
Abstract
Background
Cholesterol homeostasis and xenobiotic metabolism are complex biological processes, which are difficult to study with traditional methods. Deciphering complex regulation and response of these two processes to different factors is crucial also for understanding of disease development. Systems biology tools as are microarrays can importantly contribute to this knowledge and can also discover novel interactions between the two processes.
Results
We have developed a low density Sterolgene v0 cDNA microarray dedicated to studies of cholesterol homeostasis and drug metabolism in the mouse. To illustrate its performance, we have analyzed mouse liver samples from studies focused on regulation of cholesterol homeostasis and drug metabolism by diet, drugs and inflammation. We observed down-regulation of cholesterol biosynthesis during fasting and high-cholesterol diet and subsequent up-regulation by inflammation. Drug metabolism was down-regulated by fasting and inflammation, but up-regulated by phenobarbital treatment and high-cholesterol diet. Additionally, the performance of the Sterolgene v0 was compared to the two commercial high density microarray platforms: the Agilent cDNA (G4104A) and the Affymetrix MOE430A GeneChip. We hybridized identical RNA samples to the commercial microarrays and showed that the performance of Sterolgene is comparable to commercial arrays in terms of detection of changes in cholesterol homeostasis and drug metabolism.
Conclusion
Using the Sterolgene v0 microarray we were able to detect important changes in cholesterol homeostasis and drug metabolism caused by diet, drugs and inflammation. Together with its next generations the Sterolgene microarrays represent original and dedicated tools enabling focused and cost effective studies of cholesterol homeostasis and drug metabolism. These microarrays have the potential of being further developed into screening or diagnostic tools.
Similar content being viewed by others


Background
The level of cholesterol in mammals is tightly regulated by multiple homeostatic mechanisms which affect cholesterol biosynthesis, metabolism and uptake-excretion cycles. This regulation is achieved by different transcriptional factors, including sterol regulatory element-binding proteins (SREBP) and different members of the nuclear receptor superfamily [1, 2]. Under certain conditions (inflammation, high-cholesterol diet, etc.) cholesterol homeostasis is disrupted and can lead to atherosclerosis and other cardiovascular diseases, gallstone formation or neurological disorders. Cardiovascular diseases remain a leading cause of death in the developed world and are therefore an important research area. The central question is how different physiological processes being provoked by endogenous and exogenous sources influence cholesterol homeostasis and what the key targets of these regulations are. These findings are also of crucial importance for the development of novel hypolipidemic drugs.
Each exogenous compound entering the body requires detoxification by drug metabolism enzymes from different families. This process is also regulated by members of the nuclear receptor superfamily [3]. Recent studies have shown important crosstalk between drug metabolism and cholesterol homeostasis through nuclear receptor-mediated pathways [4]. However, this complex regulatory network is still far from being completely understood.
Microarray technology is a powerful tool and has gained broad usage in basic and medical research. It enables systemic studies of gene expression of selected genes or of the entire genome and can lead to the discovery of new genes involved in biological processes. Several commercial and academic DNA microarrays are currently available. Commercially available are often whole-genome arrays, which are not cost-effective for studies of a small number of selected genes [5]. This provides a rationale for developing small-scale arrays, enabling cost-effective studies of a limited number of genes involved in specific biochemical pathways [6–9], thus allowing more detailed studies of specific biological processes. Commercial arrays often do not include genes which may be of a particular interest to specific users [10–12]; neither do they always offer control over probe sequences. In the development process of a small-scale custom array special care is taken in the selection of the studied genes and specifically the position of their probes. The rationale for designing custom arrays is in the ease of their adaptation, e.g. addition of new genes/probes, removal of non-working probes and modification of the geometry. Owing to low production costs and small overhead with annotations the adaptations can be performed on the fly during experimentation, which is unfortunately still not the case with commercially available arrays.
We have developed Sterolgene v0 cDNA microarray as a prototype in a series of Sterolgene/Steroltalk cDNA microarrays. These dedicated arrays enable focused studies of cholesterol homeostasis and drug metabolism in mouse and in human. We have validated the Sterolgene array in studies of experimental disturbances of cholesterol homeostasis by inflammation, diet and drugs in the mouse liver. We compared the results to two commercial platforms: the Agilent cDNA (G4104A) and the Affymetrix MOE430A GeneChip. Within this paper we show that the Sterolgene array performs as well as any of the two commercial platforms in terms of detection of changes in expression of genes of our interest from the cholesterol homeostasis and drug metabolism induced by the studied factors.
Results
Production of the Sterolgene v0 microarray
The Sterolgene array contains 95 mouse genes known to be involved in cholesterol homeostasis and drug metabolism. Among these are members of the cytochrome P450 superfamily, the nuclear receptor superfamily, the ABC transporters and other transporter families, genes for cholesterol biosynthesis and SREBP signaling and some other genes linked to cholesterol homeostasis (Figure 1). The complete gene list with corresponding accession numbers is available in Additional file 1. The positions of probes have been selected according to a priori knowledge of each gene. Probes detect all gene transcripts known at the time of design. In case of major differences between transcripts more probes per gene were selected. Each gene probe is spotted in 6 replicas, three in two blocks. Each block contains 23 types of positive controls (most of them spotted in 6 replicas) and 3 types of negative controls (Figure 2). These serve as control of quality; positive controls are also used for data normalization. Level of unspecific cross-hybridization, background intensity and uniformity were used to optimize hybridization and washing protocols.

Drug metabolism and cholesterol homeostasis. Main groups of genes involved in cholesterol homeostasis and drug metabolism present on the Sterolgene v0 cDNA microarray. A scheme of the liver cell is shown together with its surroundings (blood, bile).

Sterolgene v0 microarray design. Complete array containing 12 blocks was spotted twice to increase number of replicas. Each block contains positive and negative controls to enable spatial normalization. Each gene probe is spotted three times in two blocks. Green spots – positive controls: L – Firefly luciferase, A – ArrayControls Spikes (Ambion); white spots – negative controls: C – bacterial gene probe, SB – spotting buffer, N – empty spot; grey spots – salmon sperm DNA (S); yellow spots – gene probes (G).
Normalization of Sterolgene data
Normalization of Sterolgene data was done using LOWESS fit to normalization spike in RNAs according to their average intensity (A = log2√(R*G), where R and G represent signal intensities). Figure 3A shows a MA plot of Sterolgene data and a LOWESS curve fitted to the normalization controls. However, data from normalization controls were first adjusted according to concentrations in which normalization RNAs are added to sample RNAs: log2 ratios of concentrations are subtracted from the log2 ratios of measured intensities (M = log2(R/G)). In Figure 3C the same data as in 3A is shown but with normalization controls adjusted according to their RNA concentrations. Notice the difference between the normalization curves at large A values. Such adjustment results in better fit of the normalization curve and consequently in more accurate normalization. Figures 3B and 3D show MA plots of data normalized according to the LOWESS curves shown in Figures 3A and 3C, respectively. Notice the difference in M for genes with large A values. Sterolgene data was normalized in Orange software [13] using a dedicated normalization widget, which was developed specifically for normalization of custom low-density arrays.

Normalization of Sterolgene v0 microarray data. MA plots of Sterolgene v0 microarray data illustrating its normalization. Genes are marked with crosses (+), Ambion ArrayControl spikes with circles (○) and diamonds (◇), and Firefly luciferase mRNAs with squares (□). A: data before normalization together with LOWESS curve through normalization spike in RNAs. C: the same data as shown in A, but with normalization controls adjusted according to their RNA concentrations (see text for details). B and D: data after normalization according to LOWESS curves shown in A and C, respectively. Adjusted are 4 types of Ambion spikes marked with green, orange and red circles/diamonds.
Acute TNFα inflammation and fasting affect cholesterol homeostasis and drug metabolism
The Sterolgene microarray was first evaluated in studies of how acute inflammation affects cholesterol homeostasis and drug metabolism. Mouse liver RNAs from the fasting and TNF-α/fasted experiments were hybridized to Agilent 10 K cDNA microarrays (G4104A) and Sterolgene arrays according to the recommended protocols. Differential expression was determined by two tailed Student's t-test. For Sterolgene data, probability of type I error αS = 0.05 was used. For Agilent data, a complementary probability of type I error was calculated according to Bonferroni correction for multiple testing (αA = αS*nS/nA = 0.00055 where nS and nA represent the number of genes included in Sterolgene and Agilent arrays, respectively), but final analysis was made using a more relaxed criteria αA = 0.01.
Using the Sterolgene array we show that fasting turns off most of anabolic processes due to energy conserving shifts in the liver and we observe a down-regulation of a majority of genes. These genes code for drug metabolizing enzymes (Cyp3a41, Cyp1a2, and Cyp3a25), different nuclear receptors (Pparα, Rorγ, Rorα, and Lxrα), cholesterol biosynthesis (Acat2, Mvk, Sc4mol, Cyp51a1, Nsdhl, Sc5d,) and transcriptional regulation of cholesterol biosynthesis (Srebp2) (Table 1). We also observed down-regulation of Alas1 (delta-aminolevulinate synthase 1) and Cyp11a1, which has previously been reported [14]. However, bile acid synthesis (Cyp8b1) is up-regulated by fasting. Three down-regulated (Cyp51a1, Sc4mol, Srebp2) and one up-regulated (Cyp8b1) gene were confirmed by RT-PCR [15] (Figure 4A).

Agreement of Sterolgene, Agilent and Affymetrix with RT-PCR data. Venn diagrams illustrating genes differentially expressed by Sterolgene, Agilent, Affymetrix and RT-PCR measurements. Only genes tested by RT-PCR are shown. A: fasting experiment, B: TNF-α experiment, C: phenobarbital experiment, D: cholesterol feeding experiment. Differential expression was determined using two tailed Student's t-test at probability of type I error α = 0.05 for Sterolgene-Agilent comparison (A and B) and α = 0.1 for Sterolgene-Affymetrix comparison (C and D). Differential expression in RT-PCR experiments was determined using two tailed Student's t-test at probability of type error α = 0.05.
In TNF-α treated fasted mice we observe up-regulation of cholesterol biosynthesis (Cyp51a1, Sc4mol) and down-regulation of cholesterol excretion (Cyp8b1) (Table 2). However, drug metabolism is further repressed (Cyp2f2, Cyp2j5, Cyp3a25) and this also has previously been shown [16]. Two up-regulated (Cyp51a1, Sc4mol) and one down-regulated gene (Cyp8b1) from cholesterol homeostasis have been confirmed by RT-PCR [15] (Figure 4B).
With the Agilent array we were not able to detect any change in cholesterol homeostasis and drug metabolism caused by TNF-α and fasting (Additional files 2 and 3). Agreement between both platforms is low with only one gene in common in TNF-α/fasted experiment (Cyp2f2) and with no genes in common in fasting experiment.
Additionally, we have selected only genes that are present in both platforms (we found 54 such genes) and determined their differential expression using the same p-value cut-off as for Sterolgene data (α = 0.05). The rationale behind was not to tighten the analysis conditions for Agilent data just due to the fact that the Agilent array contains more genes than the Sterolgene array. In fasting experiment we found 5 genes differentially expressed (Additional file 4) and of these Cyp1a2 was confirmed by Sterolgene and RT-PCR, Actb by RT-PCR, while Apoa1 was not confirmed by RT-PCR [15] (Figure 4A). In TNF-α/fasted experiment we found 4 genes differentially expressed (Additional file 5) and of these Cyp2f2 was confirmed by the Sterolgene (RT-PCR not performed), and Actb and Apoa1 by RT-PCR [15].
Low correspondence in differential expression may be due to the fact that of 54 genes in common only 20 showed significant signal on the Agilent arrays. For these genes we calculated Pearson's product moment correlation coefficient between log2 ratios in both experiments. Figure 5A shows a scatterplot of that data. Though the correspondence in differential expression is low, we observed a significant correlation between the platforms (r = 0.508, p = 0.001, N = 40), which is comparable to correlations reported by other cross-platform comparison studies [12, 17–19].

Scatterplots of log 2 ratios between Sterolgene, Agilent and Affymetrix. Scatterplots of log2 ratios of genes that are present in Agilent and Sterolgene arrays (A), and Affymetrix and Sterolgene arrays (B). Values from both experiments performed for each cross-platform comparison were combined in a single plot.
Phenobarbital and cholesterol feeding affect cholesterol homeostasis and drug metabolism
The Sterolgene array was also tested in studies of how phenobarbital and cholesterol feeding affect cholesterol homeostasis and drug metabolism. Mouse liver RNAs were hybridized to Affymetrix MOE430A GeneChips and Sterolgene arrays according to the recommended protocols. Differential expression was determined by two tailed Student's t-test. For Sterolgene data, probability of type I error αS = 0.1 was used. For Affymetrix data, a complementary probability of type I error was calculated according to Bonferroni correction for multiple testing (αAf = αS*nS/nAf = 0.00043 where nS and nAf represent the number of genes included in Sterolgene and Affymetrix arrays, respectively), but final analysis was made using a more relaxed criteria αA = 0.001.
In phenobarbital experiment, we detected several previously known up-regulated genes (Cyp2b and Cyp3a families, Cyp2a4 and Alas1) [20–22] using the Sterolgene array (Table 3). However, among these only Cyp2b10 was detected using Affymetrix arrays (Additional file 6). Alas1, Cyp2b10 and Cyp3a11 were confirmed by RT-PCR (Figure 4C).
In cholesterol feeding experiment, we detected down-regulation of the cholesterol biosynthesis (Fdft1, Sqle, Cyp51a1, Nsdhl) on both platforms (Table 4 and Additional file 7). However, with Sterolgene we additionally detected up-regulation of the following genes: drug-metabolizing enzyme gene Cyp3a11 and nuclear receptor Shp (Nr0b2, small heterodimeric partner) as has been previously shown [23, 24], cholesterol transporter Scp2 (Sterol carrier protein 2), and Ldlrap1 (LDL receptor adaptor protein 1). Cyp3a11 and Fdft1 were confirmed by RT-PCR (Figure 4D).
Additionally, we have selected only genes that are present in both platforms (we found 92 such genes) and determined their differential expression using the same p-value cut-off as for Sterolgene data (α = 0.1), with the same rationale as for the analysis of Agilent data. In phenobarbital experiment we found 21 genes differentially expressed (Additional file 8) and of these Alas1 and Cyp2b10 were confirmed by Sterolgene and RT-PCR, Cyp2a4 and Nr2c1 by Sterolgene (RT-PCR not performed), Scap by RT-PCR, while for Apoa1 and Scarb1 no confirmation was found (Figure 4C).
In cholesterol feeding experiment we found 41 genes differentially expressed (Additional file 9) and of these Fdft1 was confirmed by Sterolgene and RT-PCR, Acss2, Cyp51a1, Nsdhl, Pmvk and Sqle by Sterolgene (RT-PCR not performed), Cyp26a1 by RT-PCR, while for Ppara and Scap no confirmation was found (Figure 4D).
From the 92 genes that are present in both platforms we selected those that showed signal on both platforms and calculated a Pearson's product moment correlation coefficient between log2 ratios for data from both experiments. Figure 5B shows scatterplot of that data. We observed a similar correlation as for the Agilent-Steroltalk comparison (r = 0.499, p = 0.000, N = 319) and comparable to correlations reported by other cross-platform comparison studies [12, 17–19]. Since more probes per gene are available on Affymetrix array we included all in the correlation analysis.
Discussion
Systemic studies of cholesterol homeostasis and drug metabolism are still limited. In order to promote the implementation of systems biology approaches to this two research areas, we have developed the Sterolgene array. It is a highly customizable tool that together with its human version allows for detailed studies of the two biological processes in mouse and in human. The main goal of this study was to evaluate the performance of the Sterolgene array in typical studies of cholesterol homeostasis and drug metabolism in comparison with two commercial platforms (Agilent and Affymetrix). Several recent papers discuss the comparative data between different microarrays platforms [12, 17–19, 25]. Many of these studies calculated different correlation coefficients, measured reproducibility, but some have also taken into consideration the biological aspects of the comparison [12, 19, 26]. This approach was also undertaken in our evaluations of the performance of the Sterolgene array.
We have selected different factors, such as diet, drug treatment and inflammation, where scattered information existed about their effect on cholesterol homeostasis and drug metabolism (Figure 6). We analyzed the data as it is commonly done in microarray experiments. In the experiment where mice were on a high-cholesterol diet, we detected genes from cholesterol biosynthesis as down-regulated using both Sterolgene and Affymetrix arrays. However, with Sterolgene arrays, but not with Affymetrix, we detected Cyp3a family and nuclear receptor Nr0b2 as up-regulated and as previously described in [23, 24]. In the liver of mice treated with phenobarbital, we detected up-regulation of the Cyp2b family using both Sterolgene and Affymetrix arrays and as previously described in [21]. However, with Sterolgene arrays we additionally detect genes from the Cyp3a family, Cyp2a4 and Alas1 as up-regulated and as previously described in [20, 22]. Analysis of Affymetrix data using the same p-value cut-off as for the Sterolgene data showed two additional genes (Abca1, Abcg5) as differentially expressed in cholesterol feeding experiment, which were confirmed by another study [24] where also Affymetrix arrays were used. However, we were sill not able to confirm the up-regulation of Cyp3a11 and Nr0b2. In phenobarbital experiment we were able to confirm the up-regulation of Alas1 and Cyp2a4, but not Cyp3a11.

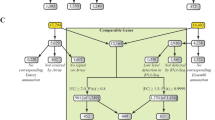
Changes in cholesterol homeostasis and drug metabolism caused by different factors in mouse liver. With Sterolgene array we successfully detected all changes in cholesterol homeostasis and drug metabolism caused by high-cholesterol diet, fasting, TNF-α, and phenobarbital (PB) treatment (solid arrows). With Agilent array (G4104A) we detected none of the changes caused by the inflammatory cytokine TNF-α and fasting (crosses). With Affymetrix MOE430A GeneChip we detected only down-regulation of the cholesterol biosynthesis by the high-cholesterol diet and induction of the Cyp2b family by the phenobarbital treatment (dashed arrows), but not the up-regulation of Cyp3a11 by high-cholesterol diet and up-regulation of Cyp3a family and Alas1 by phenobarbital treatment. In all experiments, two tailed Student's t-test was used for classification of differentially expressed genes.
Low agreement was observed between the Agilent and the Sterolgene arrays in detecting changes in gene expression, regardless of the selected p-value cut-off. In fasting and TNF-α/fasted experiments we were able to detect changes in expression of genes related to cholesterol homeostasis and drug metabolism with Sterolgene arrays, but not with Agilent arrays. In fasting experiment, we observed metabolic adaptations to energy-shortage. Energy-consuming processes as are cholesterol biosynthesis and drug metabolism are down-regulated. However, treatment with TNF-α in combination with fasting reverses these adaptations and up-regulation of cholesterol biosynthesis and down-regulation of bile acids synthesis is observed. This results in disrupted cholesterol homeostasis and higher levels of cholesterol present in the mouse liver. All these findings are in agreement with the hypothesis that pro-inflammatory factors such as TNF-α are atherogenic [27].
Cross-platform comparisons using correlation analysis showed correlations comparable to other cross-platform comparison studies [12, 17–19]. This result, together with low correspondence in differential expression (especially observed in Sterolgene-Agilent comparison) provides strong justification for development of small-scale custom arrays. As it turned out with Sterolgene data, small-scale arrays are often more sensitive than whole-genome arrays owing to the special care taken in designing probes.
Though we were able to show some important correspondence between the Steroltalk and both commercial platforms, our comparison has some limitations. First, we were limited by the availability of samples, resulting in low number of biological replicas used for comparisons. Additional samples and other diets/treatments may provide valuable information for the comparison and further insight into the performance of Sterolgene array. Second, we employed t-test based classification of differential expression as this is usually the case in most biologically-oriented studies. This may result in unstable variance (noise) estimation [26]. Other less rigid classification methods were developed, especially for microarray data analysis where we are usually dealing with small number of measurements. Although such methods might result in better correspondence between the platforms, we still decided to use a statistically-based method in order to avoid the biases that alternative approaches often introduce into results. Third, lab/platform effect, which can often be higher than the biological effect [12, 18, 28, 29] must not be disregarded in the Sterolgene-Affymetrix comparison. And most importantly, gene probes are not sequence matched across the compared platforms, which in combination with different approaches to probe design and transcript variant detection can make a major contribution to low agreement [30, 31]. Finally, labeling step and the choice of using amplification also contribute to differences [32, 33].
One of the obstacles in conducting the comparison, but not the reason for low concordance, was the lack of annotation for probes on the commercial platforms. With all the tools available for microarray annotation there is still a large portion of probes whose annotation remain unknown and need a one by one search in the public databases. Well annotated low density microarrays, such as is Sterolgene, facilitate less demanding biological interpretation and confirmation of results. Another problem exposed in early papers on cDNA microarrays is also a high rate of incorrect clones in the cDNA libraries used for their production [17, 34]. For Sterolgene array we have prepared our own cDNA library which we confirmed by sequencing.
An important advantage of small arrays is in the design of the probes. For whole-genome arrays, probes are designed (semi)automatically with a limited attention dedicated to each gene probe. With smaller arrays, more effort and a priori knowledge is invested into their design, thus enabling more accurate detection of changes in gene expression. The costs of producing and using small-scale academic arrays are still low compared to commercial ones. This is especially true for studies where a large number of microarray experiments is planned, e.g. screening process in the development of new drugs.
Conclusion
We have demonstrated that the Sterolgene array is a novel tool in studies of cholesterol homeostasis and drug metabolism and can be used for more focused and cost-effective systemic studies. Limited to our studies, we have shown that its performance is comparable to the two commercial platforms. Its application ranges from basic research of how different factors influence these two biological processes to testing effects of new cholesterol lowering drugs. It is a tool which can be easily adjusted to novel research needs and interests. We are constantly improving the array with new genes and probes; we have recently developed the third version of the array, which contains an expanded list of genes involved in atherogenesis, circadian rhythm, inflammation and metabolism of carbohydrates and lipids, together with its human version consisting of human counterparts of most of the genes, thus allowing analysis of clinical samples.
Methods
Chemicals
All chemicals were purchased from Sigma (St Louis, MI, USA) if not stated otherwise.
Mouse experiments and sample preparation
In the TNF-α (tumor necrosis factor alpha) and fasting experiments male mice C57BL/6 (Harlan, Italy) age between 10 and 12 weeks were housed in normal light-cycle room, maintained on standard rodent chow, and were allowed water and food ad libitum [15]. Three groups of mice, 5–8 animals per group, were injected i.v at the same time point either with vehicle (saline) or with human recombinant TNF-α 30 μg/animal in saline (a gift from V. Menart, Lek Pharmaceuticals d.d.) and food was withdrawn, except for the control group. After 20 h mice were sacrificed using CO2 suffocation. Total RNA was isolated from liver using TRI-reagent (Sigma, St Louis, MI, USA) and purified by RNeasy Mini Kit (Qiagen GmBH, Hilden, Germany) according to the manufacturer's protocols.
In phenobarbital treatment and cholesterol feeding experiments male mice C57BL/6 (RCC, Ittingen, Switzerland) age between 9 and 11 weeks were maintained in a 12-hour light/12-hour dark cycle and fed ad libitum standard rodent chow diet or the same diet supplemented with 1% (w/w) cholesterol (Provimi Kliba SA, Kaiseraugst, CH). Mice were divided in three groups, 8 animals per group: the control group which was injected i.p. vehicle (5% DMSO-corn oil) and maintained on standard diet; the cholesterol group which was maintained on cholesterol diet for week prior injection and injected i.p. vehicle; and the phenobarbital group which was maintained on standard diet and injected i.p. with 50 mg/kg phenobarbital sodium in a 5% DMSO-corn oil. After 10 hours, animals were sacrificed using CO2 suffocation. The livers were snap-frozen in liquid nitrogen, total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer protocol and purified by RNeasy Mini Kit (Qiagen GmBH, Hilden, Germany). The total RNAs of two animals were pooled in equal amounts resulting in four pools per group. Quality and purity of total RNAs from both studies were checked using the RNA 6000 Nano LabChip Kit on Agilent 2100 Bioanalyzator (Agilent Technologies, Pato Alto, CA, USA). All animal experiments followed the Amsterdam Protocol on Animal Protection and Welfare and were approved by the local ethics committees.
cDNA microarray production
For 95 selected genes cDNA microarray probes of a similar length (400 – 500 bp), melting temperature and located less than 1500 bp from the polyA tail were designed using ProbeWIZ software [35]. Three probes of firefly luciferase in sequential order from the 5'-end to the polyA tail have been designed as controls of the reverse transcriptase reaction and as normalization controls. Chloramphenicol acetyltransferase (CAT) and Renilla luciferase gene probes were designed as negative controls. For housekeeping genes two to three probes were designed enabling control of RNA degradation rate. PCR products were prepared and cloned using the Qiagen PCR Cloning kit (Qiagen GmBH, Hilden, Germany). Plasmids were sequenced to verify the correct insert sequence. From these plasmids PCR products were amplified (Qbiogene, Morgan Irvine, CA, USA) and purified using the QIAquick 96 PCR BioRobot purification kit (Qiagen GmBH, Hilden, Germany). PCR products were dried and dissolved in 50% formamide and 1% CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propane sulfonate) spotting buffer in final 200 ng/μl concentration. DNAs were spotted on GAPS II-coated slides (Corning Inc. Corning, NY, USA) using a GeneTAC G3 robot (Genomic Solutions, Huntington, UK) in a controlled environment (25°C to 26°C and 25–29% humidity). Each PCR product was spotted in triplicates in two blocks yielding 6 replicas. Firefly luciferase probes were spotted in each block. Commercial "spike in controls" ArrayControl Spikes (Ambion, Austin, TX, USA) were also spotted, two in each block. After spotting slides were dried for 8 hours at room temperature, protected from light. The DNA was fixed by UV cross-linking at 250 mJ/cm2 (Hoefer UVC500, Amersham Pharmacia Biotech, San Francisco, CA, USA). Slides were stored in a dry and dark place.
Probe labeling and hybridizations
In TNF-α/fasted and fasting experiments, individual samples from fasted and TNF-α-treated groups were hybridized versus a reference pool of samples from normal-fed control group using Sterolgene (3 biological replicas) and Agilent arrays (2 biological replicas, identical to those used in Sterolgene hybridizations). The hybridizations were performed in a single laboratory according to the recommended protocols.
In phenobarbital and high cholesterol feeding experiments, samples from 8 mice were pooled by two into 4 pools for each control, cholesterol fed and phenobarbital treated group. All 4 pools from each group were hybridized to Affymetrix GeneChips. 2–3 pools, identical to those used in Affymetrix hybridizations, were hybridized together with a reference pool (pool of all samples) to Sterolgene arrays. Hybridizations of each of the platforms were performed in two different laboratories according to the recommended protocols.
In Sterolgene experiments, 10 μg of total RNA was used for labeling using Agilent Fluorescent Direct Label Kit (Agilent Technologies, Pato Alto, CA, USA) according to the manufacturer's protocol. Before labeling spike RNAs were added to the total RNA in following quantities: 500 pg of Firefly luciferase mRNA (Promega, Madison, WI, USA); and one of the two mixes (reference mix and sample mix) of 8 RNAs from ArrayControl Spikes (Ambion, Austin, TX, USA). The reference spike mix was a mixture of 500 pg of each RNA. The sample spike mix was a mixture of RNAs ranging from 100 pg to 1500 pg. In the TNF-α and fasting experiment, the control group was labeled with Cy3 (PerkinElmer Life Sciences, Boston, MA, USA), the fasted and the TNF-α treated groups were labeled with Cy5 (PerkinElmer Life Sciences, Boston, MA, USA). In the phenobarbital and cholesterol experiment the control, cholesterol and phenobarbital groups were labeled with Cy5 and the reference was labeled with Cy3. Sterolgene slides were washed before hybridization at room temperature for 5 min in 2 × SSC and 0.1%SDS, 3 min in 0.2 × SSC and 2 min in MilliQ water and dried by centrifugation. Samples were denatured and hybridized in 3 × SSC and 0.2% SDS hybridization buffer at 65°C for 16 hours using humidified hybridization chambers (HybChambers, GeneMachines, San Carlos, CA, USA). After hybridization slides were washed at room temperature for 5 min in 2 × SSC and 0.5% SDS, followed by 5 min in 0.5 × SSC, then 5 min in 0.05 × SSC and dried by centrifugation. Slides were scanned using a Tecan LS200 scanner (Tecan Group Ltd., Maennedorf, Switzerland).
In Agilent experiments, 10 μg of total RNA was used for labeling by Agilent Fluorescent Direct Label Kit (Agilent Technologies, Pato Alto, CA, USA). Preparation of samples, hybridization and washing protocols were done according to Mouse cDNA Microarray Kit (Agilent Technologies, Santa Clara, CA, USA) using the mouse Agilent cDNA microarrays (G4104A). The control group was labeled with Cy3, the fasted and the TNF-α treated group were labeled with Cy5. Hybridizations were performed using hybridization chambers (HybChambers, GeneMachines, San Carlos, CA, USA) and slides scanned by Tecan LS200 scanner (Tecan Group Ltd., Maennedorf, Switzerland).
In Affymetrix experiments, total RNA was analyzed by the MOE430A GeneChip (Affymetrix, Santa Clara, CA, USA) according to manufacturer's recommendations and slides scanned by a laser scanner (Affymetrix, Santa Clara, CA, USA) as described in [36].
Data normalization and analysis
Images of Sterolgene arrays were analyzed by Array-Pro Analyzer 4.5 (Media Cybernetics, Bethesda, MD, USA). The median feature and local background intensities were extracted together with the estimates of their standard deviation. Only features with foreground to background ratio higher than 1.5 and coefficient of variation (CV, ratio between standard deviation of the background and the median feature intensity) lower than 0.5 in both channels were used for further analysis. Log2 ratios were normalized using LOWESS fit [37] to spike in control RNAs according to their average intensity. For normalization, two types of spike in controls were used: custom-made Firefly luciferase (348 such probes per array) and commercial ArrayControl Spikes (Ambion, Austin, TX, USA) (96 such probes per array). Ambion spike in RNAs were added to each sample RNAs in the following concentrations: 1:5, 1:2, 1:1, 2:1 and 3:1. Data from Ambion spike in controls were adjusted prior to fit of normalization curve by subtracting log2(1/5), log2(1/2), log2(2) and log2(3) from the log2 ratios of measured intensities according to the concentrations (Figure 3C). In phenobarbital and cholesterol-feeding experiments data were standardized (median-centered and scaled by median absolute deviation) in order to reduce inter-array variability. Mean was used to average replicated microarray measurements. Data normalization and standardization was done in Orange software [13].
Images of Agilent microarrays were analyzed using Array-Pro Analyzer 4.5 (Media Cybernetics, Bethesda, MD, USA). Features, with CV > 0.39, were filtered out and data were normalized using LOWESS fit to all genes according to their average intensity [38]. Filtration and normalization was done in BASE software [39].
Affymetrix data were normalized by the Robust Multichip Average (RMA) algorithm as described [40]. After transformation to non-logarithmic data, the expression estimates were scaled to the average expression levels in the control group analyzed using GeneSpring software (Silicon Genetics, Redwood, USA).
Classification of differentially expressed genes was done in Orange using two tailed Student's t-test for independent samples. Between the platforms genes were matched by unigene or refseq codes, or by gene symbols. Pearson's product moment correlation coefficients were calculated and scatterplots produced in SPSS 14.0. All data have been submitted to GEO (Gene expression omnibus) under accession codes: GSE6271 (Affymetrix data), GSE6317 (Agilent data), GSE6447 (Sterolgene phenobarbital and high-cholesterol diet data), and GSE6423 (Sterolgene fasting and inflammation data).
Real-time polymerase chain reaction (RT-PCR)
RT-PCR analyses were performed in triplicates on pools of 3 mice from each group in TNF-α and fasting experiments, and on 4 biological replicas in phenobarbital and high-cholesterol diet experiments. In TNF-α and fasting experiments 1 μg of total RNA was reverse-transcribed using Superscript™ II or III reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Analyses were done using Platinum® SYBR® Green qPCR SuperMix-UDG (Invitrogen, Carlsbad, CA, USA), except in case of Sc4mol where Assay-on-demand from Applied Biosystems was used, and were performed on Applied Biosystems ABI PRISM 7900 HT or 7500 (PE Applied Biosystems, Foster City, CA, USA) according to the manufacturer's protocol. In phenobarbital and high-cholesterol diet experiments 1 μg of total RNA was reverse-transcribed using MMLV reverse transcriptase (Roche Molecular Biochemicals, Rotkreuz, Switzerland). Analyses (for transcripts Cyp3a11, Cyp2b10, Alas1) were done using the TaqMan PCR Core Reagent Kit (PE Applied Biosystems, Rotkreuz, Switzerland), and performed on ABI PRISM 7700 Sequence Detection System (PE Applied Biosystems, Rotkreuz, Switzerland) according to the manufacturer's protocol. Additional analyses (for transcript Scap, Apoa1, Car, Cyp7a1, Cyp1a2, Scarb1, Ppara, Cyp26a1) were done using Platinum® SYBR® Green qPCR SuperMix-UDG (Invitrogen, Carlsbad, CA, USA) and 7500 (PE Applied Biosystems, Foster City, CA, USA) according to the manufacturer's protocol. Relative transcript levels were determined using the comparative Ct (cycle threshold) method or using 2-ddCt values [41]. Internal control used in the first set of experiments is 18s and in the second set Gapdh. Statistical significance was determined by two tailed Student's t-test (α = 0.05). Primer sequences are provided in additional material (Additional file 10).
References
Horton JD, Goldstein JL, Brown MS: SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002, 109 (9): 1125-1131. 10.1172/JCI200215593.
Repa JJ, Mangelsdorf DJ: The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000, 16: 459-481. 10.1146/annurev.cellbio.16.1.459.
Waxman DJ: P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR, and PPAR. Arch Biochem Biophys. 1999, 369 (1): 11-23. 10.1006/abbi.1999.1351.
Handschin C, Meyer UA: Regulatory network of lipid-sensing nuclear receptors: roles for CAR, PXR, LXR, and FXR. Arch Biochem Biophys. 2005, 433 (2): 387-396. 10.1016/j.abb.2004.08.030.
Yauk CL, Williams A, Boucher S, Berndt LM, Zhou G, Zheng JL, Rowan-Carroll A, Dong H, Lambert IB, Douglas GR, Parfett CL: Novel design and controls for focused DNA microarrays: applications in quality assurance/control and normalization for the Health Canada ToxArray. BMC Genomics. 2006, 7: 266-10.1186/1471-2164-7-266.
Newton SS, Bennett A, Duman RS: Production of custom microarrays for neuroscience research. Methods. 2005, 37 (3): 238-246. 10.1016/j.ymeth.2005.09.004.
Glas AM, Floore A, Delahaye LJ, Witteveen AT, Pover RC, Bakx N, Lahti-Domenici JS, Bruinsma TJ, Warmoes MO, Bernards R, Wessels LF, Van't Veer LJ: Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genomics. 2006, 7: 278-10.1186/1471-2164-7-278.
de Longueville F, Surry D, Meneses-Lorente G, Bertholet V, Talbot V, Evrard S, Chandelier N, Pike A, Worboys P, Rasson JP, Le Bourdelles B, Remacle J: Gene expression profiling of drug metabolism and toxicology markers using a low-density DNA microarray. Biochem Pharmacol. 2002, 64 (1): 137-149. 10.1016/S0006-2952(02)01055-9.
Inoue A, Yoshida N, Omoto Y, Oguchi S, Yamori T, Kiyama R, Hayashi S: Development of cDNA microarray for expression profiling of estrogen-responsive genes. J Mol Endocrinol. 2002, 29 (2): 175-192. 10.1677/jme.0.0290175.
Verdugo RA, Medrano JF: Comparison of gene coverage of mouse oligonucleotide microarray platforms. BMC Genomics. 2006, 7: 58-10.1186/1471-2164-7-58.
Perez-Iratxeta C, Andrade MA: Inconsistencies over time in 5% of NetAffx probe-to-gene annotations. BMC Bioinformatics. 2005, 6: 183-10.1186/1471-2105-6-183.
Tan PK, Downey TJ, Spitznagel EL, Xu P, Fu D, Dimitrov DS, Lempicki RA, Raaka BM, Cam MC: Evaluation of gene expression measurements from commercial microarray platforms. Nucleic Acids Res. 2003, 31 (19): 5676-5684. 10.1093/nar/gkg763.
Demsar J, Zupan B, Leban G: Orange: From Experimental Machine Learning to Interactive Data Mining. 2004, Faculty of Computer nad Information Science, University of Ljubljana, 15-
Bauer M, Hamm AC, Bonaus M, Jacob A, Jaekel J, Schorle H, Pankratz MJ, Katzenberger JD: Starvation response in mouse liver shows strong correlation with life-span-prolonging processes. Physiol Genomics. 2004, 17 (2): 230-244. 10.1152/physiolgenomics.00203.2003.
Fon Tacer K, Kuzman D, Seliskar M, Pompon D, Rozman D: TNF-{alpha} INTERFERES WITH LIPID HOMEOSTASIS AND ACTIVATES ACUTE AND PRO-ATHEROGENIC PROCESSES. Physiol Genomics. 2007
Van Ess PJ, Mattson MP, Blouin RA: Enhanced induction of cytochrome P450 enzymes and CAR binding in TNF (p55(-/-)/p75(-/-)) double receptor knockout mice following phenobarbital treatment. J Pharmacol Exp Ther. 2002, 300 (3): 824-830. 10.1124/jpet.300.3.824.
Jarvinen AK, Hautaniemi S, Edgren H, Auvinen P, Saarela J, Kallioniemi OP, Monni O: Are data from different gene expression microarray platforms comparable?. Genomics. 2004, 83 (6): 1164-1168. 10.1016/j.ygeno.2004.01.004.
Irizarry RA, Warren D, Spencer F, Kim IF, Biswal S, Frank BC, Gabrielson E, Garcia JG, Geoghegan J, Germino G, Griffin C, Hilmer SC, Hoffman E, Jedlicka AE, Kawasaki E, Martinez-Murillo F, Morsberger L, Lee H, Petersen D, Quackenbush J, Scott A, Wilson M, Yang Y, Ye SQ, Yu W: Multiple-laboratory comparison of microarray platforms. Nat Methods. 2005, 2 (5): 345-350. 10.1038/nmeth756.
Bammler T, Beyer RP, Bhattacharya S, Boorman GA, Boyles A, Bradford BU, Bumgarner RE, Bushel PR, Chaturvedi K, Choi D, Cunningham ML, Deng S, Dressman HK, Fannin RD, Farin FM, Freedman JH, Fry RC, Harper A, Humble MC, Hurban P, Kavanagh TJ, Kaufmann WK, Kerr KF, Jing L, Lapidus JA, Lasarev MR, Li J, Li YJ, Lobenhofer EK, Lu X, Malek RL, Milton S, Nagalla SR, O'Malley JP, Palmer VS, Pattee P, Paules RS, Perou CM, Phillips K, Qin LX, Qiu Y, Quigley SD, Rodland M, Rusyn I, Samson LD, Schwartz DA, Shi Y, Shin JL, Sieber SO, Slifer S, Speer MC, Spencer PS, Sproles DI, Swenberg JA, Suk WA, Sullivan RC, Tian R, Tennant RW, Todd SA, Tucker CJ, Van Houten B, Weis BK, Xuan S, Zarbl H: Standardizing global gene expression analysis between laboratories and across platforms. Nat Methods. 2005, 2 (5): 351-356. 10.1038/nmeth754.
Jover R, Hoffmann K, Meyer UA: Induction of 5-aminolevulinate synthase by drugs is independent of increased apocytochrome P450 synthesis. Biochem Biophys Res Commun. 1996, 226 (1): 152-157. 10.1006/bbrc.1996.1325.
Ueda A, Hamadeh HK, Webb HK, Yamamoto Y, Sueyoshi T, Afshari CA, Lehmann JM, Negishi M: Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol Pharmacol. 2002, 61 (1): 1-6. 10.1124/mol.61.1.1.
Wei P, Zhang J, Dowhan DH, Han Y, Moore DD: Specific and overlapping functions of the nuclear hormone receptors CAR and PXR in xenobiotic response. Pharmacogenomics J. 2002, 2 (2): 117-126. 10.1038/sj.tpj.6500087.
Gupta S, Pandak WM, Hylemon PB: LXR alpha is the dominant regulator of CYP7A1 transcription. Biochem Biophys Res Commun. 2002, 293 (1): 338-343. 10.1016/S0006-291X(02)00229-2.
Maxwell KN, Soccio RE, Duncan EM, Sehayek E, Breslow JL: Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res. 2003, 44 (11): 2109-2119. 10.1194/jlr.M300203-JLR200.
Patterson TA, Lobenhofer EK, Fulmer-Smentek SB, Collins PJ, Chu TM, Bao W, Fang H, Kawasaki ES, Hager J, Tikhonova IR, Walker SJ, Zhang L, Hurban P, de Longueville F, Fuscoe JC, Tong W, Shi L, Wolfinger RD: Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nat Biotechnol. 2006, 24 (9): 1140-1150. 10.1038/nbt1242.
Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, Luo Y, Sun YA, Willey JM, Setterquist RA, Fischer GM, Tong W, Dragan YP, Dix DJ, Frueh FW, Goodsaid FM, Herman D, Jensen RV, Johnson CD, Lobenhofer EK, Puri RK, Schrf U, Thierry-Mieg J, Wang C, Wilson M, Wolber PK, Zhang L, Slikker W, Shi L, Reid LH: The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006, 24 (9): 1151-1161. 10.1038/nbt1239.
Ohta H, Wada H, Niwa T, Kirii H, Iwamoto N, Fujii H, Saito K, Sekikawa K, Seishima M: Disruption of tumor necrosis factor-alpha gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis. 2005, 180 (1): 11-17. 10.1016/j.atherosclerosis.2004.11.016.
Wang H, He X, Band M, Wilson C, Liu L: A study of inter-lab and inter-platform agreement of DNA microarray data. BMC Genomics. 2005, 6 (1): 71-10.1186/1471-2164-6-71.
Gwinn MR, Keshava C, Olivero OA, Humsi JA, Poirier MC, Weston A: Transcriptional signatures of normal human mammary epithelial cells in response to benzo[a]pyrene exposure: a comparison of three microarray platforms. Omics. 2005, 9 (4): 334-350. 10.1089/omi.2005.9.334.
Mecham BH, Klus GT, Strovel J, Augustus M, Byrne D, Bozso P, Wetmore DZ, Mariani TJ, Kohane IS, Szallasi Z: Sequence-matched probes produce increased cross-platform consistency and more reproducible biological results in microarray-based gene expression measurements. Nucleic Acids Res. 2004, 32 (9): e74-10.1093/nar/gnh071.
Carter SL, Eklund AC, Mecham BH, Kohane IS, Szallasi Z: Redefinition of Affymetrix probe sets by sequence overlap with cDNA microarray probes reduces cross-platform inconsistencies in cancer-associated gene expression measurements. BMC Bioinformatics. 2005, 6: 107-10.1186/1471-2105-6-107.
Larkin JE, Frank BC, Gavras H, Sultana R, Quackenbush J: Independence and reproducibility across microarray platforms. Nat Methods. 2005, 2 (5): 337-344. 10.1038/nmeth757.
Ach RA, Floore A, Curry B, Lazar V, Glas AM, Pover R, Tsalenko A, Ripoche H, Cardoso F, d'Assignies MS, Bruhn L, Van't Veer LJ: Robust interlaboratory reproducibility of a gene expression signature measurement consistent with the needs of a new generation of diagnostic tools. BMC Genomics. 2007, 8: 148-10.1186/1471-2164-8-148.
Kothapalli R, Yoder SJ, Mane S, Loughran TP: Microarray results: how accurate are they?. BMC Bioinformatics. 2002, 3: 22-10.1186/1471-2105-3-22.
Nielsen HB, Knudsen S: Avoiding cross hybridization by choosing nonredundant targets on cDNA arrays. Bioinformatics. 2002, 18 (2): 321-322. 10.1093/bioinformatics/18.2.321.
Oscarson M, Zanger UM, Rifki OF, Klein K, Eichelbaum M, Meyer UA: Transcriptional profiling of genes induced in the livers of patients treated with carbamazepine. Clin Pharmacol Ther. 2006, 80 (5): 440-456. 10.1016/j.clpt.2006.08.013.
Cleveland WS, Devlin SJ: Locally weighted regression: an approach to regresion analysis by local fitting. J Am Stat Assoc. 1988, 83: 596-610. 10.2307/2289282.
Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP: Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002, 30 (4): e15-10.1093/nar/30.4.e15.
Saal LH, Troein C, Vallon-Christersson J, Gruvberger S, Borg A, Peterson C: BioArray Software Environment (BASE): a platform for comprehensive management and analysis of microarray data. Genome Biol. 2002, 3 (8): SOFTWARE0003-10.1186/gb-2002-3-8-software0003.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003, 4 (2): 249-264. 10.1093/biostatistics/4.2.249.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001, 25 (4): 402-408. 10.1006/meth.2001.1262.
Acknowledgements
The work was supported by the Slovenian Research Agency, Grants J1-6713, P1-0527, and the funds of Lek Pharmaceuticals d.d. Tadeja Režen and Klementina Fon Tacer were supported by the fellowship from the Slovenian Research Agency. The work of Adrian Roth and Urs A. Meyer was supported by the Swiss National Science Foundation. This paper and work it concerns was generated in the context of the STEROLTALK project, funded by the European Community as contract No. LSHG-CT-2005-512096 under 6th Framework Programme for Research and Technological Development in the thematic area of Life sciences, genomics and biotechnology for health. This paper reflects only the author's views and the European Community is not liable for any use that may be made of the information contained therein. We would like to acknowledge Maud Silvain (CGM-CNRS, France) for plasmid sequencing, Nicolas Agier (CNRS-CGM, France) and Anne Peyrat (LIPM-CNRS, France) for technical support. We would also like to acknowledge Nejc Jelen for the help with cloning of the Sterolgene probes and performing initial hybridizations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
TR contributed to the gene list, probe and overall microarray design, and performed cloning and preparations of probes for microarray manufacturing. TR also designed protocols for Sterolgene analysis, performed hybridizations and part of the statistical analyses. PJ developed normalization and analysis tools and performed part of the statistical analysis. KFT performed TNF-α and fasting mouse experiments, and analyzed samples using Agilent and Sterolgene microarrays. DK contributed to probe design and microarray manufacturing, and performed first bioinformatical analyses. AR and UAM contributed to the gene list and AR performed phenobarbital and high-cholesterol diet experiments on mouse and Affymetrix hybridizations under supervision of UAM. LPA cooperated in microarray production. DP contributed to the gene list and concept of Sterolgene microarrays and was involved in manufacturing process. DR contributed to the gene list and concept of Sterolgene microarrays and was a supervisor of TR and KFT. All authors have read and approved the final manuscript.
Electronic supplementary material
12864_2007_1270_MOESM2_ESM.pdf
Additional file 2: Differentially expressed genes in the mouse liver after TNF-α treatment (Agilent microarray). Differentially expressed genes in the mouse liver after TNF-α treatment as detected by the Agilent 10 K cDNA microarray (G4104A) (α = 0.01, genes in bold: α = 0.001). (PDF 48 KB)
12864_2007_1270_MOESM3_ESM.pdf
Additional file 3: Differentially expressed genes in the mouse liver after fasting (Agilent microarray). Differentially expressed genes in the mouse liver after fasting as detected by the Agilent cDNA microarray (G4104A) (α = 0.01, genes in bold: α = 0.001). (PDF 26 KB)
12864_2007_1270_MOESM4_ESM.pdf
Additional file 4: Differentially expressed genes in mouse liver after fasting using the same probability of type I error as for Sterolgene array (Agilent microarray). Differentially expressed genes as detected by Agilent 10 K cDNA microarray using the same probability of type I error as for Sterolgene data (α = 0.05). Only genes that are also present in the Steroltalk array were considered in the analysis. Genes in bold are confirmed using RT-PCR, genes in italic coincide with the results from the Sterolgene platform. (PDF 12 KB)
12864_2007_1270_MOESM5_ESM.pdf
Additional file 5: Differentially expressed genes in mouse liver after TNF-α treatment using the same probability of type I error as for Sterolgene array (Agilent microarray). Differentially expressed genes as detected by Agilent 10 K cDNA microarray using the same probability of type I error as for Sterolgene data (α = 0.05). Only genes that are also present in the Steroltalk array were considered in the analysis. Genes in bold are confirmed using RT-PCR, genes in italic coincide with the results from the Sterolgene platform. (PDF 11 KB)
12864_2007_1270_MOESM6_ESM.pdf
Additional file 6: Differentially expressed genes in the mouse liver after phenobarbital treatment (Affymetrix GeneChip). Differentially expressed genes in the mouse liver after phenobarbital treatment as detected by the Affymetrix MOE430A GeneChip (α = 0.001, genes in bold: α = 0.00043). (PDF 68 KB)
12864_2007_1270_MOESM7_ESM.pdf
Additional file 7: Differentially expressed genes in the mouse liver after cholesterol feeding (Affymetrix GeneChip). Differentially expressed genes in the mouse liver after cholesterol feeding as detected by the Affymetrix MOE430A GeneChip (α = 0.001, genes in bold: α = 0.00043). (PDF 69 KB)
12864_2007_1270_MOESM8_ESM.pdf
Additional file 8: Differentially expressed genes in mouse liver after phenobarbital treatment using the same probability of type I error as for Sterolgene array (Affymetrix GeneChip). Differentially expressed genes as detected by Affymetrix GeneChip using the same probability of type I error as for Sterolgene data (α = 0.1). Only genes that are also present in the Steroltalk array were considered in the analysis. Genes in bold are confirmed using RT-PCR, genes in italic coincide with the results from the Sterolgene platform. (PDF 20 KB)
12864_2007_1270_MOESM9_ESM.pdf
Additional file 9: Differentially expressed genes in mouse liver after cholesterol feeding using the same probability of type I error as for Sterolgene array (Affymetrix GeneChip). Differentially expressed genes as detected by Affymetrix GeneChip using the same probability of type I error as for Sterolgene data (α = 0.1). Only genes that are also present in the Steroltalk array were considered in the analysis. Genes in bold are confirmed using RT-PCR, genes in italic coincide with the results from the Sterolgene platform. (PDF 30 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Režen, T., Juvan, P., Fon Tacer, K. et al. The Sterolgene v0 cDNA microarray: a systemic approach to studies of cholesterol homeostasis and drug metabolism. BMC Genomics 9, 76 (2008). https://doi.org/10.1186/1471-2164-9-76
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-9-76