
Abstract
Background
Wolbachia are vertically transmitted bacteria known to be the most widespread endosymbiont in arthropods. They induce various alterations of the reproduction of their host, including feminization of genetic males in isopod crustaceans. In the pill bug Armadillidium vulgare, the presence of Wolbachia is also associated with detrimental effects on host fertility and lifespan. Deleterious effects have been demonstrated on hemocyte density, phenoloxidase activity, and natural hemolymph septicemia, suggesting that infected individuals could have defective immune capacities. Since nothing is known about the molecular mechanisms involved in Wolbachia-A. vulgare interactions and its secondary immunocompetence modulation, we developed a transcriptomics strategy and compared A. vulgare gene expression between Wolbachia-infected animals (i.e., “symbiotic” animals) and uninfected ones (i.e., “asymbiotic” animals) as well as between animals challenged or not challenged by a pathogenic bacteria.
Results
Since very little genetic data is available on A. vulgare, we produced several EST libraries and generated a total of 28 606 ESTs. Analyses of these ESTs revealed that immune processes were over-represented in most experimental conditions (responses to a symbiont and to a pathogen). Considering canonical crustacean immune pathways, these genes encode antimicrobial peptides or are involved in pathogen recognition, detoxification, and autophagy. By RT-qPCR, we demonstrated a general trend towards gene under-expression in symbiotic whole animals and ovaries whereas the same gene set tends to be over-expressed in symbiotic immune tissues.
Conclusion
This study allowed us to generate the first reference transcriptome ever obtained in the Isopoda group and to identify genes involved in the major known crustacean immune pathways encompassing cellular and humoral responses. Expression of immune-related genes revealed a modulation of host immunity when females are infected by Wolbachia, including in ovaries, the crucial tissue for the Wolbachia route of transmission.
Similar content being viewed by others


Background
Wolbachia are endosymbiotic α–Proteobacteria that are maternally transmitted and cause various reproductive manipulations in a wide range of invertebrate hosts (see [1] for a review). Wolbachia infection is widespread in Crustacea where species of the three main classes (Malacostraca, Ostracoda, and Maxillipoda) were found to be infected [2]. Wolbachia prevalence reaches ~60% in terrestrial isopods (order Oniscidea). In the pill bug Armadillidium vulgare, one of the most intensively studied examples, Wolbachia are responsible for inducing the development of genetic males into functional females. This is achieved by preventing the androgenic gland differentiation responsible for male development [3, 4]. Consequently, in the progenies of infected mothers the proportion of females reaches 70 to 80% according to the transmission rate of Wolbachia [5, 6]. This modification of the host sex ratio leads to a low proportion of males in the field reached 20% as evidenced by a meta-analysis of 57 populations [2]. Since Wolbachia vertical transmission is dependent on the reproductive success of their hosts, it could be expected that the infection provides fitness benefit that could promote dispersion of Wolbachia in the host population. Surprisingly, most field populations of A. vulgare are not infected by Wolbachia [2], which could reflect the conflicting relationships between the pill bug and the bacteria. As some life history traits of A. vulgare are directly impacted by Wolbachia, the low prevalence of the infected specimens in natural populations could be due to various factors that reduce the host fitness. Feminizing Wolbachia have the potential to reduce male to female ratio to values limiting mating possibilities and therefore limiting population size [7]. Furthermore, males are able to distinguish between infected and uninfected females [7]. This mating preference could lead to a sexual selection in favor of uninfected females. Rigaud and Moreau [8] also demonstrated that after multiple mating, sperm depletion in males affects fertility only in infected females. In addition, a reduced fertility and survival is recorded in Wolbachia-infected females [6, 9, 10]. However, these females had a higher reproductive investment (they produce more offspring and more eggs per clutch) so ultimately the reproductive success is similar between infected and non-infected females [6]. More recently, deleterious effects have been demonstrated on immunocompetence of infected females [10, 11]. Indeed, these females have a lower hemocyte density, a decrease in PO activity, and a more severe hemolymph septicemia that could result in a reduced life span in A. vulgare [10, 11]. This latter effect could impact host fitness including lower or higher resistance to intruders as it has been shown in many insect species [12]. For example, it has been demonstrated that Wolbachia suppress the host defence of Drosophila simulans against parasitoids [13]. Conversely, Wolbachia-induced stimulation of the host’s innate immune system has been suggested as a mechanism conferring resistance to pathogens. In D. melanogaster and D. simulans, Wolbachia protect their hosts against RNA viral infection [14–16]. This has also been demonstrated in Aedes aegypti where the injection of the life-shortening wMelPop Wolbachia strain provides resistance against the Dengue and the Chikungunya viruses as well as against Plasmodium gallinaceum and Brugia pahangi [12, 17–21]. In parallel, Wolbachia were shown to induce immune gene expression in different biological systems. For example, a Wolbachia-infected cell line displayed an overexpression of antioxidant proteins that are key components of Ae. albopictus immune response [22, 23]. Similarly, host immune genes are up-regulated in Ae. aegypti [17] and Anopheles gambiae [18] when infected by wMelPop.
Since nothing is known about the molecular mechanisms involved in Wolbachia-A. vulgare interactions and its secondary immunocompetence modulation, different Expressed Sequence Tag (EST) libraries [normalized, non-normalized, and Suppression Subtractive Hybridization (SSH) libraries] were constructed in order to generate a large transcriptomics data set. To identify genes involved in Wolbachia-host association and in host immune response, EST and SSH libraries were prepared using RNA from ovaries (i.e., the tissue involved in vertical transmission) and from A. vulgare females artificially challenged by Salmonella typhimurium. Host gene expression in Wolbachia-infected individuals was then compared to uninfected individuals by in silico and in vitro subtractions. This analysis revealed a set of potentially modulated immune genes. Expression of immune genes were investigated to examine whether the decrease of immunocompetence in the Wolbachia-infected A. vulgare may be related to modulation of the host innate immune system.
Methods
This work has been conducted in parallel in two other invertebrate models (i.e., Asobara tabida-Wolbachia and Sitophilus oryzae-SPE (Sitophilus primary endosymbiont)) in order to determine conserved and divergent immune pathways and to ascertain whether the invertebrates have selected common strategies to control their symbionts and to discriminate between symbionts and pathogens [24, 25].
Symbiotic association
Armadillidium vulgare (Crustacea Isopoda) individuals were sampled from two laboratory lineages whose Wolbachia-infection status is known. Animals infected by the feminizing Wolbachia strain (wVulC) (i.e., “symbiotic” animals) originated from Celles-sur-Belle, France. This lineage has been identified by crossing experiments as composed of all ZZ individuals: ZZ males and ZZ+Wolbachia females [2]. Uninfected individuals (i.e., “asymbiotic” animals) with genetic sex determinism (ZZ males and WZ females) originated from Nice, France [2, 5, 26]. These lines have been stably maintained in the lab since 1967 and 1991 for asymbiotic and symbiotic lineages, respectively. As A. vulgare males are never infected by Wolbachia, only females (WZ females and ZZ+Wolbachia females) were used in this study.
Bacterial challenge
Salmonella typhimurium (strain 12023G) were cultured in LB medium overnight. Dilutions were performed to obtain c104 bacteria.µL-1 (OD=0.01). Asymbiotic females were injected with 1 µL of bacterial suspension at the side of sixth pereon segment using a thin glass needle. Females were dissected at 6h, 9h, and 15h post injection. Ovaries, gut, caeca, fat tissues, hemocytes, hematopoietic organ, nerve chain, and brain were conserved in liquid nitrogen separately until total RNA extractions.
Library constructions
Seven different EST libraries were prepared from different tissues of A. vulgare (Figure 1A). Total RNA was extracted as described in [27] and treated with DNAse (TurboDNase, Ambio, Applied Biosystems), following the manufacturer’s instructions.

EST library characteristics A. Summary of the different EST libraries. Suppression Subtractive Hybridizations (SSHs) were performed with Miror Orientation Selection procedure. cDNA libraries were sequenced with or without normalization (Norm. or Non Norm. respectively). The wVulC Wolbachia strain (Celles sur Belle, France) induces feminization of genetic males and has some negative impacts in symbiotic females (see text). Immune challenge was performed through the injection of 104 Salmonella typhimurium in asymbiotic females: RNA was extracted 6h, 9h, and 15h after challenge. F = whole female tissues, Ov = ovary tissues, S = symbiotic, A = asymbiotic, C = immune challenge, NC = no immune challenge, ESTs = expressed sequence tags, Mt = mitochondrial genes, rRNA = ribosomal genes, UG = number of unigenes. B. Abundance classes of ESTs and unigenes. C. Unigenes occurrences among EST libraries. The horizontal axe represents the different EST libraries, the vertical axe represents the occurrence of unigenes within the libraries. Horizontal reading of the graph indicates the percentage of unigenes shared by several libraries. D. GO annotation results for High Scoring Pairs (HSP) coverage of 0%. GO annotation was first conducted using the Score Function (SF) of the BLAST2GO software. The GO terms selected by the annotation step were then merged with InterProScan predictions (SF + IPR). Finally, the Annex annotation was run (SF + IPR + ANNEX). E. Annotation distribution of GO terms.
Two non-normalized libraries were constructed from asymbiotic and symbiotic ovaries (AO and SO) starting with 1 µg of polyA RNAs. They were prepared using Creator SMART cDNA Library Construction kit (Clontech/BD Biosciences), following the manufacturer’s instructions. cDNA was digested by SfiI, purified (BD Chroma Spin – 400 column) and ligated into pDNRlib vector for Escherichia coli transformation. Amplified double strand cDNA (ds cDNA) was prepared using a SMART approach [28]. SMART Oligo II oligonucleotide (Clontech/BD Biosciences) and CDS primer were used for first-strand cDNA synthesis. SMART-amplified cDNA samples were further digested by RsaI endonuclease.
The SSH libraries from asymbiotic and symbiotic ovaries (SSH-A and SSH-S) were constructed starting with 20 µg of total RNA. SSH libraries from specimens challenged and not challenged by S. typhimurium (SSH-C and SSH-NC) were performed on 20.4 µg of a total RNA equally pooled from different tissues (i.e., ovaries, gut, cæca, fat tissues, hemocytes, hematopoietic organ, nerve chain, and brain) harvested at each time point. The pooled total RNA was obtained by mixing equal amounts of total RNA extracted separately for each tissue and for each time point. Subtractive hybridizations were performed using SSH method in both directions (Asymbiotic vs. Symbiotic A/S and vice-versa S/A; Not Challenged vs. Challenged NC/C and vice-versa C/NC) as described in [29, 30] using the PCR-Select cDNA Subtraction Kit (Clontech/BD Biosciences). SSH libraries were prepared by Evrogen (Moscow, Russia). The Mirror Orientation Selection (MOS) procedure was used for SSH-A/S and SSH-C/NC as described in [31] in order to reduce the number of false-positive clones in the SSH-generated libraries. Purified cDNAs from SSH-A/S and SSH-C/NC were cloned into the pAL16 vector (Evrogen) and used for E. coli transformation.
Finally, the normalized library (N) was prepared with 75 µg of a pooled total RNA from an equimolar proportion of asymbiotic and symbiotic ovaries, and 6h, 9h, and 15h challenged asymbiotic females. As for the libraries of challenged specimens, total RNA was extracted separately from the same tissues. This N library was prepared by Evrogen (Moscow, Russia). Total RNA sample was used for ds cDNA synthesis using SMART approach [28]. SMART prepared amplified cDNA was then normalized using Duplex Specific Nuclease (DSN) normalization method [32]. Normalization included cDNA denaturation/reassociation, treatment by DSN [33] and amplification of normalized fraction by PCR. Normalized cDNA was purified using QIAquick PCR Purification Kit (QIAGEN), digested with SfiI, purified (BD Chroma Spin - 1000 column) and ligated into pAL 17.3 vector (Evrogen) for E. coli transformation.
EST sequencing and data processing
All clones from the libraries were sequenced using the Sanger method (Genoscope, Evry, France) and were deposited in the EMBL database [EMBL: FQ884936 to FQ908260]. A general overview of the EST sequence data processing is given in Figure 2. Raw sequences and trace files were processed with Phred software [34] in order to remove low quality sequences (score < 20). Sequence trimming, which includes polyA tails/vector/adapter removal, was performed by cross match. Chimerical sequences were computationally digested into independent ESTs. Clustering and assembly of the ESTs were performed with TGICL [35] to obtain unique transcripts (unigenes) composed of contiguous ESTs (contigs) and unique ESTs (singletons). For that purpose, a pairwise comparison was first performed by a modified version of megaBLAST (minimum similarity 94%). Clustering was done with tclust that proceeds by a transitive approach (minimum overlap: 60bp at 20bp maximum of the end of the sequence). Assembly was done with CAP3 (minimum similarity 94%).

Sequence treatment (A) and functional annotation procedure (B).
To detect unigene similarities with other species, several BLASTs (with a high cut-off e-values) were performed against the following databases: NCBI nr [BLASTx (release: 1 March 2011); e-value < 5, HSP length > 33aa], Refseq genomic database (BLASTn, e-value < 10), Unigene division Arthropods (tBLASTx, #8 Ae. aegypti, #37 An. gambiae, #3 Apis mellifera, #3 Bombyx mori, #53 D. melanogaster, #9 Tribolium castaneum; e-value < 5), and Wolbachia sequences from Genbank (Release 164; e-value < 1e-20). Gene Ontology (GO) annotation was carried out using BLAST2GO software [36]. In the first step (mapping), a pool of candidate GO terms was obtained for each unigene by retrieving GO terms associated to the hits obtained after a BLASTx search against NCBI nr. In the second step (annotation), reliable GO terms were selected from the pool of candidate GO terms by applying the Score Function of BLAST2GO with “permissive annotation” parameters (EC-weight=1, e-value-filter=0.1, GO-weight=5, HSP/hit coverage cut-off =0%). In the third step of the annotation procedure, the pool of GO terms selected during the annotation step was merged with GO terms associated to InterPro domain (InterProScan predictions based on the longest ORF). Finally, the Annex augmentation step was run to modulate the annotation by adding GO terms coming from implicit relationships between GO terms [37].
Statistical analyses on libraries
We used the randomization procedure (with 500 random datasets) and R statistics described in [38] to detect unigenes whose transcript abundance (number of ESTs) was statistically different in AO and SO libraries (at a false discovery rate of 2.5 %). In order to extract biological processes and molecular functions statistically over-represented in SO libraries, we performed a hyper-geometrical test between GO terms from the SO library and those from the AO library, which represents the natural physiological conditions. The p-values were then adjusted using Bonferroni’s correction.
In order to perform a functional enrichment analysis of the unigenes extracted from the SSH, we used the FatiGO web tool [39] against the SO library. With respect to the GO analysis, four different levels of description (3, 4, 6, and 9) were chosen for the biological processes.
Quantitative expression by Real-Time RT-PCR
Gene expression quantification was performed in whole animal, ovaries, and immune tissues (hemocytes and hematopoietic organs pooled) of asymbiotic and symbiotic females.
RNA extractions
For the whole animal condition, each individual was crushed with pestle and mortar in liquid nitrogen. Total RNA extraction was performed from about 30 mg of powder with TRIzol® reagent according to the manufacturer's instructions (Invitrogen). For ovaries and immune tissues, total RNA extractions were performed from 25 and 50 females respectively with RNeasy Mini Kit according to the manufacturer's instructions (QIAGEN).
Real-Time RT-PCR
First-strand cDNA was synthesized with the SuperScript III kit (Invitrogen) in accordance with manufacturer’s instructions, starting from 1 µg of total RNA using random hexamer primers. For whole animal samples, 0.2 µg of 5 individual extractions were pooled in 1 µg. Three biological replicates of each sample (whole animals, ovaries, and immune tissues) were used.
For each gene, primer pairs were designed with the Real-time PCR function of PerlPrimer [40]. The Tm and the length of each primer pair were fixed at 60°C and 18-22 bp, respectively. Primers used for quantitative PCR are summarized in Additional File 1.
Quantitative RT-PCR was performed using LightCycler LC480 system (Roche) as follows: 10 min at 95°C, 45 times [10 sec at 95°C, 10 sec at 60°C, 20 sec at 72°C]. A melting curve (65°C to 97°C) was recorded at the end of each reaction in order to check that the PCR product was unique. The reaction mixture consisted of 1.25 µL of each primer (10 µM), 5 µL of Fast SYBR-Green Master Mix (Roche) and 2.5 µL of diluted cDNA (corresponding to 12.5 ng of cDNA). Standard curves were plotted using 4 dilutions (125 ng, 25 ng, 5 ng, 1.25 ng) of pooled cDNAs from whole animals and ovaries. Efficiency of the PCR reaction was calculated.
Expression data for each gene were estimated using the efficiency of the primer pair and the crossing point [41]. All gene expressions were normalized by the geometric mean of the expression level of the L8-ribosomal (RbL8) and Elongation Factor 2 (EF2) reference genes. Normalization and statistical pair-wise comparisons have been determined using REST [42].
Results
First reference transcriptome in isopods
ESTs were generated from seven high quality cDNA libraries, including four SSH libraries, two non-normalized libraries and one normalized library. Characteristics of cDNA libraries are summarized in Figure 1A. A total of 28 606 ESTs (mean length: 504 ± 170 bp) were generated which covered around 14.4 Mb. Clustering of all EST sequences was performed by TGICL [35] resulted in 10 923 unique transcripts (i.e., unigenes which covered 6.4 Mb). About 75% of the clusters contained one EST (i.e., singletons; n = 8 211) and 25% contained ESTs assembled in a consensus sequence (i.e., contigs, n = 2 712). The normalized library and the ovary libraries contained a greater proportion of contigs which is likely due to the deeper sequencing of these libraries (Figure 1C.). The average length of these unigenes was 590 ± 250 bp with a GC content of 33.5% and an average coverage of 3.5 (Figure 1B)
Functional annotation was performed on all 10 923 unigenes through BLASTx and tBLASTx similarity searches against various databases. Because of the ancient divergence between A. vulgare and the closest sequenced genomes we used a cut-off threshold of 1e-05. A total of 44% of the unigenes had BLAST similarities to known sequences, mainly from Ae. aegypti (10.5%), An. gambiae (8.7%), D. melanogaster (7%), and different malacostracans (3.1%) with an e-value lower than 1e-20 for 64.8% of the unigenes. The remaining 66% of unigenes showing no match could correspond to species-specific genes or UTR extremities of the cDNA.
Functional analysis
GO annotation was carried out using BLAST2GO software (Figures 1D, 2B). A total of 42% of unigenes were annotated after the BLAST2GO annotation procedure for High Scoring Pair (HSP) coverage of 0%. While we kept the unigenes/GO dataset corresponding to the minimum HSP coverage percentage, the mean number of GO terms assigned per unigene was low (1.18 GO term/unigene, Figure 1E).
To determine the effect of Wolbachia on host gene expression, an in silico subtraction was performed between libraries of symbiotic (SO) and asymbiotic (AO) ovaries. In these libraries, a total of 4564 unigenes have been annotated and based on the R statistics, only 6 unigenes were differentially represented: 3 unigenes were over-represented in symbiotic ovaries while 3 were over-represented in asymbiotic ovaries. Unfortunately, these unigenes could not be identified by BLAST and only one is associated to a biological function (see Additional File 2: Unigenes differentially represented between symbiotic and asymbiotic ovaries). The immune processes were over-represented in symbiotic ovaries (Table 1 and Additional File 3: Processes and functions over-represented in A. vulgare ovaries in response to Wolbachia infection, biological process levels 4 and 6). Indeed, 21 and 15 unigenes with immune gene similarities were identified in AO and SO libraries, respectively (Additional File 4: Immune unigenes present in SO, AO, SSH-S, SSH-A, SSH-C, and SSH-NC libraries).
In the same manner, two in vitro SSHs were performed by subtracting common transcripts between symbiotic and asymbiotic ovaries (SSH-S), and reciprocally (SSH-A). These SSHs were contaminated by a high proportion of mitochondrial ESTs (~40%) that were removed for further analyses. To reveal the functions over-represented, we compared each SSH to SO library by the FatiGO web tool. One biological process (vesicle transport along microtubule) and one molecular function (microtubule motor activity) were over-represented in asymbiotic ovaries (Table 2). Most of the 223 unigenes that are associated to these two GO terms belong to the kinesin-like protein family. In these two libraries, the BLAST analyses allowed the identification of 1 immune gene in SSH-S and 6 immune genes in SSH-A libraries respectively (Additional File 4: Immune unigenes present in SO, AO, SSH-S, SSH-A, SSH-C, and SSH-NC libraries).
In order to identify genes expressed in response to pathogenic bacteria, we performed SSH libraries between S. typhimurium-challenged and unchallenged asymbiotic A. vulgare females (SSH-C) and reciprocally (SSH-NC). We thus identified 31 and 29 unigenes in SSH-C and SSH-NC libraries, respectively, that are related to crustacean immune processes. In the SSH-C library these immune related unigenes exhibited a greater diversity than those of the SSH-NC library (Additional File 4: Immune unigenes present in SO, AO, SSH-S, SSH-A, SSH-C, and SSH-NC libraries).
Finally, 30 non redundant immune related unigenes were identified in libraries constructed from symbiotic/asymbiotic conditions (SO/AO, SSH-S/SSH-A) and 59 in libraries constructed from challenged/not challenged conditions (SSH-C/SSH-NC) (Additional File 3: Processes and functions over-represented in A. vulgare ovaries in response to Wolbachia infection, biological process levels 4 and 6). Among them, 28 unigenes were successfully amplified by PCR. In addition, 16 other unigenes were selected from the normalized library (N) for their putative involvement in major immune processes. Annotations were further confirmed by protein domain identification (CD Search vs the Conserved Domain Database on NCBI server [43]). If the complete domain pattern of a given protein was not found, the suffix “-like” was added to the unigene name (Table 3). Expression of these 44 genes were further analysed by RT-qPCR.
Immune gene expression
The expression of 46 candidate immune genes (Table 4 and Additional File 1: Primer pairs used for RT-qPCR quantification) were quantified in whole animal, ovaries and immune tissues of symbiotic and asymbiotic A. vulgare females. Forty four genes were selected through the procedure described above and 2 other genes were selected from previous studies [44, 45]. Twelve genes were selected from the SSH-C (11 unigenes) and SSH-NC (1 unigene) libraries in order to examine whether Wolbachia induce an immune activation as observed in a challenged condition. All the 46 selected immune genes can be placed in known crustacean immune pathways (Figure 3). We considered genes involved i) in pathogen recognition (receptors and associated signaling pathways), ii) in RNA interference (RNAi), coagulation, PO pathway, phagocytosis, apoptosis, and autophagy or iii) encoding antimicrobial peptides (AMPs) [46–50].

Pathway map for known crustacean immune functions: Armadillidium vulgare immune genes identified in this study were highlighted in pink boxes. The up and down arrows in gene boxes referred to significant up and down-regulation in symbiotic condition. AIF: Apoptosis inducing factor; ALF: Anti-lipopolysaccharide factor; LGBP: Lipopolysaccharide and β-glucan binding protein; βGBP: β-glucan binding protein; ECSIT: Evolutionary conserved intermediate in the Toll pathway; Hcy-PO: Hemocyanin with PO activity; MIP: Melanization Inhibitor Protein; PO: Phenoloxidase; PPAE: Prophenoloxidase activating enzyme; PXN: Peroxinectin; SOCS: Suppressor of cytokine signaling; SOD: Superoxide dismutase; TGase: Transglutaminase.
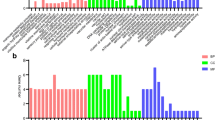
In symbiotic conditions, expression of these genes showed a general trend to a down-regulation in whole animals (37/43) and ovaries (31/44). On the contrary, 30 genes among 37 are over-expressed in immune tissues (Table 4 and Additional File 5: Expression profiles of genes studied in whole animals, ovaries, and immune tissues of A. vulgare).
Significant differential expressions in whole animals and ovaries were recorded for 16 genes, 12 of them were down-regulated and 4 up-regulated (Table 4). No significant differential expression was detected in immune tissues. Three genes involved in pathogen recognition, the C-type lectin 1, C-type lectin 2, and the C-type lectin 3 genes were differentially expressed. The C-type lectin 1 was up-regulated in ovaries whereas the C-type lectin 2 was down-regulated in the same tissue. Finally, the C-type lectin 3 was down-regulated in the whole animals. Three genes encoding AMPs were down-regulated: The armadillidin and the i-type lyzozyme genes in whole animals and the crustin3 gene in both whole animals and ovaries. One serine protease gene, the masquerade-like B, was also under-expressed in whole animals. Three genes involved in detoxification, the peroxiredoxin A and C and glutathione peroxidase, were down-regulated in ovaries whereas the thioredoxin A was up-regulated in the same tissue. In the autophagy pathway, two genes, atg7 and atg12, were under-expressed in ovaries. Among genes involved in stress response, the ferritin A and C genes were over-expressed in ovaries.
Discussion
The different EST libraries generated in this study constitute the first reference transcriptome ever obtained in the Isopoda group [51]. Among crustaceans, only the Daphnia pulex (Branchiopoda, Cladocera) genome was recently published [52] and some EST libraries were constructed from a shrimp, a crayfish, and a porcelain crab (Malacostraca, Decapoda) [53–57]. Another EST database was obtained in the marine isopod Limnoria quadripunctata, but it concerned only the hepatopancreas [58]. Thus, our result represents the eighth largest sequencing effort for any crustacean, behind the cladoceran Da. pulex and the decapods Litopenaeus vannamei and Petrolisthes cinctipes, and the sixth EST data set for any Malacostraca species [51, 57]. Few A. vulgare unigenes present similarities with crustacean ESTs. This could be in part explained by the phylogenetic distance between isopods and the crustaceans from which EST libraries or genomics data are available. However, the overlapping between libraries was low, suggesting that the sequencing effort should be increased.
The present work allowed us to identify the first immune gene repertoire from a terrestrial crustacean. Indeed, until now large scale characterizations of immune genes in crustaceans have been based on only a few model organisms, such as shrimps, crayfishes or crabs. All immune genes identified in A. vulgare are involved in canonical immune pathways (Table 4 and Figure 3): i) pathogen detection including recognition molecules such as the lectins and peroxinectins (PXN) that are able to distinguish between self and non-self particles and signal transducers; ii) immune cellular responses including opsonization molecules (e.g., PXN and masquerade-like proteins) inducing phagocytosis and cellular encapsulation; iii) immune humoral responses involving clotting and coagulation reactions, production of AMPs, generation of reactive oxygen species, detoxification processes, and the proPhenoloxidase (proPO) cascade; and iv) other pathways connected to immune responses such as antiviral immunity (RNA interference), programmed cell death (apoptosis and autophagy), and cell differentiation such as hematopoiesis [49, 50, 59, 60].
Although 40 new genes all involved in immune pathways have been identified, several key genes were lacking (Figure 3). This can be explained by three non-exclusive hypotheses: The relatively low depth of the sequencing effort, the weak annotation (44%) due to divergence between isopods and the other Arthropoda clades, and the absence of some immune genes in isopods. For example, genes encoding important innate immune receptors, such as GNBPs or Toll, and their signal transducers Imd, Dorsal, Cactus, Relish were known in different crustacean species [47, 49, 61, 62] but were not identified in A. vulgare. PO activity is detected in crustaceans, but isopods such as chelicerates seem to lack PO enzyme and the corresponding gene [11, 58, 63, 64]. In the same way, the PGRP genes have never been identified in crustacean EST libraries nor in the brine shrimp genome [47], which suggests that these genes could be absent in this clade.
A growing number of studies showed that the immune system of Wolbachia-infected animals is modulated at the molecular level [17, 18, 22]. In A. vulgare, it has recently been shown that Wolbachia impact immune cellular processes [10, 11, 65]. We show here that Wolbachia symbiosis leads to a down-regulation of some A. vulgare immune genes. Indeed, among the candidate genes tested, 72% are down-regulated in whole females, 75% in ovaries and 19% in immune tissues. Among the 46 genes analyzed, no significant differential expression was detected in the immune tissues, whereas the expression of 16 of them was significantly disturbed when Wolbachia were present in whole animals and ovaries. The impacted genes are involved in biological functions such as stress response and detoxification, autophagy, AMP synthesis, pathogen recognition, and proteolytic cascades.
Several impacted genes are involved in oxidative stress response. The production of reactive oxygen species (ROS) is one of the first lines of defence against invading microbes. High concentrations of ROS create oxidative stresses, resulting in damage to lipids, nucleic acids, and proteins and reducing life span so that complex antioxidant defence systems have evolved to minimize damaging ROS. Our study shows a down-regulation of antioxidant enzymes only in the ovaries. This result agrees with those obtained in Drosophila S2 cell line infected by Wolbachia [66] and in A. tabida - Wolbachia symbiosis [24] but not with those from the Ae. albopictus Aa23 cell line [22]. In parallel, we show an up-regulation of the thioredoxin gene that could be a response to down-regulation of other genes encoding antioxidant proteins. An alternative hypothesis is that this last gene could be induced by Wolbachia to reduce apoptosis and accelerate multiplication of gonadic cells. Indeed, in mice, this electron donor protein reduces the process of oxidant molecules but also increases cell proliferation and the inhibition of apoptosis [67].
There was a significant over-expression of Ferritins A and C in symbiotic ovaries. Ferritins are important iron sequestration proteins and play a crucial role in the iron-withholding defence system [68]. The up-regulation of ferritin genes could be an active cellular reaction for starving Wolbachia of iron, which would lead to bacterial growth limitation. Besides, this over-expression could be the result of the under-expression of the detoxification enzymes (Peroxiredoxin B and C and Glutathione peroxidase). As intracellular free iron produces ROS by the Fenton reaction in presence of H2O2, iron sequestration could reduce ROS production and thus avoid deleterious effects in the cell. Regardless, this result contrasts with that obtained in A. tabida-Wolbachia system [24, 69] where the ferritin genes were under-expressed in symbiotic condition. This down-regulation could be due to the dependence phenotype of A. tabida - Wolbachia association for the oocyte maturation, whereas our model is a facultative Wolbachia symbiosis that is not involved in host oogenesis.
Autophagy was initially reported as a bulk self-degradation mechanism for the turnover of proteins and organelles. Autophagy can be induced via PGRP-LE, which is essential in the innate bacterial recognition in Drosophila resistance against Listeria monocytogenes [70] suggesting that this biological process is involved in the innate immune response against intracellular bacteria, viruses, and parasites [70, 71]. In our study, the atg7 and atg12 genes involved in autophagy were down-regulated in ovaries. Autophagy-associated genes were down-regulated also in A. tabida-Wolbachia and S. oryzae-SPE symbioses [24, 25], which suggests that this process is critical in bacterial symbiosis. We may hypothesize that this down-regulation was an active strategy of Wolbachia to reduce their elimination by their host.
In Wolbachia-infected whole animals, three AMP genes were under-expressed (i.e., armadillidin, crustin 3, and i-type lyzozyme). Armadillidin and crustin are two Gram-positive AMPs [44, 72]. The impact of Wolbachia on AMP gene expression varies according to the host-symbiont association. In infected D. simulans and Ae. albopictus [73], and in the silkworm cell line [74], Wolbachia did not disturb AMP expression. On the contrary, attacin and diptericin genes were down-regulated in an infected D. melanogaster S2 cell line [66], whereas many AMP genes were up-regulated in the mosquitoes Ae. aegypti and An. gambiae transfected by the wMelPop strain [17–19]. In the A. tabida-Wolbachia association, the defensin, lyzozyme and hymenoptaecin genes were under-expressed [24] as well as the coleoptericin 1 gene in S.oryzae-SPE symbiosis [25, 75]. In A. vulgare, the down-regulation of AMP genes could be related to the higher septicaemia found in Wolbachia-infected animals [10, 11].
Two recognition molecules, the C-type lectins 1 and 2, were up and down-regulated, respectively, whereas gene expression of the C-type lectin 3 was not detected in ovaries. The C-type lectins are mainly carbohydrate binding proteins involved in pathogen recognition, opsonization and encapsulation response, and antiviral response [76, 77]. It has been shown that these proteins are also involved in symbiont interactions: C-type lectins were required for the symbiont acquisition in scleractinian corals [78, 79] and the marine nematode Laxus oneistus [80]. In Ae. aegypti and An. gambiae transfected with the pathogenic Wolbachia strain wMelPop, the C-type lectin genes were up-regulated [17, 18]. In A. vulgare, expression of the three C-type lectin genes presents different patterns, probably due to specific functions of each protein.
Unlike what was observed in ovaries, the C-type lectin 3 gene expression was significantly down-regulated in immune tissues of symbiotic females, which could impact pathogen recognition ability of the host. In the same way, the serine protease masquerade-like B gene was down-regulated. This protein family is involved in several biological functions such as pattern recognition, opsonization, cell adhesion activity [81], and in antiviral responses [82]. In our system, the under-expression of this masquerade-like gene could potentially impair these functions.
In symbiotic ovaries, one kinesin-related gene was down-regulated. This pattern observed by RT-qPCR was also confirmed by in silico comparison between SSH-A vs. SO libraries. Indeed GO analysis highlighted vesicle transport and microtubule motor activity as the only functions over-represented in asymbiotic ovaries. These functions were mainly associated with kinesin protein family. In D. melanogaster, kinesin-1 has been reported to be involved in wMel Wolbachia transport toward the posterior part of the oocyte [83]. In A. vulgare, the relation between kinesin and Wolbachia is still unknown. Nevertheless, the down-regulation observed in symbiotic ovaries might be a host response for limiting the movement of Wolbachia in oocytes. In the weevil S. oryzae, the primary endosymbiont SPE seems to stimulate vesicle trafficking, which emphasizes the importance of this process in host-symbiont interactions [25].
Conclusion
Our study represents the first transcriptomics approach that aims at deciphering the A. vulgare-Wolbachia interactions and it established the first reference transcriptome for isopods. In A. vulgare, Wolbachia colonize not only the ovaries but also other tissues, particularly the immune cells [65, 84]. Therefore, perturbation of the host immune gene expression could be a direct effect of the bacteria on immunity. In such a scenario, Wolbachia would not be a silent bacterium and could counteract the host immune system to survive and establish a long term association with the host. The quantification of immune-related gene expression revealed a global trend to gene under-expression in Wolbachia-infected whole animals and ovaries. Unexpected modulation of immune gene expression in ovaries could reflect a Wolbachia strategy to manipulate the crucial tissue for vertical transmission. Surprisingly, most of the immune genes (30/37) tend to be up-regulated in immune tissues. This general up-regulation could compensate the immune depressive effect of Wolbachia previously described in A. vulgare [10, 11, 65]. These results conflict with those observed in insect cell lines where Wolbachia down-regulated immune-related genes [66, 85] but are congruent with those obtained in transfected wMelpop mosquitoes [17–19]. More work needs to be done to check whether this up-regulation confers host pathogen protection as observed in Drosophila and mosquitoes [14, 15, 17, 19].
References
Werren JH, Baldo L, Clark ME: Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008, 6: 741-751. 10.1038/nrmicro1969.
Bouchon D, Cordaux R, Grève P: Feminizing Wolbachia and the evolution of sex determination in isopods. Insect symbiosis. Edited by: Bourtzis K, Miller TA. 2008, Boca Raton, FL: Taylor & Francis Group, 3: 273-294.
Cordaux R, Bouchon D, Grève P: The impact of endosymbionts on the evolution of host sex-determination mechanisms. Trends Genet. 2011, 27: 332-341. 10.1016/j.tig.2011.05.002.
Negri I, Pellecchia M, Grève P, Daffonchio D, Bandi C, Alma A: Sex and stripping: the key to the intimate relationship between Wolbachia and host?. Commun Integr Biol. 2010, 3: 110-115. 10.4161/cib.3.2.10520.
Cordaux R, Michel-Salzat A, Frelon-Raimond M, Rigaud T, Bouchon D: Evidence for a new feminizing Wolbachia strain in the isopod Armadillidium vulgare: evolutionary implications. Heredity. 2004, 93: 78-84. 10.1038/sj.hdy.6800482.
Lachat M: Impact de deux souches de Wolbachia sur les traits d'histoire de vie de leurs hôtes Armadillidium vulgare. PhD thesis. 2009, Université de Poitiers, Ecole doctorale ICBG
Moreau J, Bertin A, Caubet Y, Rigaud T: Sexual selection in an isopod with Wolbachia-induced sex reversal: males prefer real females. J Evol Biol. 2001, 14: 388-394. 10.1046/j.1420-9101.2001.00292.x.
Rigaud T, Moreau J: A cost of Wolbachia-induced sex reversal and female-biased sex ratios: decrease in female fertility after sperm depletion in a terrestrial isopod. Proc Biol Sci. 2004, 271: 1941-1946. 10.1098/rspb.2004.2804.
Lachat M, Caubet Y, Bouchon D: Does Wolbachia influence survival in starved Armadillidium vulgare?. Proceedings of the International Symposium of Terrestrial Isopod Biology ISTIB 07; Tunis. Edited by: Zimmer M, Charfi-Cheikhrouha F, Taiti S. 2008, Shaker Verlag, 125-130.
Braquart-Varnier C, Lachat M, Herbinière J, Johnson M, Caubet Y, Bouchon D, Sicard M: Wolbachia mediate variation of host immunocompetence. PLoS ONE. 2008, 3: e3286-10.1371/journal.pone.0003286.
Sicard M, Chevalier F, De Vlechouver M, Bouchon D, Grève P, Braquart-Varnier C: Variations of immune parameters in terrestrial isopods: a matter of gender, aging and Wolbachia. Naturwissenschaften. 2010, 97: 819-826. 10.1007/s00114-010-0699-2.
Cook PE, McGraw EA: Wolbachia pipientis: an expanding bag of tricks to explore for disease control. Trends Parasitol. 2010, 26: 373-375. 10.1016/j.pt.2010.05.006.
Fytrou A, Schofield PG, Kraaijeveld AR, Hubbard SF: Wolbachia infection suppresses both host defence and parasitoid counter-defence. Proc Biol Sci. 2006, 273: 791-796. 10.1098/rspb.2005.3383.
Hedges LM, Brownlie JC, O'Neill SL, Johnson KN: Wolbachia and virus protection in insects. Science. 2008, 322: 702-10.1126/science.1162418.
Teixeira L, Ferreira A, Ashburner M: The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 2008, 6: e2-
Osborne SE, Leong YS, O'Neill SL, Johnson KN: Variation in antiviral protection mediated by different Wolbachia strains in Drosophila simulans. PLoS Pathog. 2009, 5: e1000656-10.1371/journal.ppat.1000656.
Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, Lu G, Pyke AT, Hedges LM, Rocha BC, Hall-Mendelin S, Day A, Riegler M, Hugo LE, Johnson KN, Kay BH, McGraw EA, van den Hurk AF, Ryan PA, O'Neill SL: A Wolbachia symbiont in Aedes aegypti limits infection with Dengue, Chikungunya, and Plasmodium. Cell. 2009, 139: 1268-1278. 10.1016/j.cell.2009.11.042.
Kambris Z, Blagborough AM, Pinto SB, Blagrove MSC, Godfray HCJ, Sinden RE, Sinkins SP: Wolbachia stimulates immune gene expression and inhibits Plasmodium development in Anopheles gambiae. PLoS Pathog. 2010, 6: e1001143-10.1371/journal.ppat.1001143.
Bian G, Xu Y, Lu P, Xie Y, Xi Z: The endosymbiotic bacterium Wolbachia induces resistance to Dengue virus in Aedes aegypti. PLoS Pathog. 2010, 6: e1000833-10.1371/journal.ppat.1000833.
Saridaki A, Bourtzis K: Wolbachia: more than just a bug in insects genitals. Curr Opin Microbiol. 2010, 13: 67-72. 10.1016/j.mib.2009.11.005.
Walker T, Moreira LA: Can Wolbachia be used to control malaria?. Mem Inst Oswaldo Cruz. 2011, 106 (Suppl 1): 212-217.
Brennan LJ, Keddie BA, Braig HR, Harris HL: The endosymbiont Wolbachia pipientis induces the expression of host antioxidant proteins in an Aedes albopictus cell line. PLoS ONE. 2008, 3: e2083-10.1371/journal.pone.0002083.
Molina-Cruz A, DeJong RJ, Charles B, Gupta L, Kumar S, Jaramillo-Gutierrez G, Barillas-Mury C: Reactive oxygen species modulate Anopheles gambiae immunity against bacteria and Plasmodium. J Biol Chem. 2008, 283: 3217-3223.
Kremer N, Charif D, Henri H, Gavory F, Wincker P, Mavingui P, Vavre F: Influence of Wolbachia on host gene expression in an obligatory symbiosis. BMC Microbiol. 2012, 12 (Suppl 1): S7-10.1186/1471-2180-12-S1-S7.
Vigneron A, Charif D, Vallier A, Vincent-Monegat C, Gavory F, Wincker P, Heddi A: Host response to endosymbiont and pathogen in the cereal weevil Sitophilus oryzae. BMC Microbiol. 2012, 12 (Suppl 1): S14-10.1186/1471-2180-12-S1-S14.
Bouchon D, Rigaud T, Juchault P: Evidence for widespread Wolbachia infection in isopod crustaceans: molecular identification and host feminization. Proc Biol Sci. 1998, 265: 1081-1090. 10.1098/rspb.1998.0402.
Matz MV: Amplification of representative cDNA samples from microscopic amounts of invertebrate tissue to search for new genes. Methods Mol Biol. 2002, 183: 3-18.
Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD: Reverse transcriptase template switching: a smart approach for full-length cDNA library construction. Biotechniques. 2001, 30: 892-897.
Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert PD: Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci USA. 1996, 93: 6025-6030. 10.1073/pnas.93.12.6025.
Diatchenko L, Lukyanov S, Lau YF, Siebert PD: Suppression subtractive hybridization: a versatile method for identifying differentially expressed genes. Methods Enzymol. 1999, 303: 349-380.
Rebrikov DV, Britanova OV, Gurskaya NG, Lukyanov KA, Tarabykin VS, Lukyanov SA: Mirror orientation selection (MOS): a method for eliminating false positive clones from libraries generated by suppression subtractive hybridization. Nucleic Acids Res. 2000, 28: E90-10.1093/nar/28.20.e90.
Zhulidov PA, Bogdanova EA, Shcheglov AS, Vagner LL, Khaspekov GL, Kozhemyako VB, Matz MV, Meleshkevitch E, Moroz LL, Lukyanov SA, Shagin DA: Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 2004, 32: e37-10.1093/nar/gnh031.
Shagin DA, Rebrikov DV, Kozhemyako VB, Altshuler IM, Shcheglov AS, Zhulidov PA, Bogdanova EA, Staroverov DB, Rasskazov VA, Lukyanov S: A novel method for SNP detection using a new duplex-specific nuclease from crab hepatopancreas. Genome Res. 2002, 12: 1935-1942. 10.1101/gr.547002.
Ewing B, Green P: Base-calling of automated sequencer traces using Phred. ii. error probabilities. Genome Res. 1998, 8: 186-194.
Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, Tsai J, Quackenbush J: Tigr gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003, 19: 651-652. 10.1093/bioinformatics/btg034.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M: BLAST2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005, 21: 3674-3676. 10.1093/bioinformatics/bti610.
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A: High-throughput functional annotation and data mining with the BLAST2GO suite. Nucleic Acids Res. 2008, 36: 3420-3435. 10.1093/nar/gkn176.
Stekel DJ, Git Y, Falciani F: The comparison of gene expression from multiple cDNA libraries. Genome Res. 2000, 10: 2055-2061. 10.1101/gr.GR-1325RR.
Al-Shahrour F, Díaz-Uriarte R, Dopazo J: FatiGO: a web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics. 2004, 20: 578-580. 10.1093/bioinformatics/btg455.
Marshall OJ: PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics. 2004, 20: 2471-2472. 10.1093/bioinformatics/bth254.
Pfaffl MW: A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29: e45-10.1093/nar/29.9.e45.
Pfaffl MW, Horgan GW, Dempfle L: Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30: e36-10.1093/nar/30.9.e36.
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH: CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39: D225-9. 10.1093/nar/gkq1189.
Herbinière J, Braquart-Varnier C, Grève P, Strub J, Frère J, Van Dorsselaer A, Martin G: Armadillidin: a novel glycine-rich antibacterial peptide directed against gram-positive bacteria in the woodlouse Armadillidium vulgare (terrestrial isopod, crustacean). Dev Comp Immunol. 2005, 29: 489-499. 10.1016/j.dci.2004.11.001.
Herbinière J, Grève P, Strub J, Thiersé D, Raimond M, van Dorsselaer A, Martin G, Braquart-Varnier C: Protein profiling of hemocytes from the terrestrial crustacean Armadillidium vulgare. Dev Comp Immunol. 2008, 32: 875-882. 10.1016/j.dci.2008.01.007.
Jiravanichpaisal P, Lee BL, Söderhäll K: Cell-mediated immunity in arthropods: hematopoiesis, coagulation, melanization and opsonization. Immunobiology. 2006, 211: 213-236. 10.1016/j.imbio.2005.10.015.
McTaggart SJ, Conlon C, Colbourne JK, Blaxter ML, Little TJ: The components of the Daphnia pulex immune system as revealed by complete genome sequencing. BMC Genomics. 2009, 10: 175-10.1186/1471-2164-10-175.
Ghosh J, Lun CM, Majeske AJ, Sacchi S, Schrankel CS, Smith LC: Invertebrate immune diversity. Dev Comp Immunol. 2010, 35: 959-974.
Vazquez L, Alpuche J, Maldonado G, Agundis C, Pereyra-Morales A, Zenteno E: Immunity mechanisms in crustaceans. Innate Immun. 2009, 15: 179-188. 10.1177/1753425909102876.
Liu H, Wu C, Matsuda Y, Kawabata S, Lee BL, Söderhäll K, Söderhäll I: Peptidoglycan activation of the proPO-system without a peptidoglycan receptor protein (PGRP)?. Dev Comp Immunol. 2011, 35: 51-61. 10.1016/j.dci.2010.08.005.
Stillman JH, Colbourne JK, Lee CE, Patel NH, Phillips MR, Towle DW, Eads BD, Gelembuik GW, Henry RP, Johnson EA, Pfrender ME, Terwilliger NB: Recent advances in crustacean genomics. Integr Comp Biol. 2008, 48: 852-868. 10.1093/icb/icn096.
Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, Bauer DJ, Cáceres CE, Carmel L, Casola C, Choi J, Detter JC, Dong Q, Dusheyko S, Eads BD, Fröhlich T, Geiler-Samerotte KA, Gerlach D, Hatcher P, Jogdeo S, Krijgsveld J, Kriventseva EV, Kültz D, Laforsch C, Lindquist E, Lopez J, Manak JR, Muller J, Pangilinan J, Patwardhan RP, Pitluck S, Pritham EJ, Rechtsteiner A, Rho M, Rogozin IB, Sakarya O, Salamov A, Schaack S, Shapiro H, Shiga Y, Skalitzky C, Smith Z, Souvorov A, Sung W, Tang Z, Tsuchiya D, Tu H, Vos H, Wang M, Wolf YI, Yamagata H, Yamada T, Ye Y, Shaw JR, Andrews J, Crease TJ, Tang H, Lucas SM, Robertson HM, Bork P, Koonin EV, Zdobnov EM, Grigoriev IV, Lynch M, Boore JL: The ecoresponsive genome of Daphnia pulex. Science. 2011, 331: 555-561. 10.1126/science.1197761.
Jiravanichpaisal P, Puanglarp N, Petkon S, Donnuea S, Söderhäll I, Söderhäll K: Expression of immune-related genes in larval stages of the giant tiger shrimp, Penaeus monodon. Fish Shellfish Immunol. 2007, 23: 815-824. 10.1016/j.fsi.2007.03.003.
de Lorgeril J, Saulnier D, Janech MG, Gueguen Y, Bachère E: Identification of genes that are differentially expressed in hemocytes of the pacific blue shrimp (Litopenaeus stylirostris) surviving an infection with Vibrio penaeicida. Physiol Genomics. 2005, 21: 174-183. 10.1152/physiolgenomics.00281.2004.
Wongsurawat T, Leelatanawit R, Thamniemdee N, Uawisetwathana U, Karoonuthaisiri N, Menasveta P, Klinbunga S: Identification of testis-relevant genes using in silico analysis from testis ests and cDNA microarray in the black tiger shrimp (Penaeus monodon). BMC Mol Biol. 2010, 11: 55-10.1186/1471-2199-11-55.
Gorbach DM, Hu Z, Du Z, Rothschild MF: Mining ESTs to determine the usefulness of SNPs across shrimp species. Anim Biotechnol. 2010, 21: 100-103. 10.1080/10495390903323968.
Tagmount A, Wang M, Lindquist E, Tanaka Y, Teranishi KS, Sunagawa S, Wong M, Stillman JH: The porcelain crab transcriptome and pcad, the porcelain crab microarray and sequence database. PLoS ONE. 2010, 5: e9327-10.1371/journal.pone.0009327.
King AJ, Cragg SM, Li Y, Dymond J, Guille MJ, Bowles DJ, Bruce NC, Graham IA, McQueen-Mason SJ: Molecular insight into lignocellulose digestion by a marine isopod in the absence of gut microbes. Proc Natl Acad Sci USA. 2010, 107: 5345-5350. 10.1073/pnas.0914228107.
Söderhäll I, Bangyeekhun E, Mayo S, Söderhäll K: Hemocyte production and maturation in an invertebrate animal; proliferation and gene expression in hematopoietic stem cells of Pacifastacus leniusculus. Dev Comp Immunol. 2003, 27: 661-672. 10.1016/S0145-305X(03)00039-9.
Söderhäll I, Kim Y, Jiravanichpaisal P, Lee S, Söderhäll K: An ancient role for a prokineticin domain in invertebrate hematopoiesis. J Immunol. 2005, 174: 6153-6160.
Wang P, Gu Z, Huang X, Liu B, Deng X, Ai H, Wang J, Yin Z, Weng S, Yu X, He J: An immune deficiency homolog from the white shrimp, Litopenaeus vannamei, activates antimicrobial peptide genes. Mol Immunol. 2009, 46: 1897-1904. 10.1016/j.molimm.2009.01.005.
Zheng L, Hou L, Chang AK, Yu M, Ma J, Li X, Zou X: Expression pattern of a gram-negative bacteria-binding protein in early embryonic development of Artemia sinica and after bacterial challenge. Dev Comp Immunol. 2011, 35: 35-43. 10.1016/j.dci.2010.08.002.
Jaenicke E, Fraune S, May S, Irmak P, Augustin R, Meesters C, Decker H, Zimmer M: Is activated hemocyanin instead of phenoloxidase involved in immune response in woodlice?. Dev Comp Immunol. 2009, 33: 1055-1063. 10.1016/j.dci.2009.05.005.
Pless DD, Aguilar MB, Falcón A, Lozano-Alvarez E, Heimer de la Cotera EP: Latent phenoloxidase activity and N-terminal amino acid sequence of hemocyanin from Bathynomus giganteus, a primitive crustacean. Arch Biochem Biophys. 2003, 409: 402-410. 10.1016/S0003-9861(02)00615-X.
Chevalier F, Herbinière-Gaboreau J, Bertaux J, Raimond M, Morel F, Bouchon D, Grève P, Braquart-Varnier C: The immune cellular effectors of terrestrial isopod Armadillidium vulgare: meeting with their invaders, Wolbachia. PLoS ONE. 2011, 6: e18531-10.1371/journal.pone.0018531.
Xi Z, Gavotte L, Xie Y, Dobson SL: Genome-wide analysis of the interaction between the endosymbiotic bacterium Wolbachia and its Drosophila host. BMC Genomics. 2008, 9: 1-10.1186/1471-2164-9-1.
Yoshida T, Nakamura H, Masutani H, Yodoi J: The involvement of thioredoxin and thioredoxin binding protein-2 on cellular proliferation and aging process. Ann N Y Acad Sci. 2005, 1055: 1-12. 10.1196/annals.1323.002.
Ong ST, Ho JZS, Ho B, Ding JL: Iron-withholding strategy in innate immunity. Immunobiology. 2006, 211: 295-314. 10.1016/j.imbio.2006.02.004.
Kremer N, Voronin D, Charif D, Mavingui P, Mollereau B, Vavre F: Wolbachia interferes with ferritin expression and iron metabolism in insects. PLoS Pathog. 2009, 5: e1000630-10.1371/journal.ppat.1000630.
Yano T, Kurata S: Induction of autophagy via innate bacterial recognition. Autophagy. 2008, 4: 958-960.
Virgin HW, Levine B: Autophagy genes in immunity. Nat Immunol. 2009, 10: 461-470. 10.1038/ni.1726.
Smith VJ, Fernandes JMO, Kemp GD, Hauton C: Crustins: enigmatic WAP domain-containing antibacterial proteins from crustaceans. Dev Comp Immunol. 2008, 32: 758-772. 10.1016/j.dci.2007.12.002.
Bourtzis K, Pettigrew MM, O'Neill SL: Wolbachia neither induces nor suppresses transcripts encoding antimicrobial peptides. Insect Mol Biol. 2000, 9: 635-639. 10.1046/j.1365-2583.2000.00224.x.
Nakamura Y, Gotoh T, Imanishi S, Mita K, Kurtti TJ, Noda H: Differentially expressed genes in silkworm cell cultures in response to infection by Wolbachia and Cardinium endosymbionts. Insect Mol Biol. 2011, 20: 279-289. 10.1111/j.1365-2583.2010.01056.x.
Login FH, Balmand S, Vallier A, Vincent-Monégat C, Vigneron A, Weiss-Gayet M, Rochat D, Heddi A: Anti-microbial peptides keep insect endosymbionts under control. Science.
Zelensky AN, Gready JE: The C-type lectin-like domain superfamily. FEBS J. 2005, 272: 6179-6217. 10.1111/j.1742-4658.2005.05031.x.
Ao J, Ling E, Yu X: Drosophila C-type lectins enhance cellular encapsulation. Mol Immunol. 2007, 44: 2541-2548. 10.1016/j.molimm.2006.12.024.
Kvennefors ECE, Leggat W, Hoegh-Guldberg O, Degnan BM, Barnes AC: An ancient and variable mannose-binding lectin from the coral Acropora millepora binds both pathogens and symbionts. Dev Comp Immunol. 2008, 32: 1582-1592. 10.1016/j.dci.2008.05.010.
Vidal-Dupiol J, Adjeroud M, Roger E, Foure L, Duval D, Mone Y, Ferrier-Pages C, Tambutte E, Tambutte S, Zoccola D, Allemand D, Mitta G: Coral bleaching under thermal stress: putative involvement of host/symbiont recognition mechanisms. BMC Physiol. 2009, 9: 14-10.1186/1472-6793-9-14.
Bulgheresi S, Schabussova I, Chen T, Mullin NP, Maizels RM, Ott JA: A new C-type lectin similar to the human immunoreceptor dc-sign mediates symbiont acquisition by a marine nematode. Appl Environ Microbiol. 2006, 72: 2950-2956. 10.1128/AEM.72.4.2950-2956.2006.
Lee SY, Söderhäll K: Characterization of a pattern recognition protein, a masquerade-like protein, in the freshwater crayfish Pacifastacus leniusculus. J Immunol. 2001, 166: 7319-7326.
Sriphaijit T, Flegel TW, Senapin S: Characterization of a shrimp serine protease homolog, a binding protein of Yellow Head Virus. Dev Comp Immunol. 2007, 31: 1145-1158. 10.1016/j.dci.2007.03.005.
Serbus LR, Sullivan W: A cellular basis for Wolbachia recruitment to the host germline. PLoS Pathog. 2007, 3: e190-10.1371/journal.ppat.0030190.
Rigaud T, Juchault P: Success and failure of horizontal transfers of feminizing Wolbachia endosymbionts in woodlice. J Evol Biol. 1995, 8: 249-255. 10.1046/j.1420-9101.1995.8020249.x.
Hughes GL, Ren X, Ramirez JL, Sakamoto JM, Bailey JA, Jedlicka AE, Rasgon JL: Wolbachia infections in Anopheles gambiae cells: transcriptomic characterization of a novel host-symbiont interaction. PLoS Pathog. 2011, 7: e1001296-10.1371/journal.ppat.1001296.
Acknowledgements and funding
We thank Catherine Debenest, Carine Delaunay, Jerôme Lesobre and Maryline Raimond for technical assistance and Renaud Fortuner for improving the English. A. vulgare sequences were obtained in the frame of the program “Functional Genomics and Immune Signaling in Invertebrate Endosymbiosis” in collaboration with the Centre National de Séquençage, Genoscope (Evry, France). This research was funded by the CNRS UMR 6556, the Université de Poitiers and the Agence Nationale de la Recherche (“EndoSymbArt” ANR-06-BLAN-0316 and “ImmunSymbArt” ANR-2010-BLAN-170101, both coordinated by DB).
This article has been published as part of BMC Microbiology Volume 11 Supplement 1, 2012: Arthropod symbioses: from fundamental studies to pest and disease mangement. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2180/12?issue=S1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
FC performed the RT-qPCR experiments and analysis, the bioinformatics analysis, and drafted the manuscript. JHG participated in the design of experiments, prepared the libraries, and participated in the sequence analysis. DC participated in the design of experiments, carried out the EST data processing and analysis, and helped for statistical analysis of expression data. GM helped to design RT-qPCR experiments and reviewed the manuscript. FG and PW sequenced the libraries. PG, CBV and DB conceived and coordinated the study, participated in its design, and drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12866_2012_1546_MOESM3_ESM.pdf
Additional file 3: Processes and functions over-represented in A. vulgare ovaries in response to Wolbachia infection, biological process levels 4 and 6.(PDF 48 KB)
12866_2012_1546_MOESM5_ESM.pdf
Additional file 5: Expression profiles of genes studied in whole animals, ovaries, and immune tissues of A. vulgare. Gene transcripts were quantified by RT-qPCR and normalized with the expression of the ribosomal protein (RbL8) and the Elongation Factor 2 (EF2). Each bar represents the mean of three independent measurements with standard error.(PDF 132 KB)
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chevalier, F., Herbinière-Gaboreau, J., Charif, D. et al. Feminizing Wolbachia: a transcriptomics approach with insights on the immune response genes in Armadillidium vulgare. BMC Microbiol 12 (Suppl 1), S1 (2012). https://doi.org/10.1186/1471-2180-12-S1-S1
Published:
DOI: https://doi.org/10.1186/1471-2180-12-S1-S1