
Abstract
Background
For typing of Staphylococcus aureus, DNA sequencing of the repeat region of the protein A (spa) gene is a well established discriminatory method for outbreak investigations. Recently, it was hypothesized that this region also reflects long-term epidemiology. However, no automated and objective algorithm existed to cluster different repeat regions. In this study, the Based Upon Repeat Pattern (BURP) implementation that is a heuristic variant of the newly described EDSI algorithm was investigated to infer the clonal relatedness of different spa types.
For calibration of BURP parameters, 400 representative S. aureus strains with different spa types were characterized by MLST and clustered using eBURST as "gold standard" for their phylogeny. Typing concordance analysis between eBURST and BURP clustering (spa-CC) were performed using all possible BURP parameters to determine their optimal combination. BURP was subsequently evaluated with a strain collection reflecting the breadth of diversity of S. aureus (JCM 2002; 40:4544).
Results
In total, the 400 strains exhibited 122 different MLST types. eBURST grouped them into 23 clonal complexes (CC; 354 isolates) and 33 singletons (46 isolates). BURP clustering of spa types using all possible parameter combinations and subsequent comparison with eBURST CCs resulted in concordances ranging from 8.2 to 96.2%. However, 96.2% concordance was reached only if spa types shorter than 8 repeats were excluded, which resulted in 37% excluded spa types. Therefore, the optimal combination of the BURP parameters was "exclude spa types shorter than 5 repeats" and "cluster spa types into spa-CC if cost distances are less than 4" exhibiting 95.3% concordance to eBURST. This algorithm identified 24 spa-CCs, 40 singletons, and excluded only 7.8% spa types. Analyzing the natural population with these parameters, the comparison of whole-genome micro-array groupings (at the level of 0.31 Pearson correlation index) and spa-CCs gave a concordance of 87.1%; BURP spa-CCs vs. manually grouped spa types resulted in 95.7% concordance.
Conclusion
BURP is the first automated and objective tool to infer clonal relatedness from spa repeat regions. It is able to extract an evolutionary signal rather congruent to MLST and micro-array data.
Similar content being viewed by others

Background
Staphylococcus aureus, a human commensal living on the skin and mucosa, can cause a broad range of infections including endocarditis, septicemia, skin infections, soft tissue infections, and osteomyelitis. Moreover, S. aureus is the leading cause of nosocomial infections [1]. The application of several new genotypic typing methods gave many new insights into the epidemiology and population structure of S. aureus [2]. Recently, Koreen et al. investigated a collection of 36 S. aureus isolates (methicillin resistant and methicillin sensible S. aureus, MRSA and MSSA, respectively), which was recovered from 10 countries on four continents over a period of four decades as a representative of the breadth of diversity within S. aureus [3]. They used whole-genome micro-array analysis (comprising approximately 2,800 open reading frames) as typing reference to evaluate the capability of several typing techniques, among them partial S. aureus protein A (spa) gene sequencing. The spa repeat region consists of a variable number of 21–27 bp long repeats (VNTRs) varying in composition that result in different spa types.
Previously it was shown that spa typing is fast, discriminatory, and very reproducible [4, 5]. It was hypothesized by Koreen and colleagues that by manual grouping of similar spa types this region contains evolutionary signals nearly comparable to whole-genome micro-array data [3]. Until recently, however, no automated and objective algorithm existed to cluster different repeat regions. The Based Upon Repeat Pattern (BURP) implementation that is a heuristic variant of the newly described EDSI algorithm [6], was investigated in this study to infer the clonal relatedness of different spa types. We first calibrated the BURP parameters using multilocus sequence typing (MLST) data from a representative strain collection as "gold standard" and then evaluated BURP using the Koreen et al. dataset.
Methods
S. aureus strains (MRSA and MSSA) were used from our strain collection comprising 400 of the initial and most frequently to the SpaServer reported spa types [7]. From these strains, MLST sequence types (ST) were determined as previously [8]. STs that showed at least six of seven identical alleles were grouped into clonal complexes (CC) using eBURST [9]. BURP – as implemented in the StaphType software v. 1.5 (Ridom GmbH, Würzburg, Germany) – was used to cluster (spa-CC) spa types [10]. Repeat-duplication and -excision in addition to substitution and base-insertion and -deletion events were taken into account when the relatedness of different spa types was calculated. BURP offers two user-defined parameters that influence clustering: exclusion of spa types that are shorter than "x" repeats and the maximum number of costs "y" for clustering spa types into the same group. Short spa types can be excluded from further analysis because their information content is limited and no reliable evolutionary history can be inferred. The costs account for the "steps" of evolution between two different spa types, whereas the algorithm tries to minimize these steps ("parsimony assumption"). To find out the optimal combination of these two parameters, clustering of all possible combinations of both parameters (values: 1 to 10) was performed. A prerequisite was that the number of excluded spa types should be as low as possible and not exceed 10% of all investigated spa types. Subsequently, the typing concordance [11] between BURP and eBURST groupings were determined to elucidate the best parameter combination with the highest concordance on the one side and the lowest number of excluded spa types on the other. BURP calibrated in this manner was finally used to cluster the strains from the study of Koreen et al. [3].
Results and discussion
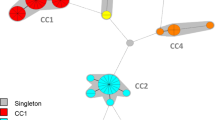
In total, the 400 investigated strains exhibited 122 different STs. The eBURST algorithm clustered the STs into 23 CCs (354 isolates) and 33 singletons (46 isolates). BURP clustering of spa types using all possible parameter combinations and subsequent comparison with eBURST CCs resulted in concordances ranging from 8.2 to 96.2%. These concordances are illustrated in Figure 1 using the Visual-XSel 9.0 software (CRGraph, München, Germany). To determine the optimal combination between the BURP parameters, a graph showing the dependence of the concordance from the minimal repeat length of included spa types (vice versa the percentage of excluded spa types) for the different costs were drawn by MS Excel XP. The overall highest concordance (96.2%) lay on the cost value 4 curve (Figure 2). Analyzing this curve, the closest integer to the first inflection point – representing the first local maximum – was chosen as the optimal combination of the BURP parameters with "exclude spa types that are shorter than 5 repeats" and "spa types are clustered if costs are less or equal than 4". In this way a concordance of 95.3% could be achieved (Figure 2). Using these parameters, BURP clustered the 400 spa types into 24 spa-CCs and 40 singletons. Only 31 (7.8%) spa types were excluded by using these parameters. In contrast, analysis of ungrouped spa types vs. eBURST CCs resulted in 92.8% concordance, only. A population snapshot of the 369 included strains after BURP grouping is displayed in Figure 3. It shows clusters of linked spa types in spa-CCs, linked doublets, and individual unlinked spa types. In Table 1, exemplarily the spa-CC004, its spa types, corresponding STs, and CCs is shown. In general, a high concordance between BURP and eBURST clustering can be observed. Of the 50 spa types that were clustered in spa-CC004, only three spa types were grouped into another CC and another three were judged as singletons by MLST.

Concordance analysis of eBURST and BURP clustering in dependence of all possible BURP parameters. A surface curve displaying the dependence of concordance (in %) between eBURST MLST CCs and BURP spa-CCs applying all possible combinations of the BURP parameters "exclude spa types that are shorter than x repeats" and "spa types are clustered if costs are less or equal than y".

High range of concordance between eBURST and BURP for optimal BURP calibration. Graph showing curves for cost integers in the high concordance range. Curves labeled "Costs: 1 to 10" represent different cost values. For the curve with the overall highest concordance (Costs: 4) the first inflection point is marked (arrow) and corresponds to the first local optimum giving a good balance between concordance and percentage of excluded spa types.

Population snapshot of the 400 S. aureus strains after BURP grouping. Population snapshot of the 400 S. aureus strains after grouping with the calibrated BURP ("exclude spa types that are shorter than 5 repeats" and "spa types are clustered if costs are less or equal than 4", 31 spa types were excluded). Clusters of linked isolates correspond to spa-CCs. Whereas eBURST uses the number of relatives (single locus variants, SLVs) to define founders and subfounders of groups, BURP sums up costs to define a founder-score for each spa type in a cluster. The spa type with the highest founder-score is defined founder of the cluster (blue color). Subfounders are the spa types with the second highest founder-score and are labeled in yellow. If two or more spa types exhibit the same highest founder-score, they are all colored in blue. For clarity, only the spa-CCs are labeled. Note that the spacing between linked spa types and between unlinked spa types and spa-CCs provides no information concerning the genetic distance between them.
Comparing whole-genome micro-array groupings (at the level of 0.31 Pearson correlation index) and spa-CCs of the 36 strains from the study of Koreen et al. using the calibrated BURP gave a concordance of 87.1% – that is in the same range as reported. BURP spa-CCs vs. manually grouped spa types resulted in 95.7% concordance.
The underlying alignment model of BURP takes repeat-duplication and -excision into account [6] – in contrast to widely-used multiple alignment strategies like ClustalW [12]. The proposed molecular mechanism of the evolution of such repeat regions is slipped-strand mispairing (SSM) during DNA duplication [13]. The presence of those evolutionary events within the spa repeat region was already detected in vivo, when sequential S. aureus isolates from long-term pulmonary S. aureus colonization/infection of cystic fibrosis patients were spa typed [14].
The high concordance of BURP spa-CCs in comparison to eBURST CCs using a diverse strain collection demonstrated that spa indeed contains long-term evolutionary signals. Recent comparisons between spa-CCs and PFGE clustering corroborated these findings [15, 16]. In future, the integration of BURP into the already established early-warning system for MRSA-outbreaks based on spa typing will help to detect clonal diversification during extended outbreaks [17].
There are some limitations using spa-CCs for long-term analysis. First, the strains must be spa-typeable. Having typed more than 8,000 isolates, however, very few isolates (approximately 0.1%) were not typeable – probably due to mutations within the primer binding regions. Second, BURP analyses are limited to spa types that pass the parameter of a certain number of repeats. However, when analyzing the SpaServer content (accessed at 12th September 2007) comprising 38,978 isolates with 2964 different spa types, only 204 (6.88%) of all spa types and 881 (2,26%) of all submitted isolates are effected, respectively. Finally, in very few instances, discrepancies can occur between spa and other typing methods as observed in this study and in two recent other publications [15, 16]. These discrepancies are most probably due to recombinational events. Large chromosomal replacements that give rise to such typing incongruences have been experimentally documented for two STs previously [18].
Conclusion
In summary, BURP is the first automated and objective tool to infer clonal relatedness from spa repeat regions. It is able to extract an evolutionary signal rather congruent to MLST and micro-array data.
Abbreviations
- BURP:
-
Based Upon Repeat Pattern
- CC:
-
clonal complex
- eBURST:
-
electronic Based Upon Related Sequence Types
- EDSI:
-
excision and duplication of repeats and substitution and indels of bases
- MLST:
-
multilocus sequence typing
- MRSA:
-
methicillin resistant S. aureus
- MSSA:
-
methicillin sensible S. aureus
- PFGE:
-
pulsed-field gel electrophoresis
- spa :
-
S. aureus protein A encoding gene
- ST:
-
sequence type
- VNTR:
-
variable number tandem repeats
References
National Nosocomial Infections Surveillance (NNIS): National Nosocomial Infections Surveillance (NNIS) System Report, data summary from January 1992 through June issued August 2003. Am J Infect Control. 2003, 31: 481-498. 10.1016/j.ajic.2003.09.002.
Feil EJ, Cooper JE, Grundmann H, Robinson DA, Enright MC, Berendt T, Peacock SJ, Smith JM, Murphy M, Spratt BG, Moore CE, Day NPJ: How clonal is Staphylococcus aureus?. J Bacteriol. 2003, 185: 3307-3316. 10.1128/JB.185.11.3307-3316.2003.
Koreen L, Ramaswamy SV, Graviss EA, Naidich S, Musser JM, Kreiswirth BN: spa typing method for discriminating among Staphylococcus aureus isolates: implications for use of a single marker to detect genetic micro- and macrovariation. J Clin Microbiol. 2004, 42: 792-799. 10.1128/JCM.42.2.792-799.2004.
Aires-de-Sousa M, Boye K, de Lencastre H, Deplano A, Enright MC, Etienne J, Friedrich AW, Harmsen D, Holmes A, Huijsdens X, Kearns A, Mellmann A, Meugnier H, Rasheed JK, Spalburg E, Strommenger B, Struelens MJ, Tenover FC, Thomas J, Vogel U, Westh H, Xu J, Witte W: High inter-laboratory reproducibility of DNA sequence-based typing of bacteria in a multicenter study. J Clin Microbiol. 2006, 44: 619-621. 10.1128/JCM.44.2.619-621.2006.
Shopsin B, Gomez M, Montgomery SO, Smith DH, Waddington M, Dodge DE, Bost DA, Riehman M, Naidich S, Kreiswirth BN: Evaluation of protein A gene polymorphic region DNA sequencing for typing of Staphylococcus aureus strains. J Clin Microbiol. 1999, 37: 3556-3563.
Sammeth M, Stoye J: Comparing tandem repeats with duplications and excisions of variable degree. IEEE/ACM Trans Comput Biol Bioinform. 2006, 3: 395-407. 10.1109/TCBB.2006.46.
Ridom SpaServer. [http://www.spaServer.ridom.de]
Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG: Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol. 2000, 38: 1008-1015.
Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG: eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004, 186: 1518-1530. 10.1128/JB.186.5.1518-1530.2004.
Harmsen D, Claus H, Witte W, Rothgänger J, Claus H, Turnwald D, Vogel U: Typing of methicillin-resistant Staphylococcus aureus in a university hospital setting by using novel software for spa repeat determination and database management. J Clin Microbiol. 2003, 41: 5442-5448. 10.1128/JCM.41.12.5442-5448.2003.
Robinson DA, Hollingshead SK, Musser JM, Parkinson AJ, Briles DE, Crain MJ: The IS1167 insertion sequence is a phylogenetically informative marker among isolates of serotype 6B Streptococcus pneumoniae. J Mol Evol. 1998, 47: 222-229. 10.1007/PL00006379.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
van Belkum A, Scherer S, van Alphen L, Verbrugh H: Short-sequence DNA repeats in prokaryotic genomes. Microbiol Mol Biol Rev. 1998, 62: 275-293.
Kahl BC, Mellmann A, Deiwick S, Peters G, Harmsen D: Variation of the polymorphic region X of the protein A gene during persistent airway infection of cystic fibrosis patients reflects two independent mechanisms of genetic change in Staphylococcus aureus. J Clin Microbiol. 2005, 43: 502-505. 10.1128/JCM.43.1.502-505.2005.
Hallin M, Deplano A, Denis O, De Mendonca R, De Ryck R, Struelens MJ: Validation of pulsed-field gel electrophoresis and spa typing for long-term, nationwide epidemiological surveillance studies of Staphylococcus aureus infections. J Clin Microbiol. 2007, 45: 127-133. 10.1128/JCM.01866-06.
Strommenger B, Kettlitz C, Weniger T, Harmsen D, Friedrich AW, Witte W: Assignment of Staphylococcus isolates to groups by spa typing, SmaI macrorestriction analysis, and multilocus sequence typing. J Clin Microbiol. 2006, 44: 2533-2540. 10.1128/JCM.00420-06.
Mellmann A, Friedrich AW, Rosenkötter N, Rothgänger J, Karch H, Reintjes R, Harmsen D: Automated DNA sequence-based early warning system for the detection of methicillin-resistant Staphylococcus aureus outbreaks. PLoS Med. 2006, 3: e33-10.1371/journal.pmed.0030033.
Robinson DA, Enright MC: Evolutionary models of the emergence of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2003, 47: 3926-3934. 10.1128/AAC.47.12.3926-3934.2003.
Acknowledgements
A. Mellmann was funded by a grant from the Deutsche Forschungsgemeinschaft (ME 3205/1-1). This work was supported by a post-doctoral fellowship of the German Academic Exchange Service to M. Sammeth.
The authors thank K. Becker, B.C. Kahl, B. Sinha (Münster, Germany), M.C. Enright (London, UK), H. Grundmann (Bilthoven, The Netherlands), A.M. Kearns (London, UK), A.C. Peterson (Lund, Sweden), A. Sabat (Warsaw, Poland), U. Vogel (Würzburg, Germany), K. Boye, H. Westh (Kopenhagen, Denmark), B. Strommenger, and W. Witte (Wernigerode, Germany) for supplying strains. Furthermore, the skillful technical assistance of I. Ramminger and U. Keckevoet is gratefully acknowledged.
This study was presented in part at the 106th General Meeting of the American Society for Microbiology, Orlando, FL, May 22–25, 2006.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
J. Rothgänger and D. Harmsen have declared a potential conflict of interest. J. Rothgänger and D. Harmsen are the developers of the Ridom StaphType software mentioned in the manuscript. The software is distributed and sold by the company Ridom GmbH that is partially owned by them. All other authors have declared that no competing interests exist.
Authors' contributions
The project was coordinated by DH. AM and CB performed the laboratory work and data analysis. MS and JS developed the EDSI algorithm. TW and JR implemented BURP. AM, TW, and DH wrote the main part of the paper. All other authors gave useful comment on the analysis of data and text of the manuscript. All authors have read and approved the final version of the manuscript.
Alexander Mellmann, Thomas Weniger contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mellmann, A., Weniger, T., Berssenbrügge, C. et al. Based Upon Repeat Pattern (BURP): an algorithm to characterize the long-term evolution of Staphylococcus aureus populations based on spa polymorphisms. BMC Microbiol 7, 98 (2007). https://doi.org/10.1186/1471-2180-7-98
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-7-98