin-binding domain of gonococcal transferrin-binding protein 2. Infect Immun. 1997, 65: 822-828.
Renauld-Mongenie G, Lins L, Krell T, Laffly L, Mignon M, Dupuy M, Delrue RM, Guinet-Morlot F, Brasseur R, Lissolo L: Transferrin-binding protein B of Neisseria meningitidis: sequence-based identification of the transferrin-Binding site confirmed by site-directed mutagenesis. J Bacteriol. 2004, 186: 850-857. 10.1128/JB.186.3.850-857.2004.
DeRocco AJ, Cornelissen CN: Identification of transferrin-binding domains in TbpB expressed by Neisseria gonorrhoeae. Infect Immun. 2007, 75: 3220-3232. 10.1128/IAI.00072-07.
Didelot X, Falush D: Inference of bacterial microevolution using multilocus sequence data. Genetics. 2007, 175: 1251-1266. 10.1534/genetics.106.063305.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol Biol Evol. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
Rokbi B, Maitre-Wilmotte G, Mazarin V, Fourrichon L, Lissolo L, Quentin-Millet MJ: Variable sequences in a mosaic-like domain of meningococcal tbp2 encode immunoreactive epitopes. FEMS Microbiol Lett. 1995, 132: 277-283. 10.1111/j.1574-6968.1995.tb07846.x.
Demerec M, Adelberg EA, Clark AJ, Hartman PE: A proposal for a uniform nomenclature in bacterial genetics. Genetics. 1966, 54: 61-76.
Rokbi B, Mazarin V, Maitre-Wilmotte G, Quentin-Millet MJ: Identification of two major families of transferrin receptors among Neisseria meningitidis strains based on antigenic and genomic features. FEMS Microbiol Lett. 1993, 110: 51-57. 10.1111/j.1574-6968.1993.tb06294.x.
Irwin SW, Averil N, Cheng CY, Schryvers AB: Preparation and analysis of isogenic mutants in the transferrin receptor protein genes, tbpA and tbpB, from Neisseria meningitidis. Mol Microbiol. 1993, 8: 1125-1133. 10.1111/j.1365-2958.1993.tb01657.x.
Renauld-Mongenie G, Poncet D, Mignon M, Fraysse S, Chabanel C, Danve B, Krell T, Quentin-Millet MJ: Role of transferrin receptor from a Neisseria meningitidis tbpB isotype II strain in human transferrin binding and virulence. Infect Immun. 2004, 72: 3461-3470. 10.1128/IAI.72.6.3461-3470.2004.
Anderson JE, Sparling PF, Cornelissen CN: Gonococcal transferrin-binding protein 2 facilitates but is not essential for transferrin utilization. J Bacteriol. 1994, 176: 3162-3170.
Gray-Owen SD, Loosmore S, Schryvers AB: Identification and characterization of genes encoding the human transferrin-binding proteins from Haemophilus influenzae. Infect Immun. 1995, 63: 1201-1210.
Luke NR, Campagnari AA: Construction and characterization of Moraxella catarrhalis mutants defective in expression of transferrin receptors. Infect Immun. 1999, 67: 5815-5819.
Staden R: The Staden sequence analysis package. Mol Biotechnol. 1996, 5: 233-241. 10.1007/BF02900361.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Perrin A, Bonacorsi S, Carbonnelle E, Talibi D, Dessen P, Nassif X, Tinsley C: Comparative genomics identifies the genetic islands that distinguish Neisseria meningitidis, the agent of cerebrospinal meningitis, from other Neisseria species. Infection and Immunity. 2002, 70: 7063-7072. 10.1128/IAI.70.12.7063-7072.2002.
Perrin A, Nassif X, Tinsley C: Identification of regions of the chromosome of Neisseria meningitidis and Neisseria gonorrhoeae which are specific to the pathogenic Neisseria species. Infection and Immunity. 1999, 67: 6119-6129.
Qvarnstrom Y, Swedberg G: Sulphonamide resistant commensal Neisseria with alterations in the dihydropteroate synthase can be isolated from carriers not exposed to sulphonamides. BMC Microbiol. 2002, 2: 34-10.1186/1471-2180-2-34.
Veron M, Thibault P, L. O: [Neisseria mucosa (Diplococcus mucosus Lingelsheim). I. Bacteriological description and study of its pathogenicity]. Ann Inst Pasteur (Paris). 1959, 97: 497-510.
Zhu P, Klutch MJ, Bash MC, Tsang RS, Ng LK, Tsai CM: Genetic diversity of three lgt loci for biosynthesis of lipooligosaccharide (LOS) in Neisseria species. Microbiology. 2002, 148: 1833-1844.
Arking D, Tong Y, DC S: Analysis of lipooligosaccharide biosynthesis in the Neisseriaceae. Journal of Bacteriology. 2001, 183: 934-941. 10.1128/JB.183.3.934-941.2001.
Branham SE: A new meningococcus-like organism (Neisseria flavescens m.sp.) from epidemic meningitis. Public Health Report. 1930, 45: 845-849.
Taha MK, Marchal C: Conservation of Neisseria gonorrhoeae pilus expression regulatory genes pilA and pilB in the genus Neisseria. Infection and Immunity. 1990, 58: 4145-4148.
Barrett SJ, Sneath PHA: A numerical phenotypic taxonomic study of the genus Neisseria. Microbiology. 1994, 140: 2867-2891.
Guibourdenche M, Popoff MY, Riou JY: Deoxyribonucleic acid relatedness among Neisseria gonorrhoeae, N. meningitidis, N. lactamica, N. cinerea and "Neisseria polysaccharea". Ann Inst Pasteur Microbiol. 1986, 137B: 177-185. 10.1016/S0769-2609(86)80106-5.
Spratt BG, Bowler LD, Zhang QY, Zhou J, Smith JM: Role of interspecies transfer of chromosomal genes in the evolution of penicillin resistance in pathogenic and commensal Neisseria species. J Mol Evol. 1992, 34: 115-125. 10.1007/BF00182388.
Zhu P, Tsang RS, Tsai CM: Nonencapsulated Neisseria meningitidis strain produces amylopectin from sucrose: altering the concept for differentiation between N. meningitidis and N. polysaccharea. J Clin Microbiol. 2003, 41: 273-278. 10.1128/JCM.41.1.273-278.2003.
Anand CM, Ashton F, Shaw H, Gordon R: Variability in growth of Neisseria polysaccharea on colistin-containing selective media for Neisseria spp. J Clin Microbiol. 1991, 29: 2434-2437.
Berger U: First isolation of Neisseria polysacchareae species nova in the Federal Republic of Germany. Eur J Clin Microbiol. 1985, 4: 431-433. 10.1007/BF02148705.
Legrain M, Rokbi B, Villeval D, Jacobs E: Characterization of genetic exchanges between various highly divergent tbpBs, having occurred in Neisseria meningitidis. Gene. 1998, 208: 51-59. 10.1016/S0378-1119(97)00646-X.
Acknowledgements
This work was funded by a Marie Curie fellowship provided by the European Commission and the Human Potential programme. The authors would like to thank D. Caugant (NIPH Norway), P. Kriz (NIPH Czech Republic), C. Ferreirós (Universidad de Santiago, Spain), JM. Alonso and MK. Taha (The Pasteur Institute, France), Y. Qvarnström (Uppsala University, Sweden), N. Bilek (Imperial College, UK), R. Tsang, (National Microbiology Laboratory, Canada) and N. Saunders (University of Oxford, UK) for the kind donation of strains.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
OBH participated in the planning of this study, performed all experimental work, data analysis and drafted the manuscript. MCJM participated in writing the manuscript. BR participated in the planning of this study, coordinated the study and assisted in writing the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Harrison, O.B., Maiden, M.C. & Rokbi, B. Distribution of transferrin binding protein B gene (tbpB) variants among Neisseriaspecies. BMC Microbiol 8, 66 (2008). https://doi.org/10.1186/1471-2180-8-66
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-8-66