
Abstract
Background
Sunflower downy mildew is a major disease caused by the obligatory biotrophic oomycete Plasmopara halstedii. Little is known about the molecular mechanisms underlying its pathogenicity. In this study we used a genomics approach to gain a first insight into the transcriptome of P. halstedii.
Results
To identify genes from the obligatory biotrophic oomycete Plasmopara halstedii that are expressed during infection in sunflower (Helianthus annuus L.) we employed the suppression subtraction hybridization (SSH) method from sunflower seedlings infected by P. halstedii. Using this method and random sequencing of clones, a total of 602 expressed sequence tags (ESTs) corresponding to 230 unique sequence sets were identified. To determine the origin of the unisequences, PCR primers were designed to amplify these gene fragments from genomic DNA isolated either from P. halstedii sporangia or from Helianthus annuus. Only 145 nonredundant ESTs which correspond to a total of 373 ESTs (67.7%) proved to be derived from P. halstedii genes and that are expressed during infection in sunflower. A set of 87 nonredundant sequences were identified as showing matches to sequences deposited in public databases. Nevertheless, about 7% of the ESTs seem to be unique to P. halstedii without any homolog in any public database.
Conclusion
A summary of the assignment of nonredundant ESTs to functional categories as well as their relative abundance is listed and discussed. Annotation of the ESTs revealed a number of genes that could function in virulence. We provide a first glimpse into the gene content of P. halstedii. These resources should accelerate research on this important pathogen.
Similar content being viewed by others


Background
Sunflower downy mildew is a major disease caused by the Oomycete Plasmopara halstedii (Farl.) Berl et de Toni. The first physiological race of this obligate parasitic oomycete has been identified by Zimmer in North America and Europe [1]. Both in compatible and incompatible interactions, host penetration occurs at the lower part of the hypocotyl [2]. Usually, about thirteen days after artificial infection of susceptible lines, the parasite invades almost all the plant tissues and is present in the cotyledons, epicotyls and leaves. In contrast, from the fifth days and onwards, a hypersensitive-like reaction develops within the hypocotyl of resistant lines and in many cases, the parasite's growth is arrested before it reaches the cotyledons [2, 3]. Molecular analysis showed that the resistance could be associated with an unusual delayed hypersensitive reaction and a systemic acquired response that take place inside the hypocotyls with the seedlings showing no apparent symptoms [3].
The establishment of the disease or the resistance is the result of the expression of defence genes in the host and virulence or pathogenicity genes in the parasite. In the sunflower, some defence-related genes whose expression varied in compatible and incompatible interactions have been characterized [3, 4]. In contrast, genes from P. halstedii potentially involved in the infectious process have not been reported yet. This may be explained by the obligate nature of the development of P. halstedii on its host.
Since completion of the Saccharomyces cerevisiae genome [5], progress on the Genome sequence information and expressed sequence tag (EST) collections from several other parasitic and symbiotic fungi that infect humans, other animals and plants are also becoming more widespread [6, 7]. More recently, the whole genome sequences of Phytophthora ramorum and Phytophthora sojae, two major oomycetes pathogens have been reported [8], providing the framework for comparative genomics studies [9] or the identification of specific gene families potentially implicated in the infectious process [10]. Similarly, the availability of the whole genome sequence of Hyaloperonospora parasitica genome should help in the discovery of similar genes in the other oomycetes [11]. The EST (Expressed Sequence Tags) approach represents a relatively simple procedure for finding genes and generating information about their expression in organisms with no genetic research history [12]. For example, in Blumeria graminis, 4908 ESTs representing 1669 individual genes have been obtained by sequencing clones from two cDNA libraries from germinating and ungerminated conidia [13]. In a different work, van der Biezen et al.[14] used the cDNA-AFLP strategy to clone 10 cDNA fragments from the obligatory biotrophic oomycete Hyaloperonospora parasitica (formerly Peronospora parasitica (Fr.)) during infection in Arabidopsis thaliana. Similarly, Casimiro et al .[15] used DD-PCR to identify 21 ESTs from H. parasitica infecting Brassica oleracea.
Here we report the cloning and analysis of 602 EST obtained by Subtractive Suppression Hybridization PCR [16] from sunflower seedlings infected by P. halstedii. In addition, the origin of these ESTs was checked by PCR using specific primers and genomic DNA isolated from P. halstedii or from sunflower.
Results
Expressed sequence tags analysis
After two rounds of subtraction hybridization, cDNAs were cloned into pGemT-easy vector and the bacteria arrayed in 96 well plates. To estimate the average size of the obtained clones, 40 clones were randomly chosen and their inserts were amplified using the SP6 and T7 universal primers present on the vector. The amplification products were then separated by agarose gel electrophoresis. The estimated sizes were between 400 and 800 bp with an average of about 500 bp. Subsequently, 602 clones were randomly chosen and single pass sequenced using the T7 universal primer. The length of good quality sequences was on average between 400 and 500. 51 sequences were of a poor quality or too short (<100 bases) thus were excluded from further analysis.
The remaining 551 sequences were compared to each other using the BlastN program [17] to identify overlapping sequences and assembled into contigs using the CAP3 program [18]. One hundred fifty three clones out of 551 were found as singletons and the remaining 398 clones formed 77 contigs containing at least 2 clones. Thus 230 unisequences were present in 551 cDNA clones analysed. The relative abundance of identical clones within the collection is shown in Table 1. The number of clones per contig ranged from 2 to 78. Half of the sequences were either unique or formed contigs containing 2 or 3 clones. Two contigs only contained respectively 52 and 78 clones, which represent approximately 24% of the total (cf. Table 1).
PCR amplification and origin of ESTs
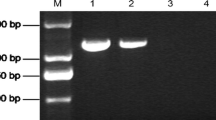
Because the 230 unisequences could originate either from P. halstedii or represent induced genes in sunflower, PCR primers were designed to amplify these gene fragments from DNA isolated either from P. halstedii sporangia or from Helianthus annuus. All the 230 primer pairs tested amplified fragments either from P. halstedii DNA or from Helianthus annuus DNA; 145 primer pairs amplified fragments only from P. halstedii DNA, indicating that these ESTs were derived from P. halstedii genes that are expressed during infection in sunflower. Figure 1 shows an example of PCR amplifications from DNA isolated from P. halstedii sporangia or from Helianthus annuus.

Examples of PCR amplification of ESTs originatingeither from sunflower (S) or from Plasmopara halstedii (P). Each primer pair was tested using DNA from either sunflower (S), or Plasmopara halstedii sporangia (P). The ESTs tested here are: 1, HSP 70 [GenBank:CB174638]; 2, Pyruvate kinase [GenBank:CB174571]; 3, Superoxyde dismutase [GenBank:CB174636]; 4, Proteasome 26 S beta subunit [GenBank:CB174617]; protein; 6, Asparagine synthase; 7, Elicitor [GenBank:CB174646]; 8, Cyclophilin B [GenBank:CB174647]. M: molecular marker. H 2 0: negative control. The PCR products were separated on 1.5% agarose gel.
Homology search
To identify homologs of the 145 ESTs derived from P. halstedii, each EST sequence was queried against the NCBI non-redundant protein database using the BLASTX algorithm [17]. Table 2 shows the proportion of EST sequences with no significant similarity to known protein sequences (> E-05), significant similarity (E-05 to E-20), or highly significant (< E-20). A total of 89 non-redundant sequences, which correspond to 60% of the 145 ESTs obtained, showed significant (< E-05) homology to sequences in the NCBI database and thus were retained for functional classification (Table 3).
Functional classification of P. halstedii ESTs
ESTs were assigned to putative cellular roles using the categories defined by Bevan et al.[19] and the Expressed Gene Anatomy Database (EGAD) [20]. Two categories (elicitor and pathogenecity, and cell defence) are added as described by Kamoun et al.[21]. A summary of the assignment of non-redundant ESTs to functional categories as well as their relative abundance is listed in Table 3. The majority of the identified cDNAs were related to protein synthesis, cell metabolism, signal transduction, and cell stress.
Protein-signature scanning and identification of putative secreted proteins
Because a large set of the ESTs (64) were predicted to code for hypothetical proteins or showed no significant homology with known proteins in the databases, we used SignalP 3.0 [22] to identify potential secreted proteins among this set. Additional information on these ESTs was obtained by protein-signature scanning. InterProScan was used for sequence comparison to the InterPro database [23]. The results of these searches are summarized in Table 4. The majority of these ESTs (47) did not display any reported motif or a signal peptide. Among those which displayed significant protein-signatures, five are predicted to contain a signal peptide, thus they may correspond to secreted proteins (Table 4). However, the average size of the ESTs was about 500 bp which is too small to allow the identification of a larger number of potentially secreted proteins.
Identification of pathogenicity-related genes
Functional annotation of the ESTs identified at least 4 that could be potentially involved in the infectious process of P. halstedii. One EST [GenBank:CB174657] showed homology with a Kazal-like serine protease inhibitor from Phytophthora infestans [24]. Alignment of these sequences (Figure 2) shows that the P. halstedii Kazal-like protein contains the conserved cysteine backbone and the motif C-X3-C-X7-C-X10-C-X6-C-X9-C defining the Kazal family signature [24].

Alignment of Kazal-like proteins from P. infestans and P. halstedii. One Kazal-like protein from P. halstedii [GenBank:CB174657] and the EPI9 Kazal-like protein from P. infestans [GenBank:AY586281.1] were aligned using ClustalX program. The asterisks indicate the conserved cysteine backbone defining the Kazal-like proteins family. The potential Peptide signal is also indicated.
A second EST [GenBank:CB174713] showed a strong homology with a Phytophthora infestans Cystatin-like protein [25]. The P. halstedii cystatin-like protein displays 42% identity with the P. infestans cystatin-like protein (Figure 3). Interestingly, as in the P. infestans protein, the P. halstedii protein contains a potential signal-peptide and conserved domains, including the N-terminal trunk (NT), first binding loop (L1) and second binding loop (L2) [25], suggesting that these proteins may have similar functions.

Alignment of Cystatin-like proteins from P. infestans and P. halstedii. The Phytophthora infestans Cystatin-like protein EPIC4 [GenBank:AY935254] was aligned with the putative P. halstedii Cystatin-like protein [GenBank:CB174713]. NT indicates the N-terminus trauncation, L1 and L2 indicate two conserved loop. The asterisk indicates the conserved Tryptophane amino acid within the L2 loop. The potential Peptide signal is also indicated.
Comparison with true fungi and other microbes for conserved virulence factors
The PHI-Base is a database containing expertly curated molecular and biological information on genes proven to affect the outcome of pathogen-host interactions [26]. Blastx search of this database identified 11 out of the 145 P. halstedii ESTs with significant similarity (E value < E-5) (Table 5). The matching hits belong both to plant and animal pathogens. Interestingly, 6 out of the eleven genes are required for pathogenicity on animals. The homologous genes are involved in a variety of cellular functions such as signal transduction (PHI:221) or cellular detoxification (PHI:410).
Comparison with other Oomycetes
Search for homologous sequences in the ESTs collections or whole genomes sequences of different oomycetes showed that 117 out of the 145 P. halstedii ESTs share similarity with sequences in at least one oomycete taxon with a BlastN E-value < E-5. When an E-value cutoff < E-20 is used, 82 P. halstedii ESTs still shared similarity with sequences in the other oomycetes. Neverthless, 11 P. halstedii showed no significant homology with any sequence in the other oomycetes or any other sequence in the public databases. Thus, these sequences are unique to P. halstedii and may represent genes specific to this pathogen. Unfortunately, these 11 ESTs are annotated as hypothetical and no obvious motif was detected within them using Interproscan and Smart programs.
Fifteen sequences were highly similar (BlastN E-value < E-20) with sequences in at least 5 out of the 8 oomycetes taxa. These sequences included 3 ESTs coding for unknown proteins ([GenBank:CB174649], [GenBank:CB174661] and [GenBank:CB174701]) and 12 ESTs coding for differents proteins such as calmodulin [GenBank:CB174628], actin [GenBank:CB174582] or ribosomal proteins ([GenBank:CB174588] and [GenBank:CB174589]).
Discussion
Many plant diseases are caused by parasitic microorganisms for which little molecular information is available. Thus, the large scale sequencing of Expressed Sequence Tags (EST) could be considered as a first step towards understanding the molecular basis of pathogenicity of these microorganisms. This approach allows rapid and exhaustive sampling of transcripts that are regulated during the infection process. For example, 704 unisequences have been identified in the wheat pathogen Mycosphaerella graminicola (Septoria tritici) [27]. In this study, we were interested in the identification of transcripts produced by P. halstedii during the infection of its host. However, a major challenge is the biotrophic nature of this parasite. To overcome this limitation, we decided to use the suppression subtractive hybridization (SSH) method [16]. One of its main advantages is that it allows the detection of low-abundance differentially expressed transcripts, such as many of those likely to be involved in signal transduction.
Redundancy
The 230 unisequences correspond to 153 clones present as singletons and 398 clones corresponding to redundant cDNA which formed 77 contigs ranging from 2 to 78 ESTs. This redundancy rate of 72% is higher than those obtained from other EST sequencing programs, for example, 49% of 1409 clone of N. crassa [28], 53% of 4809 clones from the cambial tissue of poplar [29], and 37% of 1000 clones from Phytophthora infestans [21]. This redundancy rate of 72% could be reduced by sequencing more clones after a differential screen with the most represented clones, elongation factor of P. halstedii and Asparagine synthase of Heliantus annuus which were represented in 78 and 56 copies respectively and account for up to a third of redundancy observed.
Origin of the EST
The 230 unisequences may correspond either to sunflower genes induced upon the infection by P. halstedii or sequences originating from the parasite. To overcome this difficulty, 230 primer pairs were designed and used to amplify the corresponding fragment with sunflower and P. halstedii DNA. This analysis resulted unambiguously in the identification of 145 EST originating from P. halstedii which corresponds approximately to 63% of the total of unisequences. The sequences of these primers are deposited along with the sequences of the ESTs in the Genbank. When the EST belongs to P. halstedii, amplification product was observed only when the DNA of P. halstedii extracted from infected sunflowers is used as template. Conversely, when the EST is originating from sunflower, a faint amplification product is often observed with DNA from P. halstedii (Figure 1). This is due to the contamination of sporangia with sunflower cells when being collected from infected cotyledons. Although this PCR strategy is robust, it is not suitable for high throughput sequencing project. Alternatively, in a similar work, Thara et al. [30] confirmed the fungal origin of several EST from the obligate basidiomycete Puccinia tritici by hybridization with radioactively labelled total genomic DNA. When the whole genome sequence of the host is available such as in the model plant A. thaliana, it can be exploited to distinguish between the EST originating from the host and those originating from the parasite. This strategy has been used by van der Biezen et al.[14] to identify 7 genes from the oomycete Peronospora parasitica. The G+C content of the EST also has been used to distinguish Phytophthora sojae from soybean cDNA [31]. The average G+C content of soybean EST was 46% whereas the G+C content of Phytophthora sojae was 58%, and plotting of the ESTs from infected soybean produced two distinct peaks of G+C percentage [31]. In the present study, the percentage G+C contents of ESTs from P. halstedii and from sunflower were entirely overlapping with averages of 47% and 45% respectively (data not shown), making this criterion inappropriate to uncover the origin of an EST in this pathosystem.
In contrast, all the sequences showing homology with sequences from other oomycetes such as Phytophthora species proved to be originating from P. halstedii. Therefore, the growing number of sequences produced in different oomycete EST sequencing projects and the availability of Phytophthora and H. parasitica genome sequences should facilitate the analysis of pathogen genes expressed during host-dependent stages.
Functional classification of P. halstedii ESTs
Many of the ESTs identified in this study are associated with basic metabolisms such as energy production, carbohydrates metabolism, nucleotides and protein synthesis. Protein synthesis process is highly represented which may indicate that this process is actively involved during infection. The most represented EST [GenBank:CB174619] shows significant similarity with the tef1 elongation factor from Phytophthora infestans [32], which is highly expressed during spore germination and mycelium formation [33]. Interestingly, this gene has affinity for actin and tubulin and may be involved in the regulation of the cytoskeleton [33].
The EST [GenBank:CB174624] shows homology with cyclophilin which has a cis-trans isomerase activity [34]. However, this gene has also been identified as a virulence factor in the rice blast fungus, Magnaporthe grisea [35]. It should be interesting to test whether this gene has conserved functions in different plant pathogen species as it was hypothesized by Thara et al. [30].
The homology found between one EST [GenBank:CB174646] and an elicitor from P. megasperma [36] indicates that the EST approach can be a rapid way to generate sequences potentially involved in the pathogenicity process. However, whether this gene has a similar function in P. halstedii has still to be experimentally demonstrated. Additionally, many ESTs share homology with stress-related genes such as superoxide dismutase [GenBank:CB174636] or gluthatione peroxidase [GenBank:CB174637] which may indicate that P. halstedii faces a hostile environment within the sunflower tissues and that such enzymes may detoxify compounds released by the host such as hydrogen peroxide. For example, in Mycobacterium tuberculosis, a superoxide dismutase enzyme contributes to the resistance of the parasite to the oxidative burst in macrophages [37].
Identification of P. halstedii genes potentially involved in the pathogenesis process
Two of the ESTs obtained share significant homology with protease inhibitors from P. infestans. The first one [GenBank:CB174657] contains a Kazal-like domain and is similar to the serine protease inhibitor EPI9 from P. infestans [24]. At least 35 Kazal-like Serine protease inhibitors have been reported in different oomycetes and two of these secreted proteins (EPI1 and EPI10) have been shown to interact and inhibit the apoplastic pathogenesis-related Protease P69B, a subtilisin-like serine protease of tomato [24, 38]. Interestingly, Catanzariti et al. [39] showed that the flax rust avirulence gene AvrP123-A encodes a Kazal-like protein that is recognized by P1 and P2 resistance genes in flax. The putative P. halstedii Kazal-like protein possesses all the conserved domains defining the Kazal-like family. The second P. halstedii protease inhibitor like protein [GenBank:CB174713] shares similarity with a P. infestans Cystatin-like protein [38]. Both sequences possess all the signatures sequences of the Cystatin-like protease inhibitors, including the N-terminal trunk, the first loop and the conserved Trp within the second loop. It is likely that these sequences similarities may reflect conserved physiological functions. Thus, it should be interesting to test experimentally whether theses proteins could similarly inhibit proteases of infected sunflowers.
Identification of potentially shared factors with true fungi
We exploited the recently developed PHI-base [40] a database that catalogues the phenotypes resulting from mutations in defined genes of both plant and animal pathogens [26]. Homology search using the P. halstedii ESTs identified 11 sequences with significant matches in this database. Mutation of the identified genes led to reduced virulence in the respective hosts. For instance, the P. halstedii [GenBank:CB174644] is highly similar to a thiol peroxidase (PHI:386) from the basidiomycetous fungus Cryptococcus neoformans. As peroxidases, this gene acts to remove peroxides and provide defence against oxidative damage. Mutation of this gene significantly reduced virulence in mice [41]. We have shown that resistance of sunflower to P. halstedii is associated with an oxidative-like burst within the hypocotyls [3]. Thus, it should be interesting to test whether the P. halstedii thiol peroxidase gene plays a similar role in detoxifying peroxides in sunflower. Sequence homology does not necessarily imply a conserved function, yet many animal and plant pathogens appear to utilize common signaling cascades and protective compounds during their development and pathogenesis [42].
Conclusion
In this study we have initiated an EST approach combined with SSH PCR to obtain for the first time genes from the mycelium of the obligatory oomycete P. halstedii.
Nevertheless, in the long-term process towards the identification of pathogenicity factors in P. halstedii, it should be necessary to test the physiological function of each EST by genetically transforming P. halstedii in planta, as it was developed for Erisyphe graminis f.sp. hordei [43]. Recently, a transient expression of the gfp protein has been reported in P. halstedii sporangia using electroporation and a mechanoperforation method [44]. However, the gfp expression was lost during the subsequent rounds of infection. Alternatively, it should be interesting to know to which extent the metabolic pathways are conserved among the oomycetes and whether the heterologous expression of P. halstedii genes in a transformable oomycete such as P. infestans is useful. Overall, these resources will greatly accelerate research on this important pathogen and could lead to novel perspectives for controlling the pathogenicity of sunflower downy mildew.
Methods
P. halstedii isolate and culture conditions
One isolate identified as the physiological race 300 of P. halstedii on sunflower differentials was used. This isolate was provided by Dr D. Tourvieille (INRA-Clermont-Ferrand, France). It has been collected from infected fields in the south of France in 1995 and maintained by asexual reproduction on the sunflower genotype Peredovick, susceptible to all known races of P. halstedii in a containment culture chamber [45]. Frequently, sunflower differentials are infected to assess the behaviour of the isolate. The infection method of sunflower germinated seeds and growing conditions were those described by Mouzeyar et al.[2].
Preparation of mRNA and Suppression Subtraction Hybridization library construction
Total RNA was extracted from 15-day-old infected and non-infected sunflower plants using the method described by Bogorad et al.[46] and the polyadenylated mRNA with the PolyATract mRNA Isolation System (Promega France). Using the Clontech PCR-select™ cDNA Subtraction kit [16], second-strand cDNAs are prepared from the two mRNA populations under comparison. To enrich the cDNA library with clones specifically from P. halstedii, the cDNA from infected plants was used as tester and the cDNA from non infected plants was used as driver. After subtraction, the PCR-amplified cDNA were cloned into pGemT-easy vector and transformed into E. coli JM 109 strain (Promega France).
Sunflower and P. halstedii DNA extraction
Sunflower DNA was extracted from healthy young leaf tissues by Nucleon PhytoPure kit (Amersham™). To extract P. halstedii DNA, 15-day-old infected seedlings were placed in a plastic bag for 2 days to induce sporulation on cotyledons and leaves. Zoosporangia were collected by gently washing out the cotyledons and leaves with sterile water and DNA extracted using the Nucleon PhytoPure kit (Amersham™ France).
DNA sequencing
Clones from the subtracted library were selected randomly and grown in 96-well plates. Each EST was single-pass sequenced using the Dye-Terminator method and the T7 primer (Genome Express, France).
Sequence analysis
Sequences with low quality bases (Phred score less than 20) and short sequences (< 100 nucleotides) were removed from further analysis. The vector and polylinker sequences were manually trimmed. Sequences were then assembled and arranged into unisequences using the BlastN algorithm [17] and CAP3 program [18]. Similarity searches were done using the BlastX program [17] against current version of NCBI "nr" non-redundant amino acid database.
Comparison with true fungi and other microbes for conserved virulence factors
The PHI-base containing the description of curated genes involved in pathogenicity both in animals and in plants were searched for homology with the P. halstedii ESTs [26]. The version 2.31 containing 592 proteins was downloaded and used in BlastX search using the BioEdit package v7.0.8 [47]. The hits with an E-value < E-5 were considered as significant.
Primers design and PCR amplification
Primer pairs were designed using the primer3 program [48] and used to amplify each EST from DNA isolated from P. halstedii or from DNA isolated from sunflower line. The PCR amplifications were carried out with 50 ng DNA in the presence of 0.2 mM of each dNTP, 1 U of Taq DNA polymerase (Advantage 2, Clontech), 1 × Taq polymerase buffer and 0.5 μM of each primer. PCR was carried out in a 9600 Perkin-Elmer thermocycler under the following conditions: 35 cycles of 10 s at 94°C (denaturation), 30 s at 60°C (primer annealing), and 1 min 30 s at 72°C (primer extension). PCR products were separated using standard TAE agarose gel electrophoresis.
References
Zimmer DE, Kinmann ML: Downy mildew resistance in cultivated sunflower and its inheritance. Crop Sci. 1972, 12: 749-751.
Mouzeyar S, Tourvieille De Labrouhe D, Vear F: Histopathological studies of resistance of sunflower (Helianthus annuus L.) to downy mildew (Plasmopara halstedii). J Phytopathology. 1993, 139: 289-297.
Radwan O, Mouzeyar S, Venisse J-S, Nicolas P, Bouzidi MF: Resistance of sunflower to the biotrophic oomycete Plasmopara halstedii is associated with a delayed hypersensitive response within the hypocotyls. Journal of Experimental Botany. 2005, 56: 2683-2693. 10.1093/jxb/eri261.
Mazeyrat F, Mouzeyar S, Nicolas P, Tourvieille de Labrouhe D, Ledoigt G: Cloning, sequence and characterization of a sunflower (Helianthus annuus L.) pathogen-induced gene showing sequence homology with auxin-induced genes from plants. Plant Mol Biol. 1998, 38: 899-903. 10.1023/A:1006095728033.
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG: Life with 6000 genes. Science. 1996, 274: 563-567. 10.1126/science.274.5287.546.
Tunlid A, Talbot NJ: Genomics of parasitic and symbiotic fungi. Curr Opin Microbiol. 2002, 5: 513-519. 10.1016/S1369-5274(02)00355-7.
Torto-Alalibo T, Tian M, Gajendran K, Waugh ME, van West P, Kamoun S: Expressed sequence tags from the oomycete fish pathogen Saprolegnia parasitica reveal putative virulence factors. BMC Microbiology. 2005, 5: 46-10.1186/1471-2180-5-46.
Tyler BM, Tripathy S, Zhang X, Dehal P, Jiang RHY, Aerts A, Arredondo FD, Baxter L, Bensasson D, Beynon JL, Chapman J, Damasceno CMB, Dorrance AE, Dou D, Dickerman AW, Dubchak IL, Garbelotto M, Gijzen M, Gordon SG, Govers F, Grunwald NJ, Huang W, Ivors KL, Jones RW, Kamoun S, Krampis K, Lamour KH, Lee MK, McDonald WH, Medina M, Meijer HJG, Nordberg EK, Maclean DJ, Ospina-Giraldo MD, Morris PF, Phuntumart V, Putnam NH, Rash S, Rose JKC, Sakihama Y, Salamov A, Savidor A, Scheuring CF, Smith BM, Sobral BWS, Terry A, Torto-Alalibo T, Win J, Xu Z, Zhang H, Grigoriev IV, Rokhsar DS, Boore J: Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science. 2006, 313: 1261-1266. 10.1126/science.1128796.
Jiang RHY, Tyler BM, Govers F: Comparative analysis of Phytophthora genes encoding secreted proteins reveals conserved synteny and lineage-specific gene duplications and deletions. Mol Plant Microbe Interact. 2006, 19: 1311-1321. 10.1094/MPMI-19-1311.
Meijer HJG, Van de Vondervoort PJI, Yin QY, De Koster, Klis FM, Govers F, De Groot PWJ: Identification of cell wall-associated proteins from Phytophthora, ramorum. Mol Plant Microbe Interact. 2006, 19: 1348-13458. 10.1094/MPMI-19-1348.
Holub E: Evolution of parasitic symbioses between plants and filamentous microorganisms. Current opinion in plant biology. 2006, 9: 397-405. 10.1016/j.pbi.2006.05.011.
Skinner W, Keon J, Hargreaves J: Gene information for fungal plant pathogens from expressed sequences. Curr Opin Microbiol. 2001, 4: 381-386. 10.1016/S1369-5274(00)00221-6.
Thomas SW, Rasmussen SW, Glaring MA, Rouster JA, Christiansen SK, Oliver RP: Gene identification in the obligate fungal pathogen Blumeria graminis by expressed sequence tag analysis. Fungal Genet Biol. 2001, 33: 195-211. 10.1006/fgbi.2001.1281.
Van der Biezen EA, Juwana H, Parker JE, Jones JD: cDNA-AFLP display for the isolation of Peronospora parasitica genes expressed during infection in Arabidopsis thaliana. Mol Plant Microbe Interact. 2000, 13: 895-898. 10.1094/MPMI.2000.13.8.895.
Casimiro S, Tenreiro R, Monteiro AA: Identification of pathogenesis-related ESTs in the crucifer downy mildew oomycete Hyaloperonospora parasitica by high-throughput differential display analysis of distinct phenotypic interactions with Brassica oleracea. Journal of Microbiological Methods. 2006, 66: 466-478. 10.1016/j.mimet.2006.01.015.
Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert PD: Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci USA. 1996, 93: 6025-6030. 10.1073/pnas.93.12.6025.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Huang X, Madan A: CAP3: A DNA sequence assembly program. Genome Res. 1999, 9: 868-877. 10.1101/gr.9.9.868.
Bevan M, Bancroft I, Bent E, Love K, Goodman H, Dean C, Bergkamp R, Dirkse W, Van Staveren M, Stiekema W, Drost L, Ridley P, Hudson SA, Patel K, Murphy G, Piffanelli P, Wedler H, Wedler E, Wambutt R, Weitzenegger T, Pohl TM, Terryn N, Gielen J, Villarroel R, De Clerck R, Van Montagu M, Lecharny A, Auborg S, Gy I, Kreis M, Lao N, Kavanagh T, Hempel S, Kotter P, Entian KD, Rieger M, Schaeffer M, Funk B, Mueller-Auer S, Silvey M, James R, Montfort A, Pons A, Puigdomenech P, Douka A, Voukelatou E, Milioni D, Hatzopoulos P, Piravandi E, Obermaier B, Hilbert H, Düsterhöft A, Moores T, Jones JD, Eneva T, Palme K, Benes V, Rechman S, Ansorge W, Cooke R, Berger C, Delseny M, Voet M, Volckaert G, Mewes HW, Klosterman S, Schueller C, Chalwatzis N: Analysis of 1.9 Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature. 1998, 391: 485-488. 10.1038/35140.
Kamoun S, Hraber P, Sobral B, Nuss D, Govers F: Initial assessment of gene diversity for the oomycete pathogen Phytophthora infestans based on expressed sequences. Fungal Genet Biol. 1999, 8: 94-106. 10.1006/fgbi.1999.1166.
Bendtsen JD, Nielsen H, von Heijne G, Brunak S: Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004, 340: 783-795. 10.1016/j.jmb.2004.05.028.
Zdobnov EM, Apweiler R: InterProScan – an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001, 17: 847-848. 10.1093/bioinformatics/17.9.847.
Tian M, Huitema E, Da Cunha L, Torto-Alalibo T, Kamoun S: A Kazal-like extracellular serine protease inhibitor from Phytophthora infestans targets the tomato pathogenesis-related protease P69B. J Biol Chem. 2004, 279: 26370-26377. 10.1074/jbc.M400941200.
Tian M, Win J, Song J, Van der Hoorn R, Van der Knaap E, Kamoun S: A Phytophthora infestans Cystatin-like protein targets a novel tomato Papain-like apoplastic Protease. Plant Physiol. 2007, 143: 364-377. 10.1104/pp.106.090050.
Winnenburg R, Baldwin TK, Urban M, Rawlings C, Köhler J, Hammond-Kosack KE: PHI-base: a new database for pathogen host interactions. Nucleic Acids Research. 2006, 34: D459-D464. 10.1093/nar/gkj047.
Keon J, Bailey A, Hargreaves J: A group of expressed cDNA sequences from the wheat fungal leaf blotch pathogen, Mycosphaerella graminicola (Septoria tritici). Fungal Genet Biol. 2000, 29: 118-133. 10.1006/fgbi.2000.1186.
Nelson MA, Kang S, Braun EL, Crawford ME, Dolan PL, Leonard PM, Mitchell J, Armijo AM, Bean L, Blueyes E, Cushing T, Errett A, Fleharty M, Gorman M, Judson K, Miller R, Ortega J, Pavlova I, Perea J, Todisco S, Trujillo R, Valentine J, Wells A, Werner-Washburne M, Yazzie S, Natvig DO: Expressed Sequences from Conidial, Mycelial, and Sexual Stages of Neurospora crassa. Fungal Genet Biol. 1997, 21: 348-363. 10.1006/fgbi.1997.0986.
Sterky F, Regan S, Karlsson J, Hertzberg M, Rohde A, Holmberg A, Amini B, Bhalerao R, Larsson M, Villarroel R, van Montagu M, Sandberg G, Olsson O, Teeri TT, Boerjan W, Gustafsson P, Uhlen M, Sundberg B, Lundeberg J: Gene discovery in the wood-forming tissues of poplar: analysis of 5692 expressed sequence tags. Proc Natl Acad Sci USA. 1998, 95: 13330-13335. 10.1073/pnas.95.22.13330.
Thara VK, Fellers JP, Zhou JM: In planta induced genes of Puccinia triticina. Molecular Plant Pathology. 2000, 4: 51-56. 10.1046/j.1364-3703.2003.00142.x.
Qutob D, Hraber PT, Sobral BW, Gijzen M: Comparative analysis of expressed sequences in Phytophthora sojae. Plant Physiol. 2000, 123: 243-254. 10.1104/pp.123.1.243.
van't Klooster JW, van den Berg-Velthuis G, van West P, Govers F: tef1, a Phytophthora infestans gene encoding translation elongation factor 1alpha. Gene. 2000, 249: 145-151. 10.1016/S0378-1119(00)00151-7.
Condeelis J: Elongation factor 1 alpha, translation and the cytoskeleton. Trends Biochem Sci. 1995, 20: 169-170. 10.1016/S0968-0004(00)88998-7.
Dolinski K, Muir S, Cardenas M, Heitman J: All cyclophilins and FK506 binding proteins are, individually and collectively, dispensable for viability in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1997, 94: 13093-13098. 10.1073/pnas.94.24.13093.
Viaud MC, Balhadere PV, Talbot NJ: A Magnaporthe grisea cyclophilin acts as a virulence determinant during plant infection. Plant Cell. 2002, 14: 917-930. 10.1105/tpc.010389.
Sacks W, Nurnberger T, Hahlbrock K, Scheel D: Molecular characterization of nucleotide sequences encoding the extracellular glycoprotein elicitor from Phytophthora megasperma. Mol Gen Genet. 1995, 246: 45-55. 10.1007/BF00290132.
Piddington DL, Fang FC, Laessig T, Cooper AM, Orme IM, Buchmeier NA: Cu, Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect Immun. 2000, 69: 4980-4987. 10.1128/IAI.69.8.4980-4987.2001.
Tian M, Benedetti B, Kamoun S: A second Kazal-like Protease inhibitor from Phytophthora infestans inhibits and interacts with the apoplastic pathogenesis-related protease P69B of tomato. Plant Physiol. 2005, 138: 1785-1793. 10.1104/pp.105.061226.
Catanzariti AM, Dodds PN, Lawrence GJ, Michael A, Ayliffe A, Ellis JG: Haustorially Expressed Secreted Proteins from Flax Rust Are Highly Enriched for Avirulence Elicitors. The Plant Cell. 2006, 18: 243-256. 10.1105/tpc.105.035980.
PHI-base. http://www.phi-base.org
Missall TA, Mary Ellen Pusateri ME, Lodge JK: Thiol peroxidase is critical for virulence and resistance to nitric oxide and peroxide in the fungal pathogen, Cryptococcus neoformans. Molecular Microbiology. 2004, 51: 1447-1458. 10.1111/j.1365-2958.2004.03921.x.
Sexton AC, Howlett BJ: Parallels in Fungal Pathogenesis on Plant and Animal Hosts. Eukaryotic Cell. 2006, 5: 1941-1949. 10.1128/EC.00277-06.
Chaure P, Gurr SJ, Spanu P: Stable transformation of Erysiphe graminis an obligate biotrophic pathogen of barley. Nat Biotechnol. 2000, 18: 205-207. 10.1038/72666.
Hammer TR, M Thines M, Spring O: Transient expression of gfp in the obligate biotrophic oomycete Plasmoparahalstedii using electroporation and a mechanoperforation method. Plant Pathology. 2007, 56: 177-182. 10.1111/j.1365-3059.2006.01494.x.
Tourvieille de Labrouhe D, Gulya T, Masirevic S, Penaud A, Rachid K, Viranyi F: New nomenclature of races of Plasmopara halstedii (Sunflower Downy Mildew). Proceedings of the 15th International Sunflower Conference. 2000, 61-66.
Bogorad L, Gubbins EJ, Krebbers E, Larrinua IM, Mulligan BJ, Muskavitch KMT, Orr EA, Rodermel SR, Schantz R, Steinmetz AA, de Vos G, Ye YK: Cloning and physical mapping of maize plastid genes. Meth Enzymol. 1983, 97: 524-554.
BioEdit package v7.0.8. http://www.mbio.ncsu.edu/BioEdit/bioedit.html
Rozen S, Skaletsky HJ: Primer3 on the WWW for general users and for biologist programmers. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Edited by: Krawetz S, Misener S. 2000, Humana Press, Totowa NJ, 365-386.
Acknowledgements
This study was partially supported by CETIOM (France). Some equipements were acquired from grand of the "Contrat de Plan Etat-Région Qualité des aliments". We thank F. Cambon for technical assistance. Also, we would like to thank four anonymous reviewers whose comments and suggestions have greatly improved the quality of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MFB, FP and SM, performed the experiments including construction of the cDNA library, annotation and analyses of the sequences. MFB, PN and SM wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bouzidi, M.F., Parlange, F., Nicolas, P. et al. Expressed Sequence Tags from the oomycete Plasmopara halstedii, an obligate parasite of the sunflower. BMC Microbiol 7, 110 (2007). https://doi.org/10.1186/1471-2180-7-110
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-7-110