
Abstract
Background
Helicobacter pylori has diverged in parallel to its human host, leading to distinct phylogeographic populations. Recent evidence suggests that in the current human mixing in Latin America, European H. pylori (hpEurope) are increasingly dominant at the expense of Amerindian haplotypes (hspAmerind). This phenomenon might occur via DNA recombination, modulated by restriction-modification systems (RMS), in which differences in cognate recognition sites (CRS) and in active methylases will determine direction and frequency of gene flow. We hypothesized that genomes from hspAmerind strains that evolved from a small founder population have lost CRS for RMS and active methylases, promoting hpEurope’s DNA invasion. We determined the observed and expected frequencies of CRS for RMS in DNA from 7 H. pylori whole genomes and 110 multilocus sequences. We also measured the number of active methylases by resistance to in vitro digestion by 16 restriction enzymes of genomic DNA from 9 hpEurope and 9 hspAmerind strains, and determined the direction of DNA uptake in co-culture experiments of hspAmerind and hpEurope strains.
Results
Most of the CRS were underrepresented with consistency between whole genomes and multilocus sequences. Although neither the frequency of CRS nor the number of active methylases differ among the bacterial populations (average 8.6 ± 2.6), hspAmerind strains had a restriction profile distinct from that in hpEurope strains, with 15 recognition sites accounting for the differences. Amerindians strains also exhibited higher transformation rates than European strains, and were more susceptible to be subverted by larger DNA hpEurope-fragments than vice versa.
Conclusions
The geographical variation in the pattern of CRS provides evidence for ancestral differences in RMS representation and function, and the transformation findings support the hypothesis of Europeanization of the Amerindian strains in Latin America via DNA recombination.
Similar content being viewed by others


Background
H. pylori has accompanied humans throughout evolution [1], and as humans diverged, so did H. pylori. Based on multilocus sequences (MLS), H. pylori strains can be divided into populations that are specific for the geographic origin of their human hosts [1–4]. Strains from present-day Africans include the most ancestral population hpAfrica2 from Southern Africa, hpNEAfrica from northeastern Africa and hpAfrica1 from western (sub-population hspWAfrica) and southern Africa (hspSAfrica). H. pylori from Europe, the Middle East, western Asia and India belong to the hpEurope population, and strains from Asians include hpAsia2 and hpEastAsia. The latter is subdivided into hspEAsia (from East Asians), hspAmerind (from Native Americans), and hspMaori (from Pacific islanders). About 80% of the H. pylori strains isolated from Mestizo hosts in Latin America were assigned to hpEurope and almost 20% to hspWAfrica, but no strains were assigned to hspAmerind [5]. Conversely, H. pylori strains isolated from Latin America Amerindian hosts showed multi-locus haplotypes of the hspAmerind and hpEurope populations in relatively equal proportions [2, 5].
Geographic clustering also has been shown in virulence-associated genes, such as vacA[6–8]. All H. pylori strains recovered to date from Mestizo hosts have carried European-types (s2, s1a, s1b) of vacA, while the ones recovered from Amerindian hosts exhibited similar amounts of vacA subtype s1c -clustering with East Asia-Pacific isolates- and European vacA subtype s1a and s1b[9].
We have also shown that the hpEurope strains isolated from Mestizos and Amerindians in Latin America hosts exhibit a mosaic genetic structure; they are of predominantly European ancestry, containing some introgressions from African or Asian strains [5]. Thus, this mosaicism suggests Europeanization of the DNA from the original Latin America Amerindian strains through several events of recombination [10], including transformation [11] or conjugation-like mechanisms [12]. H. pylori population dynamics is known to be shaped by DNA transformation and recombination, and the recombination rate in this bacterium is extraordinarily high [11, 13]. Since several genetically distinct H. pylori strains can co-colonize a single stomach [9, 14, 15] and since H. pylori are highly competent [16, 17], the net direction of transformation determines which genome would be invaded by foreign DNA [18]. Instead of replacement of less fit strains, allelic competition via recombination among strains seems to dominate H. pylori evolution [19–21]. Recombination, as evidenced by the mosaic genetic structure of strains recovered from Mestizo and European hosts, suggests the co-existence of at least two different haplotype-strains in a single host [14] that allows recombination and provides a mechanism of competition, in this case, allelic competition rather than strain competition.
Bacterial restriction-modification systems (RMS) confer protection against invasion by foreign DNA, for example that from bacteriophages [22], or from other bacteria [18], by cleavage of this foreign DNA. In general, RMS consist of a restriction endonuclease (RE) that recognizes and cleaves specific DNA sequences (cognate recognition sites), and a counterpart methylase that catalyses the addition of a methyl group to adenine or cytosine residues in the same cognate recognition sites, protecting it from restriction by the cognate enzyme [23]. According to their subunit composition, cofactor requirements, such as ATP, AdoMet, or/and Mg+2 and mode of action, RMS can be divided into types I, II, IIS, and III. Type II RMSs are the simplest and most widely distributed among H. pylori strains [24, 25], in which methylases and restriction enzymes act independently. Type II cognate recognition sites are often palindromic, 4–8 nt in length, with continuous (i.e. GATC) or interrupted (i.e. GCCNNNNNGGC) palindromes [26]. Similarly, Type IIS RMSs, also found in H. pylori, have independent restriction and methylation enzymes, but the endonucleases act as monomers, restriction sites are uninterrupted (4-7nt), and DNA cleavage occurs at specific distances from the recognition sites.
When cognate recognition sites are frequent, genomic or plasmid DNA can be extensively cut, impairing recombination [27]. However, cognate recognition sites also play a role in recombination, since they provide the locus for double stranded cuts suitable as substrate for recombination. Therefore, depending on the relative frequency of the cognate recognition sites, DNA restriction and methylation systems modulate the capability of DNA to recombine. As such, we hypothesized that the dominance of hpEurope strains in Latin America might be due to differences in the cognate restriction sites and active methylases between Amerindian and European strains. To test this hypothesis, we studied the frequencies of cognate recognition sites for 32 restriction enzymes in H. pylori strains that were assigned to different populations. In addition, we estimated the number of active methylases and compared transformation rates in hpEurope and hspAmerind H. pylori strains. Thus, we provide evidence of specific recombination events and mechanisms that indicate preferential receptor and donor status, respectively, in Amerindian and European strains.
Results
Observed and expected number of cognate recognition sites
We examined the published multi-locus sequences (MLS) of 110 H. pylori strains (Additional file 1: Figure S1 and Table 1) [2, 10]. The previously assigned MLS-based haplotypes were consistent with the geographic origin of their hosts: all of the H. pylori sequences from strains from European hosts were assigned to hpEurope [2, 4]; isolates from Amerindians either belonged to hpEurope or hspAmerind, and haplotypes from Mestizos were mostly hpEurope with a few hpAfrica1. We also included 19 hpAfrica1 strains from western Africa to reflect the African genetic influx to the Americas in colonial times, and 12 Korean strains (hspEAsia) to reflect the East Asian origins of Amerindians. In addition, we extracted the MLS sequences from 7 whole genomes available at the time of the analysis, including 4 from European hosts that were hpEurope (26695, HPAG1, G27, P12), one from a North American host that was hpAfrica1 (J99), and two from South American Native hosts that were hspAmerind (Shi470 and V225).
We determine the number of cognate recognition sites on the 110 MLS and 7 whole genome sequences (WGS) for 32 restriction/methylase enzymes previously reported in H. pylori. The number of cognate recognition sites per Kb on the 110 MLS and the 7 were highly consistent and comparable between the two types of sequences. To further validate that MLS are representative of the whole genome sequences, we performed a linear regression analysis. This analysis indicates a strong correlation between the observed cognate RMS sites frequencies in the 110 MLS and the seven WGS for the 32 RMS (Adjusted R2 = 0.80; p <0.001). Thus, MLS is representative of the whole genome sequences in terms of cognate RMS sites.
Of the 32 known cognate recognition sequences there were a mean (± SD) per Kb of 1.25 (± 1.26) in WGS and 1.55 (± 1.46) sites in MLS. In both WGS and MLS, the observed cognate recognition site frequencies were highly variable, ranging from 0 to 5.48 sites per Kb (Table 2). Although the distributions were relatively uniform (data not shown) along the DNA, there were several regions that showed coverage of <0.7 sites per Kb. Such sites often corresponded to "genomic islands" with G-C ratios (from 34.9% to 43.1% ± 4.1) that deviate from the intrinsic H. pylori ratio of about 39%. Expected recognition sites were calculated performing simulations on model sequences with the same length for the MLS and the WGS. These model sequences were constructed based on the average proportion of nucleotides of the actual sequences analyzed (Additional file 1: Table S1). To establish the expected frequencies of appearance of a specific recognition site by chance, we randomized the order of the nucleotides in the model sequences and enumerated the occurrence of that specific recognition site (see Methods for details). We estimated a range of 0.3 to 5.5 expected cognate recognition sites in both the MLS and WGS (R2 = 0.98, p < 0.001; Table 2). Overall, there were no significant differences in the observed or expected number of cognate restriction sites, among the haplotypes (p > 0.05).
The observed/expected (O/E) ratio indicates deviation from the expectation based on G + C ratio. O/E ratios were highly similar for the WGS and MLS (R2 = 0.87, p < 0.001), without any differences by haplotype. Analysis of the hpEurope and hspAmerind sequences showed that 10 of the 32 cognate restriction sites were underrepresented in MLS and 6 of those sites were also underrepresented in WGS (defined as O/E ≤ 0.5 and Chi Square p-value ≤ 0.005; Table 2). One exception, Hpy166III (cognate site: CCTC) was exclusively underrepresented in hpEurope MLS, but not in the hspAmerind nor in WGS. The underrepresented sites varied in their C + G content from 33.3 to 75%. Most (9) of those 10 underrepresented sites were palindromic [28–30] (Table 2). Conversely, only one cognate recognition site: Hpy99III (cognate site: GCGC), was strongly overrepresented (O/E ≥ 2 and Chi Square p-value ≤ 0.005) in both hpEurope/hspAmerind MLS and WGS (Table 2). Overall, similar results were found when analyzing hspEAsia and hspWAfrica strains (data not shown). In summary, the H. pylori genome has mostly evolved to avoid RMS cognate recognition sites. The total numbers of cognate restriction sites were not different among bacterial populations, based on H. pylori haplotypes.
Profiles of cognate RMS recognition sites
The RMS profiles delineate the specific pattern of enzymatic recognition for each sequence, and offer more detailed information than the analysis of the total number of cognate recognition sites described above. Two strains with the same total number of cognate recognition sites among the combined pool of studied enzymes usually vary in the distribution of the specific cognate recognition sites for individual restriction enzymes within that pool. We found that the profile of RMS recognition sites varied significantly in a population-dependent manner (Wilcoxon rank sum test, p < 0.005). Four RMS sites (HPy99IV, HpyCH4V, HpyF14I, and HpyF44II) showed very strong directionality in the RMS strain profile, as shown by principal coordinate analysis (PCoA) of the 110 MLS (Additional file 1: Figure S2). Another 11 cognate recognition sites (Hpy166III, HpyNI, HpyC1I, Hpy8I, HpyIV, HpyF10VI, Hpy99VIP, HpyCH4II, Hpy188III, Hpy178VII, and HpyV) also contributed significantly, explaining 47% of the haplotype-strain variation (29% and 18%, respectively) amongst strains (Additional file 1: Figure S2). The other 17 recognition sites cumulatively explain only 9% of the total variation.
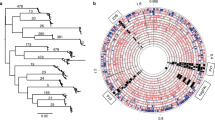
Non-parametric multidimensional scaling (NMDS), based on those 15 cognate recognition site profiles that explain most of the variation in the PCA analyses also separated the H. pylori strains in a population-dependent way (Figure 1). Both for MLS and WGS analyses, the Amerindian and Asian strains exhibit similar profiles, that are distant from European and African strains that cluster apart (Adonis, p < 0.01). In contrast to the homogeneous African and Amerindian strains, the hpEurope strains from Mestizo or Amerindian hosts showed high heterogeneity in their restriction patterns (Figure 1). These results provide evidence for a phylogenetic signal in the profile of the frequencies of the cognate recognition sites in H. pylori.

Non-parametric multidimensional scaling (NMDS) based on the RMS profile for 15 restriction endonucleases in H. pylori DNA sequences. NMDS is a visual representation of the most parsimonious distances, in terms of similarities and disparities, among the sequences. It provides a lower k-dimensional space, based on each restriction profile, which is the combination of the number of restriction sites for each of the 15 enzymes analyzed per sequence. Panel A: Analysis of 110 multilocus sequences. The restriction profile is distinct among haplotypes with the sequences clustering into groups, except for hpEurope that seems to have a more mixed restriction profile, with similarities with some hpAmerind and most hpAfrica1 strains. Panel B: Analysis of seven whole genome sequences. The restriction profile of the whole genome sequences is distinct among the H. pylori sub-groups, with hpEurope, hspAmerind, and hpAfrica1 clustering separated of each other.
A non-hierarchical analysis of the cognate recognition site profile for the same 15 RMS, with bidirectional clustering by frequency of the sites and by strain haplotype grouped RMS recognition sites (2 clusters), and strains (3 clusters, Figure 2). "Strain cluster A", with hspAmerind, hspEAsia and some hpEurope strains (from Amerindian and Mestizo hosts) has a high frequency of cognate sites for "RMS cluster 1". In contrast, the Euro-African "strain cluster C" has a low frequency of cognate sites for RMS in cluster 1, but high for "RMS cluster 2" (Figure 2). The cognate sites for RMS cluster 1 have a significantly lower G + C content compared to the cluster 2 cognate sites (59.4 ± 17.4 and 91.6 ± 20.4%, respectively. T-test = 0.002). "Strain cluster B" includes hspEAsia as well as hpEurope and hpAfrica1 from Mestizo and African hosts and shows a mosaic profile of the cognate recognition sites, consistent with the mosaic genetic structure shown in their MLS (Additional file 1: Figure S1).

Heatmap of the profile for 15 RE recognition sites on MLS DNA sequences for 110 H. pylori strains. Higher and lower frequencies of the cognate recognition sites are represented by red and blue, respectively. The upper tree showed three main strainclusters: A) Includes hspAmerind (N=25), hspEAsia (N=5), and hpEurope (N=7) strains; B) Mostly hpEurope (N=21), but also hspEAsia (N=6), and hpAfrica1 (N=2) strains; and C) hpAfrica1 (N=23), and hpEurope (N=20) strains. The hpEurope strains studied were mostly recovered from Mestizo hosts. The phylogeny on the left shows two enzyme clusters, that correlate with the A, B and C cluster-strains.
Strain-specific methylase representation
Differences in transformation rates might be due to differences in the frequency of cognate restriction sites, but also to variation in the protection conferred by active methylases belonging to the RMS. We tested the hypothesis that cognate restriction sites are more protected by active methylases in hpEurope than in hspAmerind strains. We selected 18 representative H. pylori strains; 9 were hpEurope recovered from European (n = 4), Mestizo (n = 4), and Amerindian (N = 1) hosts, and 9 were hspAmerind from Amerindian hosts (Additional file 1: Table S2). To determine methylase protection, genomic DNA from each strain was subject to digestion by each of 16 restriction endonucleases (Additional file 1: Table S3). Susceptibility to digestion indicated lack of an active methylase.
The restriction results showed a range of 5–14 active methylases (average = 8.6 ± 2.6) per H. pylori strain of the 16 examined. There were non-significant differences in the number (Wilcoxon test, p > 0.05; Figure 3, Additional file 1: Table S3) or variances (F test, p > 0.05) of active methylases between hpEurope and hspAmerind strains. The only exception was the enzyme HpaII, to which DNA from the hspAmerind strains was significantly more resistant (83%) than DNA from the hpEurope strains (42%; Wilcoxon test; p < 0.05). Overall, the results confirm that H. pylori strains conserve similar active methylase protection, regardless of their population assignment.

Total number of active methylases per strain. Both hspAmerind and hpEurope strains exhibited similar number of resistance to the 16 restriction enzymes tested.
Genetic transformation rates
To assess differences in natural competence, five H. pylori hspAmerind strains isolated from Amerindians and five hpEurope strains recovered from European (N = 4) or Mestizo (N = 1) hosts each were transformed with two plasmids: i) p801R, a plasmid with an 800 bp insertion that introduces a single-base mutation of the gene rpsL, conferring resistance to Streptomycin (StrR); or ii) pCTB8, a plasmid with a 1.2 Kb insertion with an exogenous aphA cassette that produces Kanamycin-resistant (KmR) strains [31, 32]. hspAmerind strains exhibited a significantly higher number of StrR transformants than did hpEurope strains (3×10-3vs. 5×10-5, respectively; p < 0.005). Introduction of pCTB8 showed much lower rates of transformation: very few KanR colonies (1–3) were recovered, which did not allow comparison of the transformation frequency with this plasmid between the different H. pylori populations (data not shown).
We have hypothesized that the replacement of hspAmerind strains by hpEurope strains in Latin America was mainly facilitated by the introgression of DNA from hpEurope strains into hspAmerind strains [5]. To test this hypothesis, we reproduced the encounter of hspAmerind and hpEurope H. pylori strains by co-culturing and evaluating the directionality of the DNA horizontal transfers among strains in vitro. We produced double plasmid/resistant hspAmerind and hpEurope strains by transforming the single plasmid trains described above with an additional suicide plasmid, pAD1-Cat that includes an exogenous 1.3 Kb cat cassette that elicits Chloramphenicol resistance (CmR). Thus, we obtained double resistant strains exhibiting: StrR/CmR or KmR/CmR. To evaluate the direction of the DNA transformation, we co-cultured a single plasmid strain (used as the donor) with the double plasmid/resistant strain (as the recipient).
We first assessed the ability of H. pylori hspAmerind or hpEurope strains to acquire a plasmid with a single-base mutation (p801R) from each other, co-culturing StrR strains (donor) and CmR/KmR strains (recipient). Transformants acquiring the single-base mutation from StrR strains (p801R) will exhibit a triple antibiotic resistant phenotype: StrR/CmR/KmR. The frequency of hspAmerind strains acquiring this single-base mutation from hpEurope strains was slightly higher (although not statistically significant, p value = 0.34) than hpEurope strains acquiring it from hspAmerind strains (Figure 4A). To extend our observation, we also co-cultured StrR/CmR and KmR strains. We expected that during co-culturing, transformants acquiring the single-base mutation (p801R conferring StrR) from a StrR/CmR strain will be StrR/KmR but CmS, while transformants acquiring the 1.3 Kb aphA cassette from a KmR strain will be triple antibiotic-resistant (StrR/CmR/KmR). We observed that the frequency of transformation (1×10-7-1×10-5) with a single-base mutation (p801R), was higher than the frequency of transformants that had acquired the large DNA fragment (1.3 Kb pCBT8; <2×10-8) for both the hspAmerind and hpEurope strains. Control (blank) inoculations were included in all the transformation and co-culture experiments (see Methods) to control for spontaneous mutation events. The frequency of transformation of hspAmerind strains with the single-base mutation (StrR) from hpEurope (StrR/CmR) strains was significantly higher (p value = 0.02) than that of hpEurope strains from hspAmerind strains (Figure 4B). For transformation events in which the 1.3 Kb aphA cassette is acquired from a KmR strain (pCTB8), we observed that this cassette is not a suitable genetic marker to evaluate transformation between H. pylori strains because of the low frequency of transformation (<2 × 10-8); however, the few transform colonies (2–4 colonies per plate) were predominantly hspAmerind strains acquiring the cassette from hpEurope strains. In total, these observations support that Amerindian strains are more receptive to acquiring European DNA than vice versa.

Rate of transformation in different co-culture assays among hspAmerind and hpEurope strains. The panel A, shows the rate of transformation of a single plasmid (p801R); in this case there was not significant differences when hspAmerind strains were donors (D) or recipients (R) of the DNA fragment. In the panel B, frequencies of transformation of a double plasmid (p801R+pAD1-cat) are showed. Amerindian strains exhibited higher ability to incorporate DNA from hpEurope than vice versa.
Discussion
Phylogenetic signal of H. pylori RMS cognate sites and its correlation with human evolution
Our results confirm H. pylori genomic avoidance of many cognate restriction sites [33] In some bacteria, bacteriophages mimic the avoidance pattern of cognate recognition sites of their hosts [28, 34–36] and exert selective pressure on the pattern of bacterial restriction sites [22, 37]. Since bacteriophages do not appear important in H. pylori, presumably most of the pressure came from the RMSs themselves (22). Although we did not find significant haplotype differences in the frequencies of cognate recognition sites, we found population-specific differences in the profiles of the cognate recognition sites. The relatively more recent Asian and Amerindian H. pylori strains have lower frequencies of palindromic restriction sites rich in G + C than the African strains and also than the European strains which have been shown to be hybrids between an ancestral H. pylori population (ancestral Europe 1) from Central and Western Asia and another ancestral population (ancestral Europe 2) from Northeast Africa [1, 2]. The genetic bottlenecks experienced by humans as they migrated from Africa [2, 3], might also have influenced changes in the profile of frequency of restriction words in H. pylori strains. Indeed, the more homogeneous profile of restriction word frequencies in Amerindian H. pylori strains in relation to those from African and European strains (Figure 3), is consistent with the lower genetic diversity of both Amerindian hosts and their H. pylori strains [5].
What are the implications of this phylogenetic signature for the pattern of restriction site frequency in H. pylori? That G + C-rich restriction sites were both underrepresented and overrepresented, indicates a lack of selection for total G + C-content. Given that genetic drift is expected to be functionally neutral [2, 4], we cannot discard that differences in the frequency of cognate restriction sites might be functionally relevant in H. pylori. This is consistent with the idea that RMS cognate recognition sites are important for recombination, an important force that drives the evolution of H. pylori. If modulation of natural competence occurs preferentially in one direction, this leads to genetic subversion of one of the transformed strains in a pair [18]. The results of this work suggest that the specific RMS cognate restriction site profile might lead to a recombination dynamic that favors "Europeanization" of Amerindian strains, explaining at least in part the replacement of Amerindian strains by European strains in Latin America.
In the context of human evolution, the human divergence within Africa and the worldwide divergence after the out-of-Africa migrations, were followed by genetic convergence by mixing in modern times. H. pylori strains differing in the use of cognate recognition words might have optimized fitness in the specific environment in which they evolved, but not in new host environments with different competitors. There may have been an ancestral H. pylori RMS pool, before out-of-Africa (around 60,000 years before present) followed by apparent differential selection for and avoidance of particular RMS, as H. pylori evolved with different isolated human groups. Selection against certain cognate recognition sites, particularly palindromes [26], has been shown in several bacteria and bacteriophages [38], which we again observe in H. pylori. The avoidance of specific palindromes may reflect selection pressure exerted by restriction enzymes with incomplete methylation [39], and their effects on genetic regulatory control [28, 30]. When methylation protection fails, strains that avoid specific cognate restriction sites have a fitness advantage over those with more frequent cognate sites [30]. Consistent with this hypothesis is that life forms lacking RMS, such as some DNA viruses, mitochondria, and chloroplasts, do not show palindrome avoidance [29, 30]. Differences in RMS profiles in the isolated sub-populations of H. pylori that derived from the worldwide spread of humans could reflect RMS competition, founder effects, and locale-specific selection.
The biological significance of overrepresentation of palindromic sites is harder to explain in the light of the defensive role of RMS. However, the frequent occurrence of small DNA fragments might increase recombination frequency, which may improve fitness [28, 39]. Similarly, methylation of DNA promoters and origins of replication might provide benefits for the regulation of gene expression [40] and replication [41].
This study confirms prior observations that the mean numbers of active methylases are conserved in H. pylori strains recovered from hosts of different geographical origins [42, 43], suggesting selection for an optimal RMS number across the universe of H. pylori cells [42, 44]. Such selection might be achieved by horizontal gene transfer of RMS genes among H. pylori strains, with a consequent equilibrium in the number of active methylases. RMSs have been postulated to behave as "selfish" mobile genetic elements [27, 45, 46]. Selection favors the maintenance of the system of restriction endonuclease and methylase, because loss of methylase function is lethal. However, intact methylase genes with apparently truncated restriction genes have been observed in completed H. pylori genomes, suggesting that active methylases are involved in the regulation of essential physiological processes that are independent of RMS [47]. However, the process of restriction and methylation might be a dynamic mechanism that can vary in vivo. For example, HpyI methylase (HpyIM) expression varied dramatically within H. pylori cells colonizing the gastric tissue [48].
Dominance of European over Amerindian strains
Despite a similar number of active methylases, hspAmerind strains exhibited higher rates of transformation than hpEurope strains. DNA incorporation into the chromosome during transformation can be divided into three general steps: i) DNA uptake or binding to the cell; ii) degradation of one strand of the invading DNA, and iii) recombination of the remnant DNA fragments into the genome [49, 50]. For the first step, extensive evidence supports the fact that H. pylori is highly competent in uptake of "non-self" DNA. H. pylori is genetically diverse within a single stomach niche and is subject to a very high rate of intraspecific recombination [11, 14, 51]. Proteins such as ComB4, ComB7–ComB10 of the type IV secretion system encoded by the comB genes, [52] are homologs to VirB proteins (VirB4, VirB7–VirB10) of A. tumefaciens and resemble their conjugation-like function in H. pylori DNA transformation [53]. Mutations of comB in H. pylori strains abrogate transformation [52, 54]. Whether haplotype differences in the proteins involved in DNA uptake and access to foreign DNA can affect the efficiency of DNA uptake and incorporation, remains to be tested. Step (ii) involves the degradation of one DNA strand and processing of the foreign DNA. Although H. pylori isolates from different bacterial populations exhibit a similar number of methylases, the differences in the cognate recognition sites can explain differences in the "DNA availability" as a substrate for recombination. For example, four-base cognate recognition sites are (~16-fold) more frequent that six-base recognition sites. Step (iii), homologous recombination, requires at least a single stranded break; DNA differences in the location of the homologous sites may favor higher transformation in Amerindian strains. When two H. pylori strains meet in a host’s stomach, they can recombine in an asymmetric fashion, leading to subversion of one strain by the other.
An additional explanation of European dominance might rely on host selection that seems to favor European strains, for example, host mixing with Europeans. Host selection is evidenced by the H. pylori adhesin phenotypes in relation to human blood groups. Up to 95% of "generalist" European H. pylori strains can bind A, B or O antigens whereas 60% of Amerindian strains bind only O antigens [55]. This binding-specialization of H. pylori strains coincides with the unique predominance of blood group O antigens in Amerindian hosts. Our results provide evidence that asymmetric recombination rates lead to dominance of one strain over another by means of genetic subversion. If Amerindian strains recombine at higher rates, they are more likely to become mosaic strains integrating European loci and gradually become "Europeanized".
Conclusions
In conclusion, geographical variations in the pattern of cognate recognition sites provide evidence for ancestral differences in RMS representation and possibly also in function. The higher transformation rates in Amerindian strains support the hypothesis of Europeanization of Latin American strains via recombination. A potential scenario, supported by our results is that during colonial times when Spanish conquers, African slaves, and Native Amerindians mix also did their H. pylori haplotypes, thus a new generation of H. pylori strains arise, exhibiting mosaic genetic structure result of several events of recombination among strains with different RMS profile. In this mixing, hpEurope alleles succeed dominating their incorporation into DNA from Amerindian strains (See Figure 5). Future studies are needed to evaluate differences by haplotype in competence-related function driven by comB, dprA and comH genes [56, 57].

Model of H. pylori strain dynamics in Latin America hosts. The different color of the bacteria (green and orange) represents the MLS profile and the cognate restriction profile of H. pylori strains. Ancestral strains from Europe and Latin America Amerindians share common genetic signature, both MLS [1, 2] and cognate restriction profile (as shown in our results). In colonial times where European and Amerindians mixed, we hypothesize that the new generation will acquire H. pylori from both parents. Within a single host (mestizos) allelic competition will occurs among strains and hpEurope DNA take over hspAmerind strains promoting its Europeanization (demonstrated in our co-culture results) and mosaic genetic structure.
Methods
In silicoanalysis
Sequences
We analyzed 117 DNA sequences of H. pylori strains, 110 of which were partial sequences and seven were whole genomes. The partial sequences were a string of 3,406 bp composed of ordered concatenated sequences (multilocus sequences, or MLS) from seven housekeeping genes as follows: atpA (627 bp), efp (410 bp), mutY (420 bp), ppa (398 bp), trpC (456 bp), ureI (585 bp) and yphC (510 bp) [58–60]. The MLS were from H. pylori strains from hosts from four continents: Africa, Europe, Asia, and the Americas (from Native American and Mestizo hosts). All sequences were available at the EMBL or GenBank database (http://www.ebi.ac.uk/) and/or at the MLST website for H. pylori (http://pubmlst.org/helicobacter/) [59]. Whole genome sequences (WGS ~ 1.5 Mb) of seven H. pylori were available in GenBank. Four strains were from European hosts: 26695, HPAG1, P12 and G27 (accession numbers NC_000915, NC_008086, NC_ 011333, CP001173, respectively; all hpEurope); one, J99 (NC_000921; hpAfrica1) was from the US, and two Shi470 and V225 (NC_010698; CP001582; hspAmerind) were from Native Americans from Peru and Venezuela, respectively. The MLS of the 7 strains with whole genome sequences were also taken into account for the analysis, and form part of the 110 MLS analyzed.
Haplotype assignment
All the sequences were previously analyzed and assigned to their correspondent populations [2, 5]. Neighbor joining clustering analysis [61] of all the strains was performed in MEGA 5.0. [62].
Frequency of cognate recognition sites
The observed frequency of cognate recognition sites for 32 RMS (Table 2) that have been reported in H. pylori[25, 42, 43, 63] was determined in the 110 MLS (3,406 bp) and 7 WGS (1.5-1.7 Mb) using the EMBOSS restriction program (http://emboss.sourceforge.net/), by counting the number of restriction "words", in each sequence. We determined: 1) the number of cognate recognition sites, that is the sum of all words per strain, 2) their frequency per Kb, 2) their distribution per Kb in the seven WGS, and 4) the RMS profile of each strain, which is the combination of the values for the 32 cognate recognition sites per strain. The expected frequency of cognate recognition sites was based on the actual nucleotide proportions in each WGS or MLS sequence (Additional file 1: Table S2), and determined by 1,000 simulations. The algorithm used for simulating the frequencies of cognate recognition sites was created as follows: (i) a pool of 1,000 nucleotides containing the exact proportion of each nucleotide in each genome or MLS sequence was created (the "pool-simulated sequence"); (ii) a nucleotide was randomly chosen, from the pool-simulated sequence, k times, in which k is the length of each recognition sequence; (iii) simulated words that matched the recognition sequence were counted; and steps 2, 3 were repeated l-k times, where l is the length of the whole genome or MLS sequence.
For each enzyme, observed and expected numbers of cognate recognition sites were compared (O/E ratio) values per enzyme. We estimated the Chi square of the observed and expected values of the cognate recognition sites per haplotype. Underrepresentation was defined when the O/E ratio value was lower than 0.5, and the Chi square value was significant (p values <0.005). Similarly, the sites were overrepresented in the sequences when the ratio O/E value was ≥2, and the Chi square value was significant (p values <0.005). In the case of WGS, we calculated Chi square only for the bacterial populations that contained more than one strain: hpEurope (26695, HPAG1, P12 and G27), and hspAmerind (V225 and Shi470), but not for hpAfrica1 with just one strain (J99). Differences in the frequency of observed and expected cognate recognition sites among H. pylori populations were examined using a pair-wise comparison test based on the medians (Wilcoxon rank sum test). For the 4 populations studied (hspWAfrica, hpEurope, hspEAsia, and hspAmerind), there were 6 possible pair-wise analyses. The p-value for the Wilcoxon rank sum test for each pair indicates the relationships among the haplotypes. Principal component analysis (PCoA) [64] was performed to detect patterns of cognate recognition profiles among strains. Non-parametric multidimensional scaling (NMDS), was used to visualize the variation in two dimensions [65]. NMDS does not assume linearity of the data and does not require data transformation, which represents advantages over other classical ordination methods. The ordination algorithm for NMDS clusters groups with similarities, and based on ranked similarity distances; an iterative search for the least stress position in k-dimensions is done [65].
In vitroanalysis
Bacterial strains for restriction analysis
Nine hspAmerind strains from Amerindian hosts (N = 9), and nine hpEurope strains from European (N = 4) and Mestizo (N = 5) hosts were used for this analysis. The 18 frozen cultures of H. pylori strains, maintained at -80°C, were thawed and inoculated onto Brucella agar plates supplemented with 5% blood [66]. Plates were incubated at 37°C in a microaerobic atmosphere (5% CO2) in a humid chamber for 3 to 5 days [66]. H. pylori identity was confirmed by Gram staining and detection of urease and catalase activity. DNA was extracted from H. pylori cultures using the Wizard® Genomic DNA Purification Kit (Promega, MA), with the protocol specified by the manufacturer for gram-negative bacteria.
Restriction assays
Restriction endonuclease digestions were performed on the genomic DNA from 18 strains, using 16 commercially available restriction enzymes (New England BioLabs, MA) that were sensitive to methylation of the recognition sites (Additional file 1: Table S3). These enzymes were chosen because resistance to each has been reported in at least one H. pylori strain [42]. In our experiments, we controlled for the lack of restriction activity due to presence of inhibitors or high salt, by running control DNA from an H. pylori strain with a known restriction profile [18, 42]. However, in addition to the possibility of lacking the cognate restriction sites, lack of restriction activity due to the presence of supercoiled DNA cannot be ruled out.
In the restriction assays, ~500 μg of DNA were digested with 5U of the specified endonucleases for 2 h in a final volume of 30 μl of the appropriate buffer as recommended by the manufacturer. Chromosomal DNA from E. coli DH5α, as well as the H. pylori strains HPK5 and 99–35, were used as positive controls, to assess activity of the enzymes. Digestion products were electrophoresed at 80 V for 1 h in a 1% agarose gel [42]. The number of active methylases was determined based on the sensitivity of the DNA to restriction. The variable responses to the independent digestions were dichotomous: (lack of digestion) presence of the active methylase = 1 or 0 = digestion, no active methylase. To examine the differences in the number of active methylases between the bacterial populations, Wilcoxon-sum rank test was performed.
Transformation analysis
H. pylori hspAmerind or hpEurope strains with StrR, or KmR genetic markers were obtained by transformation with plasmid p801R or pCBT8, as described [32] and listed in Table 3. Plasmid p801R contains rspL with a point mutation in position 128 (A128G substitution), which confers resistance to Streptomycin (StrR). Plasmid pCTB8 carries an aphA cassette, which is integrated into the genome on the transformation-unrelated vacA locus and confers Kanamycin resistance (KmR).
In each case, the transformants can be detected based on the resistance phenotype of the transformed cells onto selective media. In brief, H. pylori strains were inoculated and incubated at 37°C in 5% CO2[31] for 3 days. The cells were re-plated and re-incubated for 24–72 h under the same conditions, then re-suspended in 1000 μl of PBS using a sterile swab and then centrifuged at 1,500 g for 2 min. The pellet was re-suspended in 200 μl of PBS, 25 μl of the H. pylori cells were mixed with 15 μl of the plasmid at a final concentration of 30 ng/μl. The mix was plated on Brucella agar supplemented with 5% sheep blood (BAB) and incubated as described above. After 24 h, the colonies were collected with a sterile swab and diluted in series from 10-1 to 10-6 in 900 μl Brucella broth (BB). The first four dilutions were spotted on selective media: BAB + Str [20 μg/mL], or Km [10 μg/mL], depending of the phenotype to be selected. The two last dilutions were inoculated onto non-selective BAB plates. After 5 days of incubation, colony-forming units (CFU) were counted on both the selective and non-selective plates, and transformation efficiency was calculated by comparing CFU numbers on the two types of media.
CFU counts used for this analysis were over a range of 30 – 300, to maximize statistical accuracy [67]. Differences in the rates of transformation were compared using the t-test, and the variance among strains was determined using the F-test.
Horizontal DNA transfer during co-culture
To evaluate the ability of H. pylori hspAmerind or hpEurope strains to obtain DNA from each other, the co-culture assay was performed as previously described [32]. The strains and plasmids used for these experiments are listed in Table 3. In summary, in addition to the single plasmid strains explained above, we produced double-resistant hspAmerind and hpEurope strains by transforming the single resistant strains described above with an additional suicide plasmid, pAD1-Cat [32]. This suicide plasmid, which carries a ureAB fragment from H. pylori strain 60190 with a central exogenous cat cassette (1127 bp), gets incorporated into the genomic ureA locus, creating chloramphenicol resistant (CmR) strains [32].
To determine the rates of DNA transformation from a donor hspAmerind strain to a recipient hpEurope strain, a single plasmid hspAmerind strain (99–33 or 99–35) with resistance to antibiotic "X" (used as a donor) and a double plasmid hpEurope strain (08–97 or 08–100) with resistance to antibiotics "Y/Z" (used as recipient), were co-cultured; transformants were selected by double or triple antibiotic resistance: "X/Y" or "X/Y/Z", respectively. To investigate the rates of transformation from a donor hpEurope strain to a recipient hspAmerind strain, we performed the same experiment but with the reverse phenotype, i.e. donor = hpEurope with single resistance "X"; recipient = hspAmerind with double resistance "Y/Z", and transformants with double or triple antibiotic resistance: X/Y" or "X/Y/Z", were evaluated. Based on the observation that after co-culturing a StrR/CmR strain and a KmR strain, the transformants with StrR/KmR are substantially higher (>2 log10 CFU) than those with StrR/CmR/KmR, we concluded that the rate of transformation with StrR/KmR could be used to reflect the KmR strain acquiring the single-base mutation rpsL (StrR) from the StrR/CmR strain.
To test for spontaneous mutations, blank controls we included in co-culture experiments, with recipient strains (i.e. StrR/CmR resistant) plated in selective plates containing the antibiotic for the donor strains (i.e. StrR). Resistant strains due to spontaneous mutations were never observed. As described above, results were based on CFU counts. Comparisons among the rates of transformation obtained from hspAmerind and hpEurope strains were assessed by performing the Mann Whitney test. For all transformation experiments, we used the appropriate blank controls for selection. Non-transformed strains were subject to the same conditions and plated on non-selective media to confirm cell viability.
References
Moodley Y, Linz B, Bond RP, Nieuwoudt M, Soodyall H, Schlebusch CM, Bernhoft S, Hale J, Suerbaum S, Mugisha L, et al: Age of the association between Helicobacter pylori and man. PLoS Pathog. 2012, 8 (5): e1002693-10.1371/journal.ppat.1002693.
Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, Falush D, Stamer C, Prugnolle F, van der Merwe SW, et al: An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007, 445 (7130): 915-918. 10.1038/nature05562.
Nobusato A, Uchiyama I, Kobayashi I: Diversity of restriction-modification gene homologues in Helicobacter pylori. Gene. 2000, 259 (1–2): 89-98.
Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez-Perez GI, et al: Traces of human migrations in Helicobacter pylori populations. Science. 2003, 299 (5612): 1582-1585. 10.1126/science.1080857.
Baltrus DA, Guillemin K, Phillips PC: Natural transformation increases the rate of adaptation in the human pathogen Helicobacter pylori. Evolution. 2008, 62 (1): 39-49.
van Doorn LJ, Figueiredo C, Sanna R, Pena S, Midolo P, Ng EK, Atherton JC, Blaser MJ, Quint WG: Expanding allelic diversity of Helicobacter pylori vacA. J Clin Microbiol. 1998, 36 (9): 2597-2603.
Kersulyte D, Mukhopadhyay AK, Velapatino B, Su W, Pan Z, Garcia C, Hernandez V, Valdez Y, Mistry RS, Gilman RH, et al: Differences in genotypes of Helicobacter pylori from different human populations. J Bacteriol. 2000, 182 (11): 3210-3218. 10.1128/JB.182.11.3210-3218.2000.
Owen RJ, Xerry J: Geographical conservation of short inserts in the signal and middle regions of the Helicobacter pylori vacuolating cytotoxin gene. Microbiology. 2007, 153 (Pt 4): 1176-1186.
Ghose C, Perez-Perez GI, van Doorn LJ, Dominguez-Bello MG, Blaser MJ: High frequency of gastric colonization with multiple Helicobacter pylori strains in Venezuelan subjects. J Clin Microbiol. 2005, 43 (6): 2635-2641. 10.1128/JCM.43.6.2635-2641.2005.
Dominguez-Bello MG, Perez ME, Bortolini MC, Salzano FM, Pericchi LR, Zambrano-Guzman O, Linz B: Amerindian Helicobacter pylori strains go extinct, as european strains expand their host range. PLoS ONE. 2008, 3 (10): e3307-10.1371/journal.pone.0003307.
Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, Dyrek I, Achtman M: Free recombination within Helicobacter pylori. Proc Natl Acad Sci U S A. 1998, 95 (21): 12619-12624. 10.1073/pnas.95.21.12619.
Kuipers EJ, Israel DA, Kusters JG, Blaser MJ: Evidence for a conjugation-like mechanism of DNA transfer in Helicobacter pylori. J Bacteriol. 1998, 180 (11): 2901-2905.
Suerbaum S, Achtman M: Evolution of Helicobacter pylori: the role of recombination. Trends Microbiol. 1999, 7 (5): 182-184. 10.1016/S0966-842X(99)01505-X.
Israel DA, Salama N, Krishna U, Rieger UM, Atherton JC, Falkow S, Peek RM: Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc Natl Acad Sci USA. 2001, 98 (25): 14625-14630. 10.1073/pnas.251551698.
Kuipers EJ, Israel DA, Kusters JG, Gerrits MM, Weel J, van Der Ende A, van Der Hulst RW, Wirth HP, Hook-Nikanne J, Thompson SA, et al: Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart from the same host. J Infect Dis. 2000, 181 (1): 273-282. 10.1086/315173.
Morelli G, Didelot X, Kusecek B, Schwarz S, Bahlawane C, Falush D, Suerbaum S, Achtman M: Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet. 2010, 6 (7): e1001036-10.1371/journal.pgen.1001036.
Kennemann L, Didelot X, Aebischer T, Kuhn S, Drescher B, Droege M, Reinhardt R, Correa P, Meyer TF, Josenhans C, et al: Helicobacter pylori genome evolution during human infection. Proc Natl Acad Sci USA. 2011, 108 (12): 5033-5038. 10.1073/pnas.1018444108.
Aras RA, Small AJ, Ando T, Blaser MJ: Helicobacter pylori interstrain restriction-modification diversity prevents genome subversion by chromosomal DNA from competing strains. Nucleic Acids Res. 2002, 30 (24): 5391-5397. 10.1093/nar/gkf686.
Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, Pan ZJ, Suerbaum S, Thompson SA, van der Ende A, van Doorn LJ: Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol Microbiol. 1999, 32 (3): 459-470. 10.1046/j.1365-2958.1999.01382.x.
Suerbaum S, Achtman M: Helicobacter pylori: recombination, population structure and human migrations. Int J Med Microbiol. 2004, 294 (2–3): 133-139.
Furuta Y, Yahara K, Hatakeyama M, Kobayashi I: Evolution of cagA oncogene of Helicobacter pylori through recombination. PLoS ONE. 2011, 6 (8): e23499-10.1371/journal.pone.0023499.
Arber W: Host-controlled modification of bacteriophage. Annu Rev Microbiol. 1965, 19: 365-378. 10.1146/annurev.mi.19.100165.002053.
Kobayashi I: Restriction-Modification systems as a minimal forms of life from restriction endonucleases. Vol. 14: Gross HJ. 2004, Berlin, Heidelberg: Springer-Verlag
Lin LF, Posfai J, Roberts RJ, Kong H: Comparative genomics of the restriction-modification systems in Helicobacter pylori. Proc Natl Acad Sci U S A. 2001, 98 (5): 2740-2745. 10.1073/pnas.051612298.
Xu Q, Morgan RD, Roberts RJ, Blaser MJ: Identification of type II restriction and modification systems in Helicobacter pylori reveals their substantial diversity among strains. Proc Natl Acad Sci U S A. 2000, 97 (17): 9671-9676. 10.1073/pnas.97.17.9671.
Wilson GG, Murray NE: Restriction and modification systems. Annu Rev Genet. 1991, 25: 585-627. 10.1146/annurev.ge.25.120191.003101.
Kobayashi I: Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001, 29 (18): 3742-3756. 10.1093/nar/29.18.3742.
Rocha EP, Danchin A, Viari A: Evolutionary role of restriction/modification systems as revealed by comparative genome analysis. Genome Res. 2001, 11 (6): 946-958. 10.1101/gr.GR-1531RR.
Panina EM, Mironov AA, Gel’fand MS: Statistical analysis of complete bacterial genomes: palindromes and systems of restriction-modification. Mol Biol (Mosk). 2000, 34 (2): 246-252.
Gelfand MS, Koonin EV: Avoidance of palindromic words in bacterial and archaeal genomes: a close connection with restriction enzymes. Nucleic Acids Res. 1997, 25 (12): 2430-2439. 10.1093/nar/25.12.2430.
Levine SM, Lin EA, Emara W, Kang J, DiBenedetto M, Ando T, Falush D, Blaser MJ: Plastic cells and populations: DNA substrate characteristics in Helicobacter pylori transformation define a flexible but conservative system for genomic variation. Faseb J. 2007, 21 (13): 3458-3467. 10.1096/fj.07-8501com.
Zhang XS, Blaser MJ: Natural transformation of an engineered Helicobacter pylori strain deficient in type II restriction endonucleases. J Bacteriol. 2012, 194 (13): 3407-3416. 10.1128/JB.00113-12.
Pride DT, Blaser M: Identification of horizontally acquired genetic elements in Helicobacter pylori and other prokaryotes using oligonucleotide difference analysis. Genome Letters. 2002, 1 (1): 2-15. 10.1166/gl.2002.003.
Schroeder C, Jurkschat H, Meisel A, Reich JG, Kruger D: Unusual occurrence of EcoP1 and EcoP15 recognition sites and counter-selection of type II methylation and restriction sequences in bacteriophage T7 DNA. Gene. 1986, 45 (1): 77-86. 10.1016/0378-1119(86)90134-4.
Boyer HW: DNA restriction and modification mechanisms in bacteria. Annu Rev Microbiol. 1971, 25: 153-176. 10.1146/annurev.mi.25.100171.001101.
Revel HR, Luria SE: DNA-glucosylation in T-even phage: genetic determination and role in phage-host interaction. Annu Rev Genet. 1970, 4: 177-192. 10.1146/annurev.ge.04.120170.001141.
Price C, Bickle TA: A possible role for DNA restriction in bacterial evolution. Microbiol Sci. 1986, 3 (10): 296-299.
Karlin S, Burge C, Campbell AM: Statistical analyses of counts and distributions of restriction sites in DNA sequences. Nucleic Acids Res. 1992, 20 (6): 1363-1370. 10.1093/nar/20.6.1363.
Burge C, Campbell AM, Karlin S: Over- and under-representation of short oligonucleotides in DNA sequences. Proc Natl Acad Sci USA. 1992, 89 (4): 1358-1362. 10.1073/pnas.89.4.1358.
Wang MX, Church GM: A whole genome approach to in vivo DNA-protein interactions in E. coli. Nature. 1992, 360 (6404): 606-610. 10.1038/360606a0.
Campbell JL, Kleckner N: E. coli oriC and the dnaA gene promoter are sequestered from dam methyltransferase following the passage of the chromosomal replication fork. Cell. 1990, 62 (5): 967-979. 10.1016/0092-8674(90)90271-F.
Takata T, Aras R, Tavakoli D, Ando T, Olivares AZ, Blaser MJ: Phenotypic and genotypic variation in methylases involved in type II restriction-modification systems in Helicobacter pylori. Nucleic Acids Res. 2002, 30 (11): 2444-2452. 10.1093/nar/30.11.2444.
Vale FF, Vitor JM: Genomic methylation: a tool for typing Helicobacter pylori isolates. Appl Environ Microbiol. 2007, 73 (13): 4243-4249. 10.1128/AEM.00199-07.
Ando T, Ishiguro K, Watanabe O, Miyake N, Kato T, Hibi S, Mimura S, Nakamura M, Miyahara R, Ohmiya N, et al: Restriction-modification systems may be associated with Helicobacter pylori virulence. J Gastroenterol Hepatol. 2010, 25 (Suppl 1): S95-S98.
Naito T, Kusano K, Kobayashi I: Selfish behavior of restriction-modification systems. Science. 1995, 267 (5199): 897-899. 10.1126/science.7846533.
Handa N, Kobayashi I: Post-segregational killing by restriction modification gene complexes: observations of individual cell deaths. Biochimie. 1999, 81 (8–9): 931-938.
Donahue JP, Israel DA, Torres VJ, Necheva AS, Miller GG: Inactivation of a Helicobacter pylori DNA methyltransferase alters dnaK operon expression following host-cell adherence. FEMS Microbiol Lett. 2002, 208 (2): 295-301. 10.1111/j.1574-6968.2002.tb11097.x.
Takeuchi H, Israel DA, Miller GG, Donahue JP, Krishna U, Gaus K, Peek RM: Characterization of expression of a functionally conserved Helicobacter pylori methyltransferase-encoding gene within inflamed mucosa and during in vitro growth. J Infect Dis. 2002, 186 (8): 1186-1189. 10.1086/343810.
Bauman R: Microbiology. 2004, San Francisco, CA: Benjamin-Cummings Publishing Company
Lorenz MG, Wackernagel W: Bacterial gene transfer by natural genetic transformation in the environment. Microbiol Rev. 1994, 58 (3): 563-602.
Kang J, Blaser MJ: Bacterial populations as perfect gases: genomic integrity and diversification tensions in Helicobacter pylori. Nat Rev Microbiol. 2006, 4 (11): 826-836. 10.1038/nrmicro1528.
Hofreuter D, Odenbreit S, Henke G, Haas R: Natural competence for DNA transformation in Helicobacter pylori: identification and genetic characterization of the comB locus. Mol Microbiol. 1998, 28 (5): 1027-1038. 10.1046/j.1365-2958.1998.00879.x.
Smeets LC, Kusters JG: Natural transformation in Helicobacter pylori: DNA transport in an unexpected way. Trends Microbiol. 2002, 10 (4): 159-162. 10.1016/S0966-842X(02)02314-4. Response from Dirk Hofreuter and Rainer Haas, discussion 162
Chang KC, Yeh YC, Lin TL, Wang JT: Identification of genes associated with natural competence in Helicobacter pylori by transposon shuttle random mutagenesis. Biochem Biophys Res Commun. 2001, 288 (4): 961-968. 10.1006/bbrc.2001.5877.
Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, Roche N, Vikstrom S, Sjostrom R, Linden S, Backstrom A, et al: Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004, 305 (5683): 519-522. 10.1126/science.1098801.
Ando T, Israel DA, Kusugami K, Blaser MJ: HP0333, a member of the dprA family, is involved in natural transformation in Helicobacter pylori. J Bacteriol. 1999, 181 (18): 5572-5580.
Smeets LC, Bijlsma JJ, Kuipers EJ, Vandenbroucke-Grauls CM, Kusters JG: The dprA gene is required for natural transformation of Helicobacter pylori. FEMS Immunol Med Microbiol. 2000, 27 (2): 99-102. 10.1111/j.1574-695X.2000.tb01418.x.
Jolley KA, Chan MS, Maiden MC: mlstdbNet - distributed multi-locus sequence typing (MLST) databases. BMC Bioinforma. 2004, 5: 86-10.1186/1471-2105-5-86.
Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, et al: Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998, 95 (6): 3140-3145. 10.1073/pnas.95.6.3140.
Falush D, Stephens M, Pritchard JK: Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003, 164 (4): 1567-1587.
Tamura K, Nei M, Kumar S: Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA. 2004, 101 (30): 11030-11035. 10.1073/pnas.0404206101.
Tamura K, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011, 28 (10): 2731-2739. 10.1093/molbev/msr121.
Kong H, Lin LF, Porter N, Stickel S, Byrd D, Posfai J, Roberts RJ: Functional analysis of putative restriction-modification system genes in the Helicobacter pylori J99 genome. Nucleic Acids Res. 2000, 28 (17): 3216-3223. 10.1093/nar/28.17.3216.
McCune A, Grace JB, Urban DL: Analysis of ecological communities. 2001, Oregon: MJM Software Design
Clarke KR: Non-parametric multivariate analyses of changes in community structure. Austral Ecol. 1993, 18 (1): 117-143. 10.1111/j.1442-9993.1993.tb00438.x.
Buck GE, Smith JS: Medium supplementation for growth of Campylobacter pyloridis. J Clin Microbiol. 1987, 25 (4): 597-599.
Miles AA, Misra SS, Irwin JO: The estimation of the bactericidal power of the blood. J Hyg (Lond). 1938, 38 (6): 732-749. 10.1017/S002217240001158X.
Acknowledgements
This work was supported by UPR grant FIPI 880314 and by R01GM63270 from the NIH, by the Bill & Melinda Gates Foundation, and the Diane Belfer Program for Human Microbial Ecology. We thank Lihai Song and Maria Egleé Pérez for mathematical and statistical guidance, and Dr. Jason Rauscher for fruitful discussions in fundamental concepts of evolution. Part of this work was performed at New York University under the auspices of The Company of Biologists, the Faculty of Natural Science at UPR and CREST-CATEC. We thank Dr. Guillermo Perez-Perez and Edgardo Sanabria-Valentin for technical support at NYU.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All the authors declare that they have no competing interests.
Authors’ contributions
ALM designed the analysis, perform all the in silico analysis, restriction and transformation experiments, analyzed the data and perform statistics, also prepared the manuscript and figures. MS optimized the mathematical model for expected restriction sites and perform all the simulation analysis. XZ perform the co-culture experiments and participate in the manuscript preparation. PL help with the initial statistical modeling for the simulation analysis. AT and MC provided samples to the completion of the study. LB help analyzing MLST to be assigned to specific haplotype, also collaborate in the manuscript preparation. MGDB and MB participate in the experimental design, discussion of results, preparation and review of the manuscript. All authors read and approved the final manuscript.
Shrinivasrao P Mane, Xue-Song Zhang contributed equally to this work.
An erratum to this article is available at http://dx.doi.org/10.1186/1471-2180-14-122.
Electronic supplementary material
12866_2013_2090_MOESM1_ESM.pdf
Additional file 1: Table S1: Proportion of nucleotides in the H. pylori sequences analyzed. Table S2. Haplotype and origin of the strains included in the in vitro analysis of active methylases. Table S3. Distribution of active methylases in H. pylori strains, by haplotype. Figure S1. Neighbor joining clustering based on multilocus sequences of 110 H. pylori strains used in this study. The strains were grouped (Kimura-2 parameter) into four main clusters accordingly with the population assignment using STRUCTURE software: hpAfrica1 (N=25) in blue, hpEurope (N=48) in green; hspEAsia (N=12) in yellow and hspAmerind (N=25) in orange. Figure S2. PCA showing the variation among H. pylori strains. PCA is a mathematical model that transforms the data to a new coordinate system. The data is organized based on coordinates that goes from the one with the greatest variance by any projection (called the first principal component), to the second greatest variance on the second coordinate, and so on. Based on the frequency of cognate recognition sites for 32 endonucleases, H. pylori strains were separated in two coordinates. Strains are coded by haplotype: AM for hspAmerind, AS for hspEAsia, E for hpEurope, and AF for hpAfrica1. The number that follow the haplotype code indicate the sequence number (e.g. hspAmerind, N=25= AM1, AM2… AM25). Zero (0) indicates no variation. Arrows in red indicate the direction of the variation for each of the 32 restriction sites analyzed; longer arrows indicate that the variation of the restriction profile for a given, is far from zero (more variable). Differences in the RMS profile were mainly due to 15 cognate recognition sites for: HpyCH4V, HpyF14I, Hpy99IV, Hpy166III, HpyF44II, HpyNI, HpyC1I, Hpy8I, HpyIV, HpyF10VI, Hpy99VIP, HpyCH4II, Hpy188III, Hpy178VII, HpyV endonucleases; which explained 29% and 18% of the variation in component 1 and 2, respectively. (PDF 1 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Maldonado-Contreras, A., Mane, S.P., Zhang, XS. et al. Phylogeographic evidence of cognate recognition site patterns and transformation efficiency differences in H. pylori: theory of strain dominance. BMC Microbiol 13, 211 (2013). https://doi.org/10.1186/1471-2180-13-211
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-13-211