
Abstract
Background
The species Pectobacterium carotovorum includes a diverse subspecies of bacteria that cause disease on a wide variety of plants. In Morocco, approximately 95% of the P. carotovorum isolates from potato plants with tuber soft rot are P. carotovorum subsp. carotovorum. However, identification of this pathogen is not always related to visual disease symptoms. This is especially true when different pathogen cause similar diseases on potato, citing as an example, P. carotovorum, P. atrosepticum and P. wasabiae. Numerous conventional methods were used to characterize Pectobacterium spp., including biochemical assays, specific PCR-based tests, and construction of phylogenetic trees by using gene sequences. In this study, an alternative method is presented using a gene linked to pathogenicity, in order to allow accuracy at subspecies level. The pmrA gene (response regulator) has been used for identification and analysis of the relationships among twenty nine Pectobacterium carotovorum subsp. carotovorum and other Pectobacterium subspecies.
Results
Phylogenetic analyses of pmrA sequences compared to ERIC-PCR and 16S rDNA sequencing, demonstrated that there is considerable genetic diversity in P. carotovorum subsp. carotovorum strains, which can be divided into two distinct groups within the same clade.
Conclusions
pmrA sequence analysis is likely to be a reliable tool to identify the subspecies Pectobacterium carotovorum subsp. carotovorum and estimate their genetic diversity.
Similar content being viewed by others


Background
Pectobacterium carotovorum subsp. carotovorum (P. carotovorum subsp. carotovorum) is a plant-pathogenic enterobacterium which belongs to the soft-rot group of Pectobacterium. It has the ability to cause serious damage worldwide on a numerous types of plants in field and storage stage [1]. In Morocco, approximately 95% of the P. carotovorum isolated from potato plants with tuber soft rot are P. carotovorum subsp. carotovorum[2]. This bacteria produce a wide variety of plant cell wall-degrading enzymes, causing maceration of different plant organs and tissues [1, 3]. Many of its virulence genes have been identified, including genes encoding degradative enzymes, diverse regulatory systems, and the type III secretion system [4]. Pectobacterium spp. is a complex taxon consisting of strains with a range of different phenotype, biochemical, host range and genetic characteristics. Several methods were used to characterize this taxon, including biochemical assays and construction of phylogenetic trees by using gene sequences. For example, Toth and his collaborators [4–8] have shown that there are five major clades of Pectobacterium (formerly E. carotovorum): atrosepticum, betavasculorum, carotovorum, odoriferum, and wasabiae. Their analysis did not include P. brasiliensis which form individual clade [9]. Recently, other authors [10, 11] were interested in molecular typing methods. These methods are increasingly used in the analysis of P. carotovorum subsp. carotovorum relatedness in order to identify their transmission routes and to assess its biodiversity. They have demonstrated a high diversity of polymorphism between these subspecies.
To survive, colonize and cause disease, plant-pathogenic bacteria modulate expression of their genes often using two-component signal transduction systems (TCS). These systems typically consist of two conserved components, a sensor histidine kinase and a response regulator [12]. P. carotovorum subsp. carotovorum employs different two-component systems for controlling production of virulence determinants [13–16]. PmrA-PmrB is one example of TCS for plant pathogenic bacteria, which affects production of extracellular enzymes, virulence and bacterial survival in potato tubers as well as in Arabidopsis leaves and generally in planta[17]. The main target genes of this TCS encode products with sequence similarity to DNA binding response regulators and autophosphorylatable histidine kinases.
The pmrA locus is required for resistance to the cationic peptide antibiotic polymyxin B and to other plant-derived antimicrobial peptides in Pectobacterium. It controls the production of proteins that mediate the modification of the lipopolysaccharide (LPS) core and lipid A [17–19]. The changes in LPS structure leads to reduction of the negative charges at cell surface and hence altered interactions with iron and cationic peptides [20]. This gene was found in almost all Enterobacteriaceae[20]. In P. carotovorum subsp. carotovorum, pmrA gene encodes a protein of 222 amino acid (aa) that reveals 59.7% of identity to pmrA of Salmonella and BasR of E. coli. Its inactivation in P. carotovorum subsp. carotovorum does not reduce the maceration ability of the bacterium on potato tuber but nevertheless remains essential for survival under adverse environmental conditions [16, 20, 21]. Phylogenies built with single genes have been used already to examine the relationships of the plant-pathogenic enterobacteria [22–25]. In this study, pmrA sequence analysis was used to identify the Pectobacterium carotovorum subsp. carotovorum and to estimate their genetic diversity. In addition, in at least one other system, this analysis was better correlated with Enterobacterial Repetitive Intergenic Consensus PCR (ERIC-PCR) assays and phylogenies built from 16S rDNA genes [10].
Results and discussion
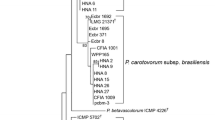
Twenty-nine isolates from soft-rotted potato tubers (Table 1) were used in this study. They have been identified by biochemical and phenotypic tests ([2] and Additional file 1 Table S1). A part of the strains were already confirmed as P. carotovorum subsp. carotovorum using ERIC-PCR [2, 10]. However, all strains yielded a 434 bp DNA fragment in PCR with the Y1 and Y2 specific primers for pectate lyase (pel) genes of Pectobacterium spp. [26, 27] and a 666pb with specifics primers for pmrA of Pectobacterium carotovorum subsp. carotovorum (F0145 and E2477 [16]) (Figure 1). Our purpose in this study was to develop a tool with a high specificity to detect typical Pectobacterium carotovorum subsp. carotovorum isolated in Morocco and that could serve as a tool to evaluate the genetic diversity of these subspecies. To investigate the utility of pmrA-PCR as a method of identification, the dendrogram built (Figure 2A) from well-characterized strains was used to illustrate the clustering of subspecies, on the basis of a single-gene (pmrA) and analysis of 16 s rRNA gene sequences of Pectobacterium spp. (Figure 2B,C). Our phylogenetic tree (Figure 2A) revealed a high diversity among the subspecies tested with a maximum identity to the pmrA gene of strain WPP14 (AB447882.1), ranging from 95 to 99%. Moreover, phylogenetic distance between all strains is 0,02 suggesting that all Pectobacterium carotovorum subsp. carotovorum circulating in Morocco, have their origin from the United States [28, 29]. Following numerical analysis of the 29 pmrA sequences by Neighbor-Joining (NJ) and UPGMA, the taxa were divided into two groups (clusters I to II), the similarity value between the two main clusters was about 96%. However, both clusters were represented by six different sequences (Figure 2A) and over 50% of the strains were included in the cluster I. Detailed scrutiny of the results given by the NJ method showed that all P. carotovorum subsp. carotovorum formed only one clade with 99% bootstrap. However, to verify the genetic diversity within our subspecies, the sequence alignment with maximum composite likelihood method (ML) were used. A comparison of 13 different pmrA sequences (Figure 3) revealed 0.05 as estimated value of the shape parameter for the discrete Gamma Distribution. The intraspecies comparison of DNA sequence identity is determined by the BLAST algorithm for P. carotovorum subsp. carotovorum strains for pmrA gene. This finding suggests that there is considerable genetic diversity in P. carotovorum subsp. carotovorum strains, which is in accordance with previous works reported by different authors [9, 10, 23, 28]. Also, the multiple sequence alignment of these sequences revealed conserved regions at different stretches. These regions could be used for designing degenerate primers or probes for PCR-based amplification or hybridization-based detection of pmrA sequences from different subspecies of P. carotovorum. Furthermore, within the genus Pectobacterium, there are five major clades forming a polyphyletic group: P. atrosepticum, P. betavasculorum, P. carotovorum subsp. carotovorum, P. odoriferum[23], and P. wasabiae. These analyses did not include strains (P. brasiliensis[27]). Our phylogeny (Figure 4) places all the strains previously identified using biochemical and phenotypic methods in the group P. carotovorum subsp. carotovorum, noting that, some potato strains collected in different years and in widely different locations were grouped closely in the same group. It places also P. brasiliensis more similar to P. carotovorum subsp. carotovorum than to P. atrosepticum (E. carotovora subsp. atroseptica SCRI1043) and P. wasabiae WPP163, knowing that the level of similarity between the two pmrA sequences subspecies P. atrosepticum and P. carotovorum subsp. carotovorum is 98.19%. Many others phylogenetic analysis revealed that not all subspecies of P. carotovorum were grouped in a single, robust clade identified by all methods [9, 29]. This was a strong indication that the different subspecies of P. carotovorum could indeed belong to different species. Despite the fact that some authors have concluded that the phylogenies built with single genes do not have many informative characters, and they “may not accurately reflect interspecies taxonomic relatedness” [22], our current phylogenetic analysis of pmrA sequences was clearly sufficient to determine whether all of these subspecies can be placed in the same subspecies or to split into two different subspecies. Noting that, the pmrA gene sequences have several advantages, including being effectively a single-copy gene, highly conserved in P. carotovorum subsp. carotovorum and easy to amplify. Therefore, the sequencing and analysis sequence data for the pmrA region of P. carotovorum subsp. carotovorum strains could be a reliable tool for detection of pathogens. Moreover, pmrA sequence analysis has shown a high genetic diversity among the isolates P. carotovorum subsp. carotovorum. The same results have been reported by other studies [2, 5, 9, 23, 29] using several phylogenetic analyses seeking to understand the relationship among these nominal subspecies.

Amplification of a specific 666 bp band in 7 strains generated using pmrA F0145 and E2477 primers. Abbreviation: M, 100 bp DNA Step Ladder (1 kbp); C + (positive control), P116C2; C-, negative control 1, P111C2; 2, P111C3; 3, P111C4; 4, P211C1; 5, P211C2; 6, P211C3 and 7, P211C4.

Phylogenetic tree based on a comparison of pmrA sequences (A) and 16S rRNA (B) for Pectobacterium carotovorum subsp . carotovorum. (C) Accession numbers of 16S rRNA sequences used for sequence alignments and construction of phylogenetic tree. The branching pattern was generated by the Neighbor-Joining method [31]. The numbers at the nodes indicate the levels of bootstrap support based on a Neighbor-Joining analysis of 500 resampled data sets. The evolutionary distances were computed using the Maximum Composite Likelihood method [32] and are in the units of the number of base substitutions per site. The generation of tree was conducted in MEGA5 [33].

Nucleic acid sequence alignment of pmrA gene among various strains of Pectobacterium carotovorum subsp. carotovorum . P. carotovorum subsp. carotovorum pmrA gene for response regulator PmrA (AB447882.1) available in GenBank was downloaded from NCBI. The alignments were performed using the ClustalW program [31]. The identical Nucleic acid in equivalent positions are indicated by dots and generated using the MEGA 5 program [32].

Compressed subtree sequenced data for pmrA gene of 8 subspecies of Enterobacteriaceae based upon Neighbor-Joining method [[33]]. Subtrees presented in Figure 2 are compressed into black triangle. The numbers at the nodes indicate the levels of bootstrap support based on a Neighbor-Joining analysis of 500 resampled data sets. The evolutionary distances were computed using the Maximum Composite Likelihood method [34] and are in the units of the number of base substitutions per site. The generation of tree was conducted in MEGA5 [32].
Conclusions
Our pmrA gene sequence analysis, linked to pathogenicity studies, could be used to identify and monitor the diversity of the P. carotovorum subsp. carotovorum subspecies.
Methods
Sample handling and isolate bacteria
During the years 2003 to 2011, different potato fields and the most important potato storages were controlled in Morocco and several samples were collected from plants with soft rot disease. Nutrient agar, King’s B agar, Crystal Violet Pectate (CVP) and LPGA medium (5 g/L yeast extract, 5 g/L peptone, 5 g/L glucose 15 g/L agar) were used to isolate the suspected bacteria. The 29 strains used in this study are isolated from different geographic Moroccan regions and had been stored in 20% glycerol at −20°C [2, 30]. Table 1 shows the strains whose sequences were determined in this study and the reference strains used for comparison when phylogenetic trees were constructed. Table 1 includes the strain designations and the GenBank accession numbers for the pmrA sequences.
Biochemical and physiological tests
In order to identify Pectobacterium spp., the strains were grown at 27°C for 24 h on agar plates and they were tested for Gram staining, catalase, oxidase, nitrate production, reductase activity, pectinolytic activity on Sutton medium, and absence pigmentation of the strains in the King B medium (Difco) [2]. Identification of confirmed Pectobacterium spp. isolates to species and subspecies was conducted on the basis of biochemical tests (indole production from tryptophan, lecithinase activity and acid production from α-methyl glucoside, trehalose, sorbitol, melibiose, lactose). All tests were carried out at 27°C for 24 h and compared with the standard strains (see Additional file 1 Table S1 for the fourteen strains used only in this study) [2, 10].
DNA extraction and PCR amplification
Bacterial cultures from frozen stocks were grown onto LPGA medium and suspended in sterile H2O. The concentration was adjusted to 108 CFU.ml-1. DNA was extracted from bacterial suspension as described by Terta et al. [2]. The precipitated DNA then was quantified using a NanoDrop 8000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA), adjusted to 100 ng.μl-1 and stored at 4°C. All PCR amplifications were performed using the following primers: pmr A F0145 (5’-TACCCTGCAGATGAAATTATTGATTGTTGAAGAC-3’) and E2477 (5’-TACCAAGCTTTGGTTGTTCCCCTTTGGTCA-3’) as described by Hyytiäinen et al. 2003 [16]. A 25 μl PCR mix contained: 1 μl DNA, 0.5 U Taq DNA polymerase, 2.5 μl 10 × PCR buffer, 2.5 mM each of dNTPs, 2.5 mM MgCl2, 0.5 μM of each primer. DNA amplification was performed on Veriti® Thermal Cycler (Applied Biosystems) under the following conditions: 5 min at 94°C for initial denaturation, 35 cycles of 1 min at 94°C for, 1 min at 55°C and 2 min 72°C, followed by a final elongation step of 10 min at 72°C. PCR products (6 μl) were separated by gel electrophoresis in 1.8% agarose gels in TBE buffer. Following staining with ethidium bromide, the gels were viewed and photographed under UV Transilluminator. Fragment sizes were determined by comparison to a 100 bp DNA Ladders.
Sequencing of pmrA and phylogenetic analysis
The PCR-amplified fragments of pmrA were purified and the sequencing reactions were performed with a Big-Dye Terminator v3.1 (Applied Biosystems). The pmrA sequences which we determined and the sequences of the reference strains of members of the family Enterobacteriaceae obtained from the GenBank databases were analyzed. The pmrA sequences were first aligned by using the Clustal W program [34], and then the alignments were corrected by hand. Evolutionary trees for the data set were inferred by using the Neighbor-Joining program of MEGA [31, 33]. The stability of relationships was assessed by performing bootstrap analyses of the Neighbor-Joining data based on 500 resamplings. The entire sequences corresponding to positions 4317866-4318532 of the reference sequence of the subspecies.
Nucleotide sequence accession numbers
The pmrA sequences which we determined have been deposited in the GenBank database under the accession numbers shown in Table 1.
References
Perombelon MCM, Kelman A: Ecology of the Soft Rot Erwinias. Annu Rev Phytopathol. 1980, 18 (1): 361-387. 10.1146/annurev.py.18.090180.002045.
Terta M, El Karkouri A, Ait M’hand R, Achbani E, Barakate M, Amdan M, Annajar B, El Hassouni M, Val F, Bouteau F: Occurrence OF Pectobacterium carotovorum strains isolated from potato soft rot in Morocco. Cell Mol Biol (Noisy-le-Grand). 2010, 56 (Suppl): OL1324-1333.
Norman-Setterblad C, Vidal S, Palva ET: Interacting signal pathways control defense gene expression in Arabidopsis in response to cell wall-degrading enzymes from Erwinia carotovora. Mol Plant Microbe Interact. 2000, 13 (4): 430-438. 10.1094/MPMI.2000.13.4.430.
Toth IK, Bell KS, Holeva MC, Birch PRJ: Soft rot erwiniae: from genes to genomes. Mol Plant Pathol. 2003, 4 (1): 17-30. 10.1046/j.1364-3703.2003.00149.x.
Toth IK, Avrova AO, Hyman LJ: Rapid identification and differentiation of the soft rot erwinias by 16S-23S intergenic transcribed spacer-PCR and restriction fragment length polymorphism analyses. Appl Environ Microbiol. 2001, 67 (9): 4070-4076. 10.1128/AEM.67.9.4070-4076.2001.
Avrova AO, Hyman LJ, Toth RL, Toth IK: Application of Amplified Fragment Length Polymorphism Fingerprinting for Taxonomy and Identification of the Soft Rot Bacteria Erwinia carotovora and Erwinia chrysanthemi. Appl Environ Microbiol. 2002, 68 (4): 1499-1508. 10.1128/AEM.68.4.1499-1508.2002.
Bell KS, Sebaihia M, Pritchard L, Holden MTG, Hyman LJ, Holeva MC, Thomson NR, Bentley SD, Churcher LJC, Mungall K: Genome sequence of the enterobacterial phytopathogen Erwinia carotovora subsp. atroseptica and characterization of virulence factors. Proc Natl Acad Sci USA. 2004, 101 (30): 11105-11110. 10.1073/pnas.0402424101.
Toth IK, Pritchard L, Birch PRJ: Comparative genomics reveals what makes an enterobacterial plant pathogen. Annu Rev Phytopathol. 2006, 44 (1): 305-336. 10.1146/annurev.phyto.44.070505.143444.
Ma B, Hibbing ME, Kim HS, Reedy RM, Yedidia I, Breuer J, Glasner JD, Perna NT, Kelman A, Charkowski AO: Host range and molecular Phylogenies of the soft rot enterobacterial genera Pectobacterium and dickeya. Phytopathology. 2007, 97 (9): 1150-1163. 10.1094/PHYTO-97-9-1150.
Terta M, Azelmat S, M’hand R, Achbani E, Barakate M, Bouteau F, Ennaji M: Molecular typing of Pectobacterium carotovorum isolated from potato tuber soft rot in Morocco. Ann Microbiol. 2012, 7: 1-7.
Tavasoli E, Marefat AR, Hassanzadeh N: Identity and genetic diversity of Pectobacterium spp., causal agents of potato soft rot in Zanjan, Iran. Edited by: Journals A. 2011, Academic Journals, 329-336.
Stock AM, Robinson VL, Goudreau PN: TWO-COMPONENT SIGNAL TRANSDUCTION. Annu Rev Biochem. 2000, 69 (1): 183-215. 10.1146/annurev.biochem.69.1.183.
Frederick RD, Chiu J, Bennetzen JL, Handa AK: Identification of a Pathogenicity Locus, rpfA, in Erwinia carotovora subsp. carotovora That Encodes a Two-Component Sensor-Regulator Protein. Mol Plant Microbe Interact. 1997, 10 (3): 407-415. 10.1094/MPMI.1997.10.3.407.
Eriksson ARB, Andersson RA, Pirhonen M, Palva ET: Two-Component Regulators Involved in the Global Control of Virulence in Erwinia carotovora subsp. carotovora. Mol Plant Microbe Interact. 1998, 11 (8): 743-752. 10.1094/MPMI.1998.11.8.743.
Flego D, Marits R, Eriksson ARB, Koiv V, Karlsson MB, Heikinheimo R, Palva ET: A two-component regulatory system, pehR-pehS, controls endopolygalacturonase production and virulence in the plant pathogen Erwinia carotovora subsp carotovora. Mol Plant Microbe Interact. 2000, 13 (4): 447-455. 10.1094/MPMI.2000.13.4.447.
Hyytiainen H, Sjoblom S, Palomaki T, Tuikkala A, Palva ET: The PmrA-PmrB two-component system responding to acidic pH and iron controls virulence in the plant pathogen Erwinia carotovora ssp carotovora. Mol Microbiol. 2003, 50 (3): 795-807. 10.1046/j.1365-2958.2003.03729.x.
Hyytiäinen H: Regulatory networks controlling virulence in the plant pathogen Erwinia carotovora ssp. carotovora. 2005, University of Helsinki: Department of Biological and Environmental Sciences FoB, 57-ISBN 952-10-2485-2
Helander IM, Kilpeläinen I, Vaara M: Increased substitution of phosphate groups in lipopolysaccharides and lipid A of the polymyxin-resistant pmrA mutants of Salmonella typhimurium: a 31P-NMR study. Mol Microbiol. 1994, 11 (3): 481-487. 10.1111/j.1365-2958.1994.tb00329.x.
Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI: PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol Microbiol. 1998, 27 (6): 1171-1182. 10.1046/j.1365-2958.1998.00757.x.
Wösten MMSM, Groisman EA: Molecular Characterization of the PmrA Regulon. J Biol Chem. 1999, 274 (38): 27185-27190. 10.1074/jbc.274.38.27185.
Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI: Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect Immun. 2000, 68 (11): 6139-6146. 10.1128/IAI.68.11.6139-6146.2000.
Brown EW, Davis RM, Gouk C, Van der Zwet T: Phylogenetic relationships of necrogenic Erwinia and Brenneria species as revealed by glyceraldehyde-3-phosphate dehydrogenase gene sequences. Int J Syst Evol Microbiol. 2000, 50 (6): 2057-2068. 10.1099/00207713-50-6-2057.
Gardan L, Gouy C, Christen R, Samson R: Elevation of three subspecies of Pectobacterium carotovorum to species level: Pectobacterium atrosepticum sp. nov., Pectobacterium betavasculorum sp. nov. and Pectobacterium wasabiae sp. nov. Int J Syst Evol Microbiol. 2003, 53 (2): 381-391. 10.1099/ijs.0.02423-0.
Hauben L, Moore ERB, Vauterin L, Steenackers M, Mergaert J, Verdonck L, Swings J: Phylogenetic position of phytopathogens within the Enterobacteriaceae. Syst Appl Microbiol. 1998, 21 (3): 384-397. 10.1016/S0723-2020(98)80048-9.
Samson R, Legendre JB, Christen R, Saux MF-L, Achouak W, Gardan L: Transfer of Pectobacterium chrysanthemi (Burkholder et al. 1953) Brenner et al. 1973 and Brenneria paradisiaca to the genus Dickeya gen. nov. as Dickeya chrysanthemi comb. nov. and Dickeya paradisiaca comb. nov. and delineation of four novel species, Dickeya dadantii sp. nov., Dickeya dianthicola sp. nov., Dickeya dieffenbachiae sp. nov. and Dickeya zeae sp. nov. Int J Syst Evol Microbiol. 2005, 55 (4): 1415-1427. 10.1099/ijs.0.02791-0.
Darrasse A, Priou S, Kotoujansky A, Bertheau Y: PCR and restriction fragment length polymorphism of a pel gene as a tool to identify Erwinia carotovora in relation to potato diseases. Appl Environ Microbiol. 1994, 60 (5): 1437-1443.
Duarte V, De Boer SH, Ward LJ, De Oliveira AMR: Characterization of atypical Erwinia carotovora strains causing blackleg of potato in Brazil. J Appl Microbiol. 2004, 96 (3): 535-545. 10.1111/j.1365-2672.2004.02173.x.
Yap MN, Barak JD, Charkowski AO: Genomic diversity of Erwinia carotovora subsp carotovora and its correlation with virulence. Appl Environ Microbiol. 2004, 70 (5): 3013-3023. 10.1128/AEM.70.5.3013-3023.2004.
Glasner JD, Marquez-Villavicencio M, Kim HS, Jahn CE, Ma B, Biehl BS, Rissman AI, Mole B, Yi X, Yang CH: Niche-specificity and the variable fraction of the pectobacterium pan-genome. Mol Plant Microbe Interact. 2008, 21 (12): 1549-1560. 10.1094/MPMI-21-12-1549.
Terta M, Kettani-Halabi M, Ibenyassine K, Tran D, Meimoun P, M’Hand RA, El-Maarouf-Bouteau H, Val F, Ennaji MM, Bouteau F: Arabidopsis thaliana cells: a model to evaluate the virulence of pectobacterium carotovorum. Mol Plant Microbe Interact. 2010, 23 (2): 139-143. 10.1094/MPMI-23-2-0139.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R: Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23 (21): 2947-2948. 10.1093/bioinformatics/btm404.
Tamura K, Nei M, Kumar S: Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA. 2004, 101 (30): 11030-11035. 10.1073/pnas.0404206101.
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4 (4): 406-425.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S:MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011, 28 (10): 2731-2739. 10.1093/molbev/msr121.
Acknowledgements
This work was supported by the Agronomic Research for Development Project PRAD N° 07-07 and the “Agence Universitaire de la Francophonie” (AUF).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MK-H designed the study, performed the experiments, data analyses and wrote the paper, MT and MA participated in the sample preparation and preliminary examination, EE participated in the design of the study, FB drafted the manuscript, MME coordinated the study, designed and participated in manuscript preparation. All authors read and approved the manuscript.
Electronic supplementary material
12866_2012_2068_MOESM1_ESM.docx
Additional file 1: Table S1: Phenotypic characteristics of the strains of Pectobacterium isolated from potato in comparison with standard isolate. (DOCX 17 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kettani-Halabi, M., Terta, M., Amdan, M. et al. An easy, simple inexpensive test for the specific detection of Pectobacterium carotovorum subsp. carotovorum based on sequence analysis of the pmrA gene. BMC Microbiol 13, 176 (2013). https://doi.org/10.1186/1471-2180-13-176
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-13-176