
Abstract
Background
Teleost fish have seven paralogous clusters of Hox genes stemming from two complete genome duplications early in vertebrate evolution, and an additional genome duplication during the evolution of ray-finned fish, followed by the secondary loss of one cluster. Gene duplications on the one hand, and the evolution of regulatory sequences on the other, are thought to be among the most important mechanisms for the evolution of new gene functions. Cichlid fish, the largest family of vertebrates with about 2500 species, are famous examples of speciation and morphological diversity. Since this diversity could be based on regulatory changes, we chose to study the coding as well as putative regulatory regions of their Hox clusters within a comparative genomic framework.
Results
We sequenced and characterized all seven Hox clusters of Astatotilapia burtoni, a haplochromine cichlid fish. Comparative analyses with data from other teleost fish such as zebrafish, two species of pufferfish, stickleback and medaka were performed. We traced losses of genes and microRNAs of Hox clusters, the medaka lineage seems to have lost more microRNAs than the other fish lineages. We found that each teleost genome studied so far has a unique set of Hox genes. The hoxb7a gene was lost independently several times during teleost evolution, the most recent event being within the radiation of East African cichlid fish. The conserved non-coding sequences (CNS) encompass a surprisingly large part of the clusters, especially in the HoxAa, HoxCa, and HoxDa clusters. Across all clusters, we observe a trend towards an increased content of CNS towards the anterior end.
Conclusion
The gene content of Hox clusters in teleost fishes is more variable than expected, with each species studied so far having a different set. Although the highest loss rate of Hox genes occurred immediately after whole genome duplications, our analyses showed that gene loss continued and is still ongoing in all teleost lineages. Along with the gene content, the CNS content also varies across clusters. The excess of CNS at the anterior end of clusters could imply a stronger conservation of anterior expression patters than those towards more posterior areas of the embryo.
Similar content being viewed by others


Background
Genome duplications [1, 2] and regulatory evolution [3–5] are thought to be two major genomic evolutionary mechanisms that are, at least partly, responsible for the increased diversity of vertebrates compared to their chordate relatives. Genome and gene duplications can provide the raw material on which evolution can act since they lead to redundant gene copies that are freed up to evolve novel gene functions [1, 6].
Sequence data from complete genomes of tetrapods such as mouse, frog and human as well as from invertebrates such as Caenorhabditis elegans and Drosophila melanogaster show that many gene families tend to be larger in vertebrates [7–9]. Synteny data demonstrated that the most likely scenario for the increased size of gene families are two consecutive rounds of genome duplication, the so-called 2R-hypothesis [10–12]. Genomic data from zebrafish and pufferfish showed that many genes were duplicated before the divergence of those two species representing the major fish orders Neoteleostei and Ostariophysii [13–15]. More recently, syntenic data from zebrafish, medaka and pufferfish further confirmed the existence of an additional genome duplication event within the ray-finned fish lineage, the fish-specific genome duplication (FSGD) [16–18]. The most recent comparative genomic analyses also support the FSGD and found that the majority of genes was duplicated around 320–350 mya [19, 20]. Moreover, studies on individual nuclear genes and gene families propose a timing of the duplication preceding the diversification of teleosts [15, 21–23].
One of the earliest and best-studied examples for duplicated chromosomal regions is the clusters of Hox genes [21, 24, 25]. Hox genes are transcription factors, characterized by their DNA binding domain, the homeodomain. They were first discovered in Drosophila as the target of homeotic mutation, meaning the change of the segmental identity, as in the bithorax phenotype [26]. One special feature of Hox genes is their arrangement in genomic clusters. While invertebrates have a single cluster that can be interrupted as in Drosophila species [27] or dispersed through the genome as in urochrodates [28, 29] and nematodes [30], tetrapods such as human or frogs all have four clusters [reviewed in [31, 32]], as do cartilaginous fish [33]. Even invertebrates closely related to vertebrates, such as the cephalochordate Branchiostoma [25, 34] have a single cluster, which in the case of the sea urchin, is also rearranged [35]. Due to the fish-specific genome duplication, extant fish have seven Hox clusters, with alternate cluster loss in Ostariophysi (HoxDb in zebrafish) [36] and Acanthopterygii (HoxCb in pufferfish, medaka, cichlid) [18, 37–40]. The additional clusters, however, are not exactly equivalent with the homologous genes of tetrapods, but have experienced independent losses of genes [31], making the teleost clusters much more variable in gene content than those of tetrapods. So far, all of the fish that have been studied showed differences in gene content among their Hox clusters [18, 31, 37, 41].
Individual gene loss after gene or genome duplication events is common and can occur even long after the duplication [42–44]. Interestingly, some functional categories such as signal transducers and transcriptional regulators tend to retain more members than most gene families created by duplication [45–47]. The reasons and mechanisms for these differences in rate of gene loss among different functional groups remain incompletely understood, but current theories propose a link of equimolar amounts of different regulatory genes (gene balance hypothesis) [48].
The other main genomic source for evolutionary change is thought to be the evolution of regulatory sequences, so called regulatory evolution [49]. Also for this major type of evolutionary change, Hox genes are an often-studied example [50–54]. The clustered nature of Hox genes facilitates comparison of orthologous and paralogous sequences and the high degree of conservation allows for identification and detailed analyses of evolutionary events in regulatory sequences. Vertebrate Hox clusters are almost free of repetitive elements [55] which adds further tractability to the study of regulatory evolution. Hox genes play an important role in the specification of the primary body axis [56] as well as in later ontogenetic processes demanding highly specific regulation such as limb development [57].
Conserved Non-coding Sequences (CNS) in Hox clusters have been intensely studied previously, both in terms of content and cluster identity [53, 58, 59] as well as their evolutionary rates in duplicated clusters [50, 54]. The intergenic regions of Hox clusters are enriched for CNS and it has been argued that this abundance of cis- and trans-regulatory elements is the main reason for cluster conservation since neighboring genes share regulatory elements. However, it is unclear how strong this "gluing effect" of regulatory elements is for the cohesion of Hox genes in clusters since Hox clusters in at least some invertebrates can be split without apparent loss of function [27]. One possible source of the higher plasticity of the invertebrate cluster is the presence of repetitive DNA while in vertebrates, there is strong selection acting against it [55].
Actinopterygian (ray-finned) fishes not only encompass more than half of all vertebrate species (about 27,000) [60], but also display a huge variety of body shapes. One particular species-rich, monophyletic group of derived teleosts is the Euteleostei, currently ranked as one of the four subdivisions of the Teleostei, along with the more basal groups, Osteoglossomorpha, Elopomorpha, and Clupeomorpha [61–65]. The Euteleostei comprise approximately 25,000 species, of which 17,000 are Neoteleosts (e.g., pufferfishes, medaka, cichlids, and stickleback) and 8,000 Ostariophysi (e.g. zebrafish)[60]. Among the Neoteleostei, most species are classified as Perciformes (about 10,000 species), this however is a polyphyletic assembly, with at least five lineages [62]. One family of the Perciformes are cichlid fishes (Family Cichlidae), with more than 2,500 species; almost ten percent of all fish species are cichlids. Of particular interest is the immense species richness of the adaptive radiations of the East African Lakes Victoria, Malawi and Tanganyika which are made up each of several hundred endemic species each [66, 67]. The species flocks of Lake Victoria as well as of Lake Malawi are monophyletic and hundreds of species arose within less than 100,000 years in the case of the Lake Victoria species flock – the fastest known rates of speciation [66, 68].
One of the most intriguing questions now is, whether there is a genetic basis for this astonishing speciation rate and the enormous morphological diversity cichlids show. Since these events occur very rapidly, changes involving regulatory pathways are likely to be involved. Hox clusters provide a good starting point for a genomic investigation of this kind due to their clustered structure, which can be easily homologized with other species, and because of their known high content of CNS [59], combined with their key role in early and later development.
During the last few years, complete genomic sequences have become available for many species, including several teleost fishes. Some species were selected either for their small genome size (e.g., the pufferfishes Takifugu rubripes and Tetraodon nigroviridis) [18, 69] or because they are model organisms of developmental research (Danio rerio, Oryzias latipes) or speciation (Gasterosteus aculeatus)[70]. The construction of large insert libraries such as those in BAC or fosmid vectors, make it possible to study genomic regions also for species for which a genome project is (not yet) available [23, 51, 71].
In this study, we sequenced the Hox-cluster containing BAC clones from the East African cichlid Astatotilapia burtoni [72]. We performed a phylogenetic analysis with concatenated coding sequences and investigated gene and microRNA loss in the clusters as well as content of conserved non-coding sequences in the A. burtoni clusters in comparison with all available teleost Hox gene clusters. The clusters show a tendency to preserve a higher amount of CNS towards the 'anterior' end of the cluster, the region which is fundamentally involved in development of the head [73, 74], while the 'posterior' part contains more variation in regulatory elements.
Results
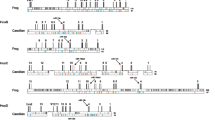
We screened the BAC library of Astatotilapia burtoni [72] for clones containing Hox clusters using specific-probes. Fragments spanning the intron were used as probes to avoid non-specific cross-reactions. Positive clones obtained by these screens were checked with specific primers for the 5' and 3' most genes of a cluster (e.g. evx-2 and hoxd3a) to confirm that they contain complete clusters. Clones containing all Hox genes of a cluster were shotgun sequenced. In this way, we obtained seven BAC clones which contain complete HoxAa, HoxAb, HoxBb, HoxCa, HoxDa and HoxDb clusters as well as the 5' part of the HoxBa cluster spanning the region from hoxb13a to hoxb5a (Figure 1).

Sequenced BAC clones and the annotated genes drawn to scale. Hox and Evx genes are shown in color, neighboring genes are drawn in black. Abbreviations used are according to [38], the surrounding genes are identical to those found in Takifugu rubipes. The HoxBa cluster is incomplete, sequence data stops at 12 kb downstream of hoxb5a. Sequence data for the remaining four Hox genes and the non-coding regions of remaining parts of the cluster have been gathered by PCR, indicating that the clustered structure still exists.
The clone 116-M8 (149.6 kb) contained the complete HoxAa cluster from hoxa13a to hoxa1a plus the related evx1 gene. 5' to evx1 we also identified the complete coding sequence of Hibadha (3-hydroxyisobutyrate dehydrogenase a). This is in agreement with the gene order in the Takifugu rubripes genome. The complete Hox containing region of the HoxAa cluster (including evx1) spans 86 kb.
For the HoxAb cluster, we selected clone 150-O18 (164.6 kb) for sequencing. The genes in this genomic region are tightly packed; this is true for the Hox genes as well as the surrounding genes. Also here the adjacent genes are orthologous to pufferfish sequences (cpv1 (carboxypeptidase vitellogenic-like), creb5b (cAMP responsive element binding protein 5), jazf1b (juxtaposed with another zinc finger protein 1), tax1bp1b (Tax1 binding protein 1), hibadhb (3-hydroxyisobutyrate dehydrogenase b), and skap2 (Src family associated phosphoprotein 2)). The HoxAb cluster is a small cluster both in terms of number of genes as well as intergenic regions and has a size of only 27 kb.
The HoxBa cluster is the largest in the genome of A. burtoni and we sequenced clone 170-E12 (160.1 kb), which contains the 5' part of the cluster from hoxb13a to hoxb5a. Despite intense screening of the BAC library with probes for the 3'genes (hoxb4a, hoxb3a, hoxb2a, hoxb1a), we could not identify a BAC clone containing this region in our library. We were able to amplify those genes from genomic DNA however. Therefore, they are not lost from the genome of Astatotilapia burtoni, but were apparently not contained in our BAC library. Similar to Gasterosteus aculeatus, Astatotilapia burtoni also has a large intergenic region (63 kb) containing repetitive elements between hoxb13a and hoxb9a. While this region looks "normal" in both pufferfish species as well as in medaka and zebrafish, this region in tetrapods also appears to be "decaying". So far, no hoxb13 gene from frog could be identified [31, 75] and in the human cluster, two non-Hox genes are situated between HoxB13 and HoxB9 (PRAC, LOC729146). The increase in size of the HoxBa cluster relative to that of the other Hox clusters also affects the size of its intergenic and the intronic regions. Although the hoxb7a gene of Astatotilapia burtoni was identified and annotated without problems, the coding sequence contains a stop codon at the beginning of the coding sequence, rendering it a pseudogene. However, in the other cichlid species studied so far, Oreochromis niloticus, hoxb7a has a completely intact coding sequence indicating that the non-functionalization of this gene in A. burtoni occurred within only the approximately last seven million years since the two species last shared a common ancestor.
The paralogous cluster of the "giant" HoxBa, is the "dwarf" HoxBb cluster, which was identified within BAC clone 34-B18 (152.2 kb). It contains only four Hox genes (hoxb6b, hoxb5b, hoxb3b, and hoxb1b) and spans only about 20 kb. Apart from the Hox genes themselves, the genes surrounding the HoxBb cluster are also densely packed. The clone contains also a partial sequence of ndp52 (nuclear domain 10 protein 52) and the complete sequences of TTLL6 (tubulin tyrosine ligase-like family member 6), scap1 (Src family associated phosphoprotein 1), snx11 (sorting nexin 11), cbx1 (chromobox-like 1), nfe2l1 (nuclear factor erythroid derived 2-like 1), and cdk5rap3 (CDK5 regulatory subunit associated protein 3) (Figure 1).
Clone 103-K21 (182.8 kb) contains the complete HoxCa cluster and three additional genes 3' of the cluster (cbx5 (chromobox-like 5), hnrpa1 (heterogeneous nuclear ribonucleoprotein A1), and nfe2 (nuclear factor erythroid-derived 2)). Also here the order of the neighbouring genes is the same as in Takifugu rubripes. This also confirms that there are no further Hox genes downstream of hoxc3a in cichlids, while in zebrafish hoxc1a was retained. The complete length of the HoxCa cluster in Astatotilapia burtoni from hoxc13a to hoxc3a is 91 kb.
The HoxDa cluster was sequenced from clone 32-B18 (182.2 kb). This Hox cluster only spans 53 kb from evx2 to hoxd3a, and the surrounding sequences contain only one more gene, lnpa (lunapark a).
From clone 19-E16 (186.2 kb), the sister cluster HoxDb was sequenced. Two upstream genes (chrna1b (cholinergic receptor nicotinic alpha polypeptide) and lnpb (lunapark b)) confirm that there are only two Hox genes in this cluster, hoxd9b and hoxd4b, and that hoxd11b, which is present in HoxDb clusters of stickleback and the two pufferfishes was lost in cichlids. Downstream of hoxd4b, we identified the complete coding region of mtx2 (metaxin 2).
In general, the neighboring genes that were identified were orthologous to those in Takifugu rubripes. From other fish genomes, the neighbouring genes were not identified in such detail. Based on blast hits adjacent to the Hox genes, it appears that gene order is conserved generally in teleost species.
Phylogenetic analyses
Since there is no prior phylogenetic study including all of the model organisms that were part of our study, we performed a phylogenetic analysis in order to be able to trace gene loss events and the evolutionary history of the Hox clusters in an accurate phylogenetic framework. Based on alignments of coding regions, we selected 24 Hox genes (Table 1) for which orthologs had been identified both in human and Xenopus tropicalis and the complete dataset for seven teleost species: two cichlids (Astatotilapia burtoni, Oreochromis niloticus), medaka (Oryzias latipes), stickleback (Gasterosteus aculeatus), two pufferfishes (Tetraodon nigroviridis, Takifugu rubripes) and zebrafish (Danio rerio). When two teleost paralogs were available, we were careful to choose the more slowly evolving copy based on a preliminary tree to reduce the potentially detrimental effects of introducing noise into the dataset. We excluded positions that could not be aligned and concatenated 24 genes for a complete dataset of nine species and 20,009 basepairs. Modelgenerator [76] identified GTR + G (alpha = 0.53) as the best fitting model. With these parameters we ran PhyML [77] with 500 bootstrap replicates and MrBayes 3.1 for 1,000,000 generations, sampling every 10th generation and with a burn-in of 5000. We obtained a fully resolved tree with maximal support for all nodes using both methods (Figure 2).

Maximum likelihood tree based on 20,009 nucleotide positions of Hox genes. Values above branches are Maximum Likelihood bootstraps; two asterisks indicate posterior probabilities of 1.00 as obtained by MrBayes 3.1.
Danio rerio, the only ostariophysian species for which a complete set of Hox clusters is currently available, is the sister group of the Neoteleosts, hence all other species included in this study. Within the Neoteleosts, two clades were recovered: firstly the pufferfishes, which form a monophyletic group and secondly, a clade consisting of stickleback, medaka and the cichlids, with a sister group relationship of O. latipes and the two cichlid species. The close relationship of cichlids with medaka has been previously described based on nuclear genes [78] and on ESTs [79].
Gene loss and loss of microRNAs in the teleost Hox clusters
We identified 46 functional coding sequences for Hox genes and one recent pseudogene in Astatotilapia burtoni. Based on the tree obtained, we traced events of gene loss and loss of microRNAs among these major fish model systems (Figure 3). The most salient gene losses that can be traced with confidence without complete data on basal teleosts and non-duplicated actinopterygians happened after the divergence of the Ostariophysii and Neoteleostei while most gene losses probably immediately followed the FSGD. Based on Danio rerio, the Ostariophysii have lost seven genes since the last hypothetical common ancestor with the Neoteleosts. During the evolution of the Neoteleosts eight Hox genes were lost. If we assume a divergence time of 290–304 mya between Ostariophysii and Neosteleostei [80, 81] and an age of the genome duplication of 320–350 mya [19, 20], at least thirty-one genes were lost within the 50 mya following the FSGD [31] and only 7–8 during the last 300 mya. This corroborates previous findings of high initial gene loss rates immediately following a large scale duplication event [82–84].

Hox cluster of teleost model fish species and the event of gene loss plotted on a phylogeny. Hox and Evx genes are shown as arrows, pseudogenes are shown without coloration and missing delineation indicates missing sequence data of most likely existing genes. MicroRNAs are drawn as small diamonds and were added according to our analyses. Data for H. sapiens were copied from [86] and the mir-10-db of Danio rerio according to [85].
The pufferfish lineage lost three genes in the common lineage leading to Takifugu and Tetraodon (Figure 3). Gasterosteus aculeatus has the most complete set Hox genes, only the hoxb6b gene has not been identified so far, most likely due to a large sequence gap in the genomic contig, leaving open the possibility that it was not been lost. The loss of hoxd11b in both medaka and cichlids supports their monophyletic grouping in a parsimony framework. Both species also lost the hoxb7a gene, but due to independent events, as the existence of a functional hoxb7a gene in another cichlid species (Oreochromis niloticus) implies.
The sets of microRNAs in ostariophysian and neoteleost Hox clusters are similar but not identical. An equivalent to the mir196-Cb could not be identified in neoteleosts, which have lost the entire HoxCb cluster. So far, the assembly of the D. rerio genome is still incomplete and the HoxCb cluster is not contained in a single contig, therefore the identification of its neighboring genes and thus, the corresponding regions in Neoteleosts is not yet possible. D. rerio retains the mir10-Db copy between the lunapark b and metaxin2 genes, even though the hox genes in this genomic region have been lost [85]. In medaka, we were not able to identify mir196-Ab, mir196-Ba and mir10-Bb, even though sequences were complete and without gaps in these intergenic regions. Therefore these microRNAs might have been lost in the medaka lineage. In contrast to a previous study, we were able to identify mir196-Ab and mir196-Ba in the zebrafish clusters [86], probably due to increased sequence quality of the genomic sequence.
Analyses of Conserved Non-coding Sequences (CNS)
We performed analyses of CNS using the program Tracker [52] with orthologous teleost Hox clusters. The datasets analyzed included 3 kb of additional sequence on both ends of the cluster. This rule was only changed for HoxCa clusters of both pufferfishes, where we used the complete genomic sequence up to the next downstream gene, cbx5, in order to be able include the pseudogene hoxc3a in this analysis. In the analysis of the HoxBb clusters we included upstream sequences to the end of the TTLL6 coding sequence to identify possible conserved sequences that surround hoxb8b in Danio rerio and may still exist in other fish, where this gene was lost. Also for HoxDb clusters in Oryzias latipes and Astatotilapia burtoni, the 5' region was extended until the beginning of the lnb gene, since we were not able to find a gene or pseudogene of hoxd11b with other methods in this species and we wanted to include any possible CNSs. For the pufferfishes, the 3' overlap had to be shortened because the intergenic region between hoxd4b and mtx2 is shorter than 3 kb. For a visual analysis, we also constructed mVista plots [87] based on LAGAN alignments [88] that are provided in Additional files 1, 2, 3, 4, 5, 6, 7. MicroRNAs are marked in green.
For the HoxAa cluster, we obtained a total of 192 footprint cliques (FCs, clusters of conserved footprints forming a single alignment), which add up to a total length of 34.4 kb (37%) in A. burtoni (total cluster length 92.2 kb) (Figure 4). The sequence included all hox genes plus evx1. For the most part the identified CNS are teleost specific, i.e. present in all fish species included, (14%) or neo-teleost specific (15%, present in all species except zebrafish) (Figure 4). One of the teleost specific cliques contains mir196-Aa [see Additional File 1]. However, we found more CNS shared only between A. burtoni and Gasterosteus aculeatus than between A. burtoni and Oryzias latipes. This is in interesting disagreement with the phylogenetic hypothesis (Figure 2). Comparisons of the lengths of CNS relative to sequence length in intergenic regions along one cluster show a tendency to increase towards the anterior end of the cluster (Figure 5).

Percentage of CNS within intergenic regions of the Hox clusters of Neoteleost fishes. Starting from the complete length of analyzed sequence, we calculated the relative amounts of genes (including introns), PFC (as identified by Tracker) and marked the remaining sequence as "junk". The footprint cliques were further divided as shared by all six fish species included (teleost), shared by all species except zebrafish (neoteleost), shared by medaka, cichlid and stickleback (Ol-Ab-Ga) or shared by cichlid and stickleback (Ab-Ga). Against our expectations there were usually no or only very few cliques shared only between cichlid and medaka except for HoxDb.

"Proportional" analyses of the Hox clusters of Astatotilapia burtoni. Large error bars for anterior regions of HoxCa cluster are explained by missing data from the pufferfish, which lost the hoxc3a gene.
The HoxAb cluster is shorter (total length in A. burtoni: 33.2 kb) than the HoxAa cluster, contains only five genes and 53 PFCs could be identified, which cover a total of 7.6 kb (23%) of the A. burtoni HoxAb cluster. The average content of cliques in intergenic regions is lower than in the HoxAa cluster (Figure 4). While the proportion of coding sequence of the entire Hox cluster is about the same as in the HoxAa cluster, the CNS make up a much smaller portion of the total cluster length (37% in the HoxAa cluster, but only 23% in HoxAb cluster). Most of the CNSs are evolutionarily conserved and shared between all teleosts or at least between the neoteleosts. Also here we find more similarities between cichlid and stickleback than between cichlid and medaka. The footprint clique containing mir196-Ab did not identify a orthologous sequence for medaka, suggesting a lineage-specific loss of this microRNA in medaka [see Additional File 2].
For the HoxBa cluster, we included the partial cluster of A. burtoni (total length 135.0 kb) that we obtained in this study. For the statistical analyses we also compared data from stickleback (total length 233.3 kb) since it showed more similarities in terms of CNS to the cichlid than medaka. For all species except A. burtoni, the compared HoxBa cluster sequence included the sequence from hoxb13a to hoxb1a. In total, the analyses identified 311 footprint cliques, adding up to a total length of 13.3 kb in A. burtoni (10%) and 26.7 kb in G. aculeatus (11%). Comparing the relative proportions of coding sequences, CNS and non-conserved non-coding sequences are similar between G. aculeatus and A. burtoni, implying that the partial cluster is representative of the rest of this Hox gene cluster. Also, the distributions of amounts of CNS in the intergenic regions along the cluster are similar in both species (Figure 4). In both species, there is a long stretch of sequence between hoxb13a and hoxb9a that does not contain any footprint but gives BLAST hits to repetitive elements from the same species. The analyses revealed the existence of mir196-Ba in all teleosts – except for medaka – and mir10-Ba was found in all species but A. burtoni, due to missing sequence data [see Additional File 3]. While in terms of gene number the cluster is still complete, the CNS content is low (in comparison to other clusters), which is probably also an effect of the large intergenic regions.
The small HoxBb cluster had a total length of 29.2 kb, Tracker identified 70 FC, and the CNSs covered a total of 8.7 kb in A. burtoni. In contrast to its big "sister" HoxBa cluster, the CNSs make up a higher percentage of the cluster than the coding regions and the distribution of CNSs in the intergenic regions display a high density of conserved sites. The extremely short intergenic region between hoxb6b and hoxb5b results in the high peak seen in Figure 5d. Interestingly, the mir10-Bb could be identified in all species except again for O. latipes [see Additional File 4]. It is interesting to note that the medaka Hox clusters seems to lose microRNAs apparently more frequently than other fish lineages.
The HoxCa cluster contains 10 genes and has a complete length of 96.5 kb in A. burtoni. 38% of the clusters are CNS (37.0 kb) identified in the Tracker analysis, and make up more than the complete length of coding sequences (22%). The CNSs were mainly teleost and neoteleost-specific, and we could identify two microRNAs in all species: mir196-Ca and mir10-Ca [see Additional File 5]. In comparison with other clusters, the CNS density is high and there is also a slight increase in CNS length towards the anterior end of the cluster (Figure 5f).
For the HoxDa cluster, we also included the evx-2 gene and we could identify 113 footprint cliques. The sequence has a complete length of 53.9 kb in A. burtoni and the CNS take up 18.2 kb (34%) of it. Most of these are teleost specific (21%). The distribution shows a peak 5' of evx-2, where a highly conserved sequence has been described before [89]. Otherwise there is a noticeable trend for more CNS towards the anterior end of the HoxDa cluster [see Additional File 6].
The HoxDb cluster fragment analyzed for A. burtoni spanned 38.8 kb, reaching from the end of lnpb to 3 kb downstream of hoxd4b. We obtained 60 footprint cliques, the conserved sequences in A. burtoni have a total length of 5.3 kb (14%). The CNS are mainly neoteleost specific, due to the loss of the Danio rerio Hoxdb cluster, teleost specific CNS could not be detected. Our analysis also found the mir10-db in all species [see Additional File 7].
In order to be able to recognize general trends of sequence conservation of CNS in Hox clusters, we calculated the percentage of CNS for each intergenic region. Figure 5 shows the average of that percentage within the neoteleosts. We excluded zebrafish from this calculation, since the values were much lower due to evolutionary distance and the different gene setup of clusters made it difficult to include these data. Based on our analyses, the clusters with the highest percentage of CNS in their intergenic regions are HoxAa, HoxBb, HoxCa, and HoxDa (Figure 5a,d–f). It might have been expected therefore that HoxAa, HoxCa and HoxDa are also the clusters where most of the genes are conserved, while HoxBb only has retained four and yet, surprisingly, retained large sets of conserved CNS. Here, the high percentage of CNS (Figure 5d) is most likely caused by the high "gene density" of the cluster and the short intergenic regions. Whereas, the long intergenic regions of the HoxBa cluster "dilute" the CNS in this genomic region (Figure 5c). The anterior part of the HoxCa gene is very heterogenic in terms of CNS content due to gene loss in the pufferfishes (Figure 5e). HoxAb and HoxDb have also lost genes (by comparison to their paralogous clusters) and, correspondingly, CNS (Figure 5b,g). A general interesting trend that is observed in all clusters (except HoxAb) is a maximal peak of CNS towards the anterior end of the clusters (Figure 5).
Discussion
We screened the BAC library of A. burtoni for Hox-positive clones and identified clones with complete clusters by PCR of the 5' and 3' most genes. Since obtained sequence data included surrounding genes for most clusters, we can be certain that our analysis misses only maximally four genes of the HoxBa cluster. Through PCR, sequence data for those genes (hoxb4a, hoxb3a, hoxb2a and hoxb1a) and partial non-coding data of this region were also obtained and, therefore, we can safely assume that these genes are clustered also in A. burtoni. Our analyses of the non-coding area of the partial HoxBa cluster of A. burtoni in comparison with the complete cluster of G. aculeatus shows that the features of this Hox cluster of these two species are similar. The Hox gene content is almost identical to that of Oreochromis niloticus [40]. There are two exceptions: we were not able to identify any trace of a hoxd11b gene in the BAC library of A. burtoni. A previously described sequence, which claimed to be from a cichlid [40] is almost identical to Tetraodon nigroviridis (169 of 171 base pairs identical), including a part of the additional intron this gene has acquired. This suggests that this is not an Oreochromis sequence (AY757355) as claimed by Santini and Bernardi [40] but rather is indeed simply previously published Tetraodon nigroviridis sequence instead. We strongly suspect that a large portion of the Oreochromis sequences of that study [40] were taken from an unpublished data set of the Meyer laboratory which was not collected by S. Santini but published fraudulently under her name. The proposed cichlid genome projects on Oreochromis and Astatotilapia will also aid in the clarification of this matter.
The other difference between the cichlids is the existence of a stop codon in the sequence of hoxb7a of A. burtoni while the coding sequence of Oreochromis niloticus is still intact [[40], Hoegg et al. unpublished data]. This implies not only that hoxb7a was lost independently in different lineages of fish such as in the lineages leading to pufferfish or medaka, but also in at least part of the cichlid fish radiation, suggesting that it is not essential and can be lost easily and repeatedly. However, some selective forces apparently did prevent it from being deleted for probably hundreds of millions of years since the fish-specific genome duplication [20]. More detailed analyses of its expression, the exact phylogenetic timing of gene loss and its possible implications for speciation in haplochromine cichlids will be needed to investigate this further. Differences in gene content of the Hox gene clusters, the essential developmental toolkit, that differentiate two species of closely related African cichlid fish species is a rather unexpected finding.
We performed a phylogenetic analysis based on a dataset of 24 Hox genes from seven fish species and two outgroup species (human and frog) and obtained a single, highly supported tree (Figure 2), which shows a monophyletic group of G. aculeatus, O. latipes and the cichlids A. burtoni and O. niloticus. The close relationship of Beloniformes (O. latipes) and Perciformes (A. burtoni/O. niloticus) is in agreement with recent molecular phylogenies [78, 79] and rejects the monophyly of Smegmamorpha, a clade that contains Beloniformes and Gasterosteiformes but not Perciformes. Even though the Order Perciformes is not monophyletic itself [62], more data including more species will be required to resolve the complete phylogeny. This will be necessary in the future to make assumptions about genomic evolution in the neoteleost fishes within the correct phylogenetic framework. A new phylogeny and a new classification of the highly diverse clade Percomorpha is especially required. For our species set used, we are confident in the tree, especially since it is also fully congruent with the inferred gene loss patterns (Figure 2). Only the position of G. aculeatus cannot be determined with certainty by the parsimony approach of gene losses.
Also the Hox phylogeny indicates a close relationship of cichlids and medaka to the exclusion of other orders in the Percomorpha such as the pufferfishes and the sticklebacks. Interestingly, the analyses of CNS, however, show consistently a higher similarity between G. aculeatus and A. burtoni than between O. latipes and A. burtoni (Figure 4). CNS shared only between O. latipes and A. burtoni where consistently too few to show, except for HoxDb. Also, of nine miRNAs contained in the other neoteleostean species, three were lost in the O. latipes cluster (Figure 3). In general, we find a high variability of gene content within teleost fish, no two of the species examined so far had the same gene content in their clusters. This might be due to the redundancy that was created by the FSGD, and that still permits gene loss without major consequences on the bodyplan. In tetrapods, it is assumed that the Hox gene setup is more conserved. However, data from the frog Xenopus tropicalis [31] and from the coelacanth Latimeria menadoensis [90] show that there is variation also among sarcopterigians. When more tetrapod lineages are examined, it will become more clear if and how much more variable fish clusters are.
The finding of greater similarity between sticklebacks and cichlids in regulatory elements suggests however, that the medaka genome evolves at a higher rate, at least in non-coding sequences. Why this should be so deserves further attention. The coding sequences also show a slightly accelerated rate of evolution in comparison to other neoteleost species (Figure 2). The increase in evolutionary rate rather seems to be a Hox-specific trend; a study on differential rates of duplicate genes more often identified slower evolutionary rates in medaka rather than accelerated ones [44]. Since the sequences of the O. latipes Hox clusters are not directly taken from a genome sequencing project but from sequenced BAC clones with only few, small gaps, possible artifacts due to potential assembly problems of the medaka genome can be excluded.
The analyses of CNS of A. burtoni showed as well that the major part, or at least the longest part, of the potential regulatory elements is conserved between all teleost species included in this study, or at least between neoteleost species. This can also be seen as an indication that the analysis parameters have been chosen correctly, are conservative and do not tend to overestimate the number of phylogenetic footprints. This also indicates that even though the gene content in Hox clusters in fish is more variable than previously thought, the main regulatory elements are highly conserved. That gene loss and gain even in such important genes such as Hox genes, even among relatively closely related species, might imply that the putative paramount importance of regulatory evolution in bringing about phenotypic change during evolution is not as great as generally believed.
We could also show that there is a trend towards more CNS in the anterior portion of the cluster (Figure 5) which has been described before for the HoxAa cluster based on a different analytical method [59]. Knockout studies on other vertebrates showed that a complete knockdown for paralogy group 1 in Xenopus results in serious developmental defects [91], and mutations in HoxA1 in humans are linked with Bosley-Salih-Alorainy syndrome effecting delayed development, eye movement and formation of the cranial nerve VIII [92]. Several mutations for posterior genes have been described in HoxA13 (Hand-foot-genital syndrome [93]) and HoxD13 (Synpolydactyly [94]). This might imply that anterior Hox genes are usually buffered by functional redundancy of paralogs and any mutation in the CNS in those anterior Hox-clusters would be expected to be severe and most likely lethal. Therefore, the regulation of anterior Hox genes would be expected to be more important for the patterning and survival of an embryo than those of posterior genes, which do not affect the brain and head development to the same extent. Similar conclusions were drawn from the observation that among the vertebrates, sequence divergence between posterior genes is higher than between more anterior genes, an effect termed "laxitas terminalis" [95]. The authors of this study suggest that posterior genes are not linked to basal vertebrate functions but rather fulfill lineage specific functions. In a comparison among all posterior Hox genes among more distantly related phyla, it was found that deuterostome posterior genes are evolving faster than their protostome counterparts as well as the deuterostome anterior genes [96]. This "posterior flexibility" also indicates a higher conservation of the anterior part of the clusters.
Our study also shows that different Hox clusters are evolving with distinct patterns in different evolutionary lineages, even though an overall evolutionary trend can be observed: that after duplication, one cluster retains more genes and also the regulatory elements that go with them while the paralogous cluster loses genes and conserved elements concomitantly more rapidly and possibly more easily due to relaxed constraints. Our data also demonstrate clearly that the loss of Hox genes in teleost clusters is an ongoing process that occurred even within the last seven million years within the cichlid family (hoxb7a). A close comparison among different closely related genes might also provide insight into species-specific differences and the potential influence of regulatory evolution on different cichlid species.
Conclusion
Hox clusters in fish are more variable in gene content than expected and also, each cluster has its own characteristics in terms of absolute length and content of CNS. While genes have continuously been lost, somewhat surprisingly most microRNAs remained unchanged (with the notable exception in the medaka lineage). The CNSs form a large portion of Hox clusters, usually even more basepairs than the coding regions and are, typically, conserved over very long evolutionary time spans. Their distribution is not constant along the cluster but the maximum frequency of occurrence is usually towards the anterior end, implying stronger selection on the anterior Hox gene expression patterns, while the more posterior Hox genes are more free to vary.
Methods
DNA extraction and PCR
DNA was extracted from muscle tissue or fins from specimens stored at -80°C following a standard phenol-chloroform protocol. PCRs were performed in 25 μl reactions using 0.5–1 units of RedTaq (Genaxxon, Germany) and the corresponding reaction buffer (10 mM Tris-HCI (pH 9.0 at 25°C), 50 mM KCl, 1.5 mM MgCl2, 0.1% Triton X-100), 1 mM additional MgCl2, 0.6 mM dNTPs (Genaxxon, Germany), 0.4 μM primers and for long fragments 0.1 unit of Pwo polymerase (Fermentas, Germany) was added. PCR used an initial denaturation step at 94°C for 3 minutes, followed by 35 cycles with 20 sec at 94°C, 40 seconds at 58°C and 2.5 minutes at 68°C, and a final extension step at 68°C for 7 minutes. PCR products were checked on 1% agarose gels running in 1× TAE buffer containing 0.05% ethidium bromide. PCR fragments were purified directly via spin columns (PEQLAB, Germany) or were cut from preparative agarose gels (1%) using the gel extraction kit (QIAGEN, Germany).
Primer design
Primers were designed in conserved regions of the coding sequence, preferably the forward primer at the beginning of the first exon and the reverse primer in the second exon outside the homeodomain, so the PCR fragment would cover the intron and yield specific fragments for screening of the BAC library. (Primer sequences are provided in Additional file 8).
Screening of the Astatotilapia burtoni BAC library and plasmid preps
The BAC library was spotted on four filters containing 18,432 clones each. The screening using chemiluminescence was conducted according to Lang et al. (2006). Positive clones were picked from the library and grown overnight in Luria Broth Base medium (Invitrogen™) containing 12.5 μg/ml chloramphenicol. Plasmids were isolated using a modification of a standard plasmid miniprep protocol [97]. Gene content of the BAC clones were confirmed by PCR for 5' and 3' most Hox/Evx genes. For size estimation, BACs were digested with NotI and ran on a pulse-field gel apparatus.
Shotgun sequencing of BAC clones
BAC clone DNA was isolated from each preparation, and then sheared into random fragments of approximately 3 kb by repeated passage through a narrow aperture using a Hydroshear device. These fragments were repaired to blunt ends using T4 polymerase and Klenow fragment, and then a narrow distribution of sizes was selected from an agarose gel. These fragments were ligated into plasmid vector, introduced into E. coli by electroporation and then plated on nutrient agar. A random selection of these clones was processed for sequencing reads from each end using rolling circle amplification of the plasmids, sequencing reactions using BigDye terminators (ABI), cleanup using solid phase reversible immobilization (SPRI), then sequence determination on an ABI 3730 × l automated DNA sequencer.
Sequence assembly
Raw sequences were trimmed for vector sequences and sequence quality was scored with Phred. Contigs were assembled automatically using Sequencer™ using a minimal overlap of 17 nucleotides and a minimal identity of 85% and refined and corrected manually. Sequence gaps were closed by PCR using sequence specific primers designed with Primer3.
Annotation
Genes were annotated manually by pairwise BLAST and based on alignments of available sequences from other species. We also performed BLAST searches against the EST sequences available for A. burtoni as well as two other haplochromine cichlids (Haplochromis chilotes, Haplochromis sp. 'red tail sheller') [98], especially for a better annotation of adjacent non-Hox genes. BAC clone sequences were submitted to GenBank (accession numbers EF594310–EF594316).
Database searches and phylogenetic analyses
Complete Hox cluster sequences were downloaded from GenBank (Homo sapiens, Oryzias latipes, Takifugu rubripes) [99], the Joint Genome Institute (Xenopus tropicalis) [100], Ensembl (Danio rerio (Zv6), Gasterosteus aculeatus (BROAD S1)) [101], and Genoscope (Tetrodon nigroviridis) [102] [for accession numbers see Additional file 9]. Coding sequences were aligned based on their amino acid sequences with their respective orthologs using ClustalW as implemented in Bioedit. Regions that could not be aligned with confidence were omitted from the phylogenetic analyses [the final alignment is given in Additional file 10]. For the concatenated datasets, only genes that were available for both tetrapod outgroup species were used (H. sapiens and X. tropicalis) as well as for all seven fish species (A. burtoni, O. niloticus, O. latipes, G. aculeatus, T. rubripes, T. nigroviridis, D. rerio). For genes with two paralogs in fish we selected the slower evolving copy to avoid additional noise in the dataset. We performed a Maximum Likelihood analysis using PhyML [77]with 500 bootstrap replicates as well as an analysis based on Bayesian Inference with the MrBayes 3.1 [103] software for 1,000,000 generations and a burn-in of 5,000 with sampling every 10th generation.
Analyses of non-coding sequences
Genomic regions were prepared for analyses including 3 kb of sequence upstream of the first Hox gene and downstream of the 3'-most Hox gene. For HoxBb clusters, sequences between ndp52 (5') and scap1 (3') were used since D. rerio also has a Hoxb8b gene, which was lost in all neoteleosts for which this genomic information is available, since we wanted to avoid losing sequence information. In both pufferfish HoxCa clusters, sequences up to the next downstream gene (cbx5) were used because this lineage has lost hoxc3a. For species that had lost the hoxd11b gene (Astatotilapia burtoni, Oryzias latipes) sequence data until lnb (lunapark b) were included. For an overview as well as for a visual display, we used mVISTA [87] based on LAGAN multiple alignments [88]. For more detailed analyses, we used the program Tracker [52], using more stringent than default parameters (minimal BlastZ score 2000, minimum identity 85%) since the sequences analyzed here are more closely related than those used in previous studies. Phylogenetic footprint cliques obtained through Tracker were checked carefully for double hits of the same alignments and microsatellites that were eliminated. Footprint cliques containing microRNAs were identified.
References
Ohno S: Evolution by gene duplication. 1970, New York: Springer-Verlag
Sidow A: Gen(om)e duplications in the evolution of early vertebrates. Curr Opin Genet Dev. 1996, 6: 715-722. 10.1016/S0959-437X(96)80026-8.
Carroll SB, Grenier JK, Weatherbee SD: From DNA to diversity. 2001, Abingdon: Blackwell Science
Levine M, Tjian R: Transcription regulation and animal diversity. Nature. 2003, 424: 147-151. 10.1038/nature01763.
Davidson EH, McClay DR, Hood L: Regulatory gene networks and the properties of the developmental process. Proc Natl Acad Sci USA. 2003, 100: 1475-1480. 10.1073/pnas.0437746100.
Stephens SG: Possible significance of duplication in evolution. Advanced Genetics. 1951, 4: 247-265.
Meyer A, Schartl M: Gene and genome duplications in vertebrates: the one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr Opin Cell Biol. 1999, 11: 699-704. 10.1016/S0955-0674(99)00039-3.
Furlong RF, Holland PWH: Were vertebrates octoploid?. Phil Trans R Soc Lond B Biol Sci. 2002, 357: 531-544. 10.1098/rstb.2001.1035.
Holland PW: More genes in vertebrates?. J Struct Funct Genomics. 2003, 3: 75-84. 10.1023/A:1022656931587.
Dehal P, Boore JL: Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005, 3: e314-10.1371/journal.pbio.0030314.
Larhammar D, Lundin LG, Hallbook F: The human Hox-bearing chromosome regions did arise by block or chromosome (or even genome) duplications. Genome Res. 2002, 12: 1910-1920. 10.1101/gr.445702.
Lundin LG, Larhammar D, Hallbook F: Numerous groups of chromosomal regional paralogies strongly indicate two genome doublings at the root of the vertebrates. J Struct Funct Genomics. 2003, 3: 53-63. 10.1023/A:1022600813840.
Taylor JS, Braasch I, Frickey T, Meyer A, Van de Peer Y: Genome duplication, a trait shared by 22,000 species of ray-finned fish. Genome Res. 2003, 13: 382-390. 10.1101/gr.640303.
Taylor JS, Van de Peer Y, Braasch I, Meyer A: Comparative genomics provides evidence for an ancient genome duplication event in fish. Phil Trans R Soc Lond B Biol Sci. 2001, 356: 1661-1679. 10.1098/rstb.2001.0975.
Hoegg S, Brinkmann H, Taylor JS, Meyer A: Phylogenetic timing of the fish-specific genome duplication correlates with the diversification of teleost fish. J Mol Evol. 2004, 59: 190-203. 10.1007/s00239-004-2613-z.
Naruse K, Fukamachi S, Mitani H, Kondo M, Matsuoka T, Kondo S, Hanamura N, Morita Y, Hasegawa K, Nishigaki R, Shimada A, Wada H, Kusakabe T, Suzuki N, Kinoshita M, Kanamori A, Terado T, Kimura H, Nonaka M, Shima A: A detailed linkage map of medaka, Oryzias latipes: comparative genomics and genome evolution. Genetics. 2000, 154: 1773-1784.
Postlethwait JH, Woods IG, Ngo-Hazelett P, Yan YL, Kelly PD, Chu F, Huang H, Hill-Force A, Talbot WS: Zebrafish comparative genomics and the origins of vertebrate chromosomes. Genome Res. 2000, 10: 1890-1902. 10.1101/gr.164800.
Jaillon O, Aury J-M, Brunet F, Petit J-L, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A, Nicaud S, Jaffe D, Fisher S, Lutfalla G, Dossat C, Segurens B, Dasilva C, Salanoubat M, Levy M, Boudet N, Castellano S, Anthouard V, Jubin C, Castelli V, Katinka M, Vacherie B, Biemont C, Skalli Z, Cattolico L, Poulain J: Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004, 431: 946-957. 10.1038/nature03025.
Christoffels A, Koh EG, Chia JM, Brenner S, Aparicio S, Venkatesh B: Fugu genome analysis provides evidence for a whole-genome duplication early during the evolution of ray-finned fishes. Mol Biol Evol. 2004, 21: 1146-1151. 10.1093/molbev/msh114.
Vandepoele K, De Vos W, Taylor JS, Meyer A, Van de Peer Y: Major events in the genome evolution of vertebrates: Paranome age and size differs considerably between ray-finned fishes and land vertebrates. Proc Natl Acad Sci USA. 2004, 101: 1638-1643. 10.1073/pnas.0307968100.
Crow KD, Stadler PF, Lynch VJ, Amemiya C, Wagner GP: The "fish-specific" Hox cluster duplication is coincident with the origin of teleosts. Mol Biol Evol. 2006, 23: 121-136. 10.1093/molbev/msj020.
de Souza FS, Bumaschny VF, Low MJ, Rubinstein M: Subfunctionalization of expression and peptide domains following the ancient duplication of the proopiomelanocortin gene in teleost fishes. Mol Biol Evol. 2005, 22: 2417-2427. 10.1093/molbev/msi236.
Mulley JF, Chiu CH, Holland PW: Breakup of a homeobox cluster after genome duplication in teleosts. Proc Natl Acad Sci USA. 2006, 103: 10369-10372. 10.1073/pnas.0600341103.
Acampora D, D'Esposito M, Faiella A, Pannese M, Migliaccio E, Morelli F, Stornaiuolo A, Nigro V, Simeone A, Boncinelli E: The human HOX gene family. Nucleic Acids Res. 1989, 17: 10385-10402. 10.1093/nar/17.24.10385.
Garcia-Fernandez J, Holland PW: Archetypal organization of the amphioxus Hox gene cluster. Nature. 1994, 370: 563-566. 10.1038/370563a0.
Lewis EB: A gene complex controlling segmentation in Drosophila. Nature. 1978, 276: 565-570. 10.1038/276565a0.
Negre B, Casillas S, Suzanne M, Sanchez-Herrero E, Akam M, Nefedov M, Barbadilla A, de Jong P, Ruiz A: Conservation of regulatory sequences and gene expression patterns in the disintegrating Drosophila Hox gene complex. Genome Res. 2005, 15: 692-700. 10.1101/gr.3468605.
Seo HC, Edvardsen RB, Maeland AD, Bjordal M, Jensen MF, Hansen A, Flaat M, Weissenbach J, Lehrach H, Wincker P, Reinhardt R, Chourrout D: Hox cluster disintegration with persistent anteroposterior order of expression in Oikopleura dioica. Nature. 2004, 431: 67-71. 10.1038/nature02709.
Ikuta T, Yoshida N, Satoh N, Saiga H: Ciona intestinalis Hox gene cluster: Its dispersed structure and residual colinear expression in development. Proc Natl Acad Sci USA. 2004, 101: 15118-15123. 10.1073/pnas.0401389101.
Aboobaker A, Blaxter M: Hox gene evolution in nematodes: novelty conserved. Curr Opin Genet Dev. 2003, 13: 593-598. 10.1016/j.gde.2003.10.009.
Hoegg S, Meyer A: Hox clusters as models for vertebrate genome evolution. Trends Genet. 2005, 21: 421-424. 10.1016/j.tig.2005.06.004.
Meyer A, Van de Peer Y: From 2R to 3R: evidence for a fish-specific genome duplication (FSGD). Bioessays. 2005, 27: 937-945. 10.1002/bies.20293.
Powers TP, Amemiya CT: Evolutionary plasticity of vertebrate Hox gene. Curr Genom. 2004, 5: 459-472. 10.2174/1389202043349048.
Mahadevan NR, Horton AC, Gibson-Brown JJ: Developmental expression of the amphioxus Tbx1/10 gene illuminates the evolution of vertebrate branchial arches and sclerotome. Dev Genes Evol. 2004, 214: 559-566. 10.1007/s00427-004-0433-1.
Cameron RA, Rowen L, Nesbitt R, Bloom S, Rast JP, Berney K, Arenas-Mena C, Martinez P, Lucas S, Richardson PM, Davidson EH, Peterson KJ, Hood L: Unusual gene order and organization of the sea urchin hox cluster. J Exp Zoolog B Mol Dev Evol. 2006, 306: 45-58. 10.1002/jez.b.21070.
Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, Westerfield M, Ekker M, Postlethwait JH: Zebrafish hox clusters and vertebrate genome evolution. Science. 1998, 282: 1711-1714. 10.1126/science.282.5394.1711.
Kurosawa G, Takamatsu N, Takahashi M, Sumitomo M, Sanaka E, Yamada K, Nishii K, Matsuda M, Asakawa S, Ishiguro H, Miura K, Kurosawa Y, Shimizu N, Kohara Y, Hori H: Organization and structure of hox gene loci in medaka genome and comparison with those of pufferfish and zebrafish genomes. Gene. 2006, 370: 75-82. 10.1016/j.gene.2005.11.015.
Lee AP, Koh EG, Tay A, Brenner S, Venkatesh B: Highly conserved syntenic blocks at the vertebrate Hox loci and conserved regulatory elements within and outside Hox gene clusters. Proc Natl Acad Sci USA. 2006, 103: 6994-6999. 10.1073/pnas.0601492103.
Malaga-Trillo E, Meyer A: Genome duplications and accelerated evolution of Hox genes and cluster architecture in teleost fishes. Am Zool. 2001, 41: 676-686. 10.1668/0003-1569(2001)041[0676:GDAAEO]2.0.CO;2.
Santini S, Bernardi G: Organization and base composition of Tilapia Hox genes: implications for the evolution of Hox clusters in fish. Gene. 2005, 346: 51-61. 10.1016/j.gene.2004.10.027.
Amores A, Suzuki T, Yan Y-L, Pomeroy J, Singer A, Amemiya C, Postlethwait JH: Developmental roles of pufferfish hox clusters and genome evolution in ray-fin fish. Genome Res. 2004, 14: 1-10. 10.1101/gr.1717804.
Aury JM, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Segurens B, Daubin V, Anthouard V, Aiach N, Arnaiz O, Billaut A, Beisson J, Blanc I, Bouhouche K, Camara F, Duharcourt S, Guigo R, Gogendeau D, Katinka M, Keller AM, Kissmehl R, Klotz C, Koll F, Le Mouel A, Lepere G, Malinsky S, Nowacki M, Nowak JK, Plattner H: Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006, 444: 171-178. 10.1038/nature05230.
Taylor JS, Van de Peer Y, Meyer A: Genome duplication, divergent resolution and speciation. Trends Genet. 2001, 17: 299-301. 10.1016/S0168-9525(01)02318-6.
Steinke D, Salzburger W, Braasch I, Meyer A: Many genes in fish have species-specific asymmetric rates of molecular evolution. BMC Genomics. 2006, 7: 20-10.1186/1471-2164-7-20.
Blomme T, Vandepoele K, De Bodt S, Simillion C, Maere S, Van de Peer Y: The gain and loss of genes during 600 million years of vertebrate evolution. Genome Biol. 2006, 7: R43-10.1186/gb-2006-7-5-r43.
Maere S, De Bodt S, Raes J, Casneuf T, Van Montagu M, Kuiper M, Van de Peer Y: Modeling gene and genome duplications in eukaryotes. Proc Natl Acad Sci USA. 2005, 102: 5454-5459. 10.1073/pnas.0501102102.
De Bodt S, Maere S, Van de Peer Y: Genome duplication and the origin of angiosperms. Trends Ecol Evol. 2005, 20: 591-597. 10.1016/j.tree.2005.07.008.
Teichmann SA, Babu MM: Gene regulatory network growth by duplication. Nature Genetics. 2004, 36: 492-496. 10.1038/ng1340.
Carroll SB: Evolution at two levels: on genes and form. PLoS Biol. 2005, 3: e245-10.1371/journal.pbio.0030245.
Wagner GP, Takahashi K, Lynch V, Prohaska SJ, Fried C, Stadler PF, Amemiya C: Molecular evolution of duplicated ray finned fish HoxA clusters: Increased synonymous substitution rate and asymmetrical co-divergence of coding and non-coding sequences. J Mol Evol. 2005, 60: 665-676. 10.1007/s00239-004-0252-z.
Chiu C-h, Dewar K, Wagner GP, Takahashi K, Ruddle F, Ledje C, Bartsch P, Scemama J-L, Stellwag E, Fried C, Prohaska SJ, Stadler PF, Amemiya CT: Bichir HoxA cluster sequence reveals surprising trends in ray-finned fish genomic evolution. Genome Res. 2004, 14: 11-17. 10.1101/gr.1712904.
Prohaska SJ, Fried C, Flamm C, Wagner GP, Stadler PF: Surveying phylogenetic footprints in large gene clusters: applications to Hox cluster duplications. Mol Phylogenet Evol. 2004, 31: 581-604. 10.1016/j.ympev.2003.08.009.
Prohaska SJ, Fried C, Amemiya CT, Ruddle FH, Wagner GP, Stadler PF: The shark HoxN cluster is homologous to the human HoxD cluster. J Mol Evol. 2004, 58: 212-217. 10.1007/s00239-003-2545-z.
Wagner GP, Fried C, Prohaska SJ, Stadler PF: Divergence of conserved non-coding sequences: Rate estimates and relative rate tests. Mol Biol Evol. 2004, 21: 2116-2121. 10.1093/molbev/msh221.
Fried C, Prohaska S, Stadler PF: Exclusion of repetitive DNA elements from gnathostome Hox clusters. J Exp Zool B Mol Dev Evol. 2004, 302: 165-173.
Kessel M, Gruss P: Murine developmental control genes. Science. 1990, 249: 374-379. 10.1126/science.1974085.
Nelson CE, Morgan BA, Burke AC, Laufer E, DiMambro E, Murtaugh LC, Gonzales E, Tessarollo L, Parada LF, Tabin C: Analysis of Hox gene expression in the chick limb bud. Development. 1996, 122: 1449-1466.
Chiu C-h, Amemiya C, Dewar K, Kim CB, Ruddle FH, Wagner GP: Molecular evolution of the HoxA cluster in the three major gnathostome lineages. Proc Natl Acad Sci USA. 2002, 99: 5492-5497. 10.1073/pnas.052709899.
Santini S, Boore JL, Meyer A: Evolutionary conservation of regulatory elements in vertebrate hox gene clusters. Genome Res. 2003, 13: 1111-1122. 10.1101/gr.700503.
Nelson JS: Fishes of the world. 2006, Hoboken, New Jersey: John Wiley & Sons
Nelson JS: Fishes of the world. 1994, New York: Wiley
Miya M, Takeshima H, Endo H, Ishiguro NB, Inoue JG, Mukai T, Satoh TP, Yamaguchi M, Kawaguchi A, Mabuchi K, Shirai SM, Nishida M: Major patterns of higher teleostean phylogenies: a new perspective based on 100 complete mitochondrial DNA sequences. Mol Phylogenet Evol. 2003, 26: 121-138. 10.1016/S1055-7903(02)00332-9.
De Pinna MCC: Teleostean monophyly. Interrelationship of Fishes. Edited by: Stiassny MLJ, Parenti LR, Johnson GJ. 1996, San Diego: Academic Press, 147-162.
Arratia G: The monophyly of teleostei and stem-group teleosts. Mesozoic fish 2: Systematics and fossil record. Edited by: Arratia G, Schultze H. 1999, Munich: Pfeil
Inoue JG, Miya M, Tsukamoto K, Nishida M: Mitogenomic evidence for the monophyly of elopomorph fishes (Teleostei) and the evolutionary origin of the leptocephalus larva. Mol Phylogenet Evol. 2004, 32: 274-286. 10.1016/j.ympev.2003.11.009.
Salzburger W, Mack T, Verheyen E, Meyer A: Out of Tanganyika: genesis, explosive speciation, key-innovations and phylogeography of the haplochromine cichlid fishes. BMC Evol Biol. 2005, 5: 17-10.1186/1471-2148-5-17.
Salzburger W, Meyer A: The species flocks of East African cichlid fishes: recent advances in molecular phylogenetics and population genetics. Naturwissenschaften. 2004, 91: 277-290. 10.1007/s00114-004-0528-6.
Verheyen E, Salzburger W, Snoeks J, Meyer A: Origin of the superflock of cichlid fishes from Lake Victoria, East Africa. Science. 2003, 300: 325-329. 10.1126/science.1080699.
Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, Gelpke MD, Roach J, Oh T, Ho IY, Wong M, Detter C, Verhoef F, Predki P, Tay A, Lucas S, Richardson P, Smith SF, Clark MS, Edwards YJ, Doggett N, Zharkikh A, Tavtigian SV, Pruss D, Barnstead M, Evans C: Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002, 297: 1301-1310. 10.1126/science.1072104.
McKinnon JS, Rundle HD: Speciation in nature: the threespine stickleback model systems. Trends Ecol Evol. 2002, 17: 480-488. 10.1016/S0169-5347(02)02579-X.
Braasch I, Salzburger W, Meyer A: Asymmetric evolution in two fish-specifically duplicated receptor tyrosine kinase paralogons involved in teleost coloration. Mol Biol Evol. 2006, 23: 1192-1202. 10.1093/molbev/msk003.
Lang M, Miyake T, Braasch I, Tinnemore D, Siegel N, Salzburger W, Amemiya CT, Meyer A: A BAC library of the East African haplochromine cichlid fish Astatotilapia burtoni. J Exp Zoolog B Mol Dev Evol. 2006, 306: 35-44. 10.1002/jez.b.21068.
McClintock JM, Kheirbek MA, Prince VE: Knockdown of duplicated zebrafish hoxb1 genes reveals distinct roles in hindbrain patterning and a novel mechanism of duplicate gene retention. Development. 2002, 129: 2339-2354.
Hadrys T, Punnamoottil B, Pieper M, Kikuta H, Pezeron G, Becker TS, Prince V, Baker R, Rinkwitz S: Conserved co-regulation and promoter sharing of hoxb3a and hoxb4a in zebrafish. Dev Biol. 2006, 297: 26-43. 10.1016/j.ydbio.2006.04.446.
Mannaert A, Roelants K, Bossuyt F, Leyns L: A PCR survey for posterior Hox genes in amphibians. Mol Phylogenet Evol. 2006, 38: 449-458. 10.1016/j.ympev.2005.08.012.
Keane TM, Naughton TJ, McInerney JO: ModelGenerator: amino acid and nucleotide substitution model selection. 2004, National University of Ireland, Maynooth, Ireland
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704. 10.1080/10635150390235520.
Chen W-J, Orti G, Meyer A: Novel evolutionary relationship among four fish model systems. Trends Genet. 2004, 20: 424-431. 10.1016/j.tig.2004.07.005.
Steinke D, Salzburger W, Meyer A: Novel relationships among ten fish model species revealed based on a phylogenomic analysis using ESTs. J Mol Evol. 2006, 62: 772-784. 10.1007/s00239-005-0170-8.
Peng Z, He S, Wang J, Wang W, Diogo R: Mitochondrial molecular clocks and the origin of the major Otocephalan clades (Pisces: Teleostei): A new insight. Gene. 2006, 370: 113-124. 10.1016/j.gene.2005.11.024.
Kumazawa Y, Yamaguchi M, Nishida M: Mitochondrial molecular clocks and the origin of euteleostean biodiversity: Familial radation of perciforms may have predated the cretaceous/tertiary boundary. The Biology of Biodiversity. Edited by: Kato M. 1999, Hong Kong: Springer-Verlag, 35-52.
Brunet FG, Crollius HR, Paris M, Aury JM, Gibert P, Jaillon O, Laudet V, Robinson-Rechavi M: Gene loss and evolutionary rates following whole-genome duplication in teleost fishes. Mol Biol Evol. 2006, 23: 1808-1816. 10.1093/molbev/msl049.
Bowers JE, Chapman BA, Rong J, Paterson AH: Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003, 422: 433-438. 10.1038/nature01521.
Kellis M, Birren BW, Lander ES: Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004, 428: 617-624. 10.1038/nature02424.
Woltering JM, Durston AJ: The zebrafish hoxDb cluster has been reduced to a single microRNA. Nat Genet. 2006, 38: 601-602. 10.1038/ng0606-601.
Tanzer A, Amemiya CT, Kim CB, Stadler PF: Evolution of microRNAs located within Hox gene clusters. J Exp Zoolog B Mol Dev Evol. 2005, 304: 75-85. 10.1002/jez.b.21021.
Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I: VISTA : visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000, 16: 1046-1047. 10.1093/bioinformatics/16.11.1046.
Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, Green ED, Sidow A, Batzoglou S: LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13: 721-731. 10.1101/gr.926603.
Sabarinadh C, Subramanian S, Tripathi A, Mishra RK: Extreme conservation of noncoding DNA near HoxD complex of vertebrates. BMC Genomics. 2004, 5: 75-10.1186/1471-2164-5-75.
Koh EG, Lam K, Christoffels A, Erdmann MV, Brenner S, Venkatesh B: Hox gene clusters in the Indonesian coelacanth, Latimeria menadoensis. Proc Natl Acad Sci USA. 2003, 100: 1084-1088. 10.1073/pnas.0237317100.
McNulty CL, Peres JN, Bardine N, van den Akker WM, Durston AJ: Knockdown of the complete Hox paralogous group 1 leads to dramatic hindbrain and neural crest defects. Development. 2005, 132: 2861-2871. 10.1242/dev.01872.
Tischfield MA, Bosley TM, Salih MA, Alorainy IA, Sener EC, Nester MJ, Oystreck DT, Chan WM, Andrews C, Erickson RP, Engle EC: Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat Genet. 2005, 37: 1035-1037. 10.1038/ng1636.
Mortlock DP, Innis JW: Mutation of HOXA13 in hand-foot-genital syndrome. Nat Genet. 1997, 15: 179-180. 10.1038/ng0297-179.
Muragaki Y, Mundlos S, Upton J, Olsen BR: Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science. 1996, 272: 548-551. 10.1126/science.272.5261.548.
van der Hoeven F, Sordino P, Fraudeau N, Izpisua-Belmonte JC, Duboule D: Teleost HoxD and HoxA genes: comparison with tetrapods and functional evolution of the HOXD complex. Mech Dev. 1996, 54: 9-21. 10.1016/0925-4773(95)00455-6.
Ferrier DE, Minguillon C, Holland PW, Garcia-Fernandez J: The amphioxus Hox cluster: deuterostome posterior flexibility and Hox14. Evol Dev. 2000, 2: 284-293. 10.1046/j.1525-142x.2000.00070.x.
Sambrook J, Fritsch EF, Maniatis T: Molecular cloning – a laboratory manual. 1989, Cold Spring Harbour: Cold Spring Harbor Laboratory Press, 2:
The TIGR Gene Indices. [http://compbio.dfci.harvard.edu/tgi/cgi-bin/tgi/Blast/index.cgi]
National Center for Biotechnology Information. [http://www.ncbi.nlm.nih.gov]
JGI Database. [http://www.jgi.doe.gov/]
Ensembl Genome Browser. [http://www.ensembl.org/index.html]
Genoscope. [http://www.genoscope.cns.fr/externe/tetranew/]
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001, 17: 754-755. 10.1093/bioinformatics/17.8.754.
Acknowledgements
This work was supported by grants of the Deutsche Forschungsgemeinschaft and the University of Konstanz to AM, and the Landesgraduiertenförderung Baden-Württemberg to SH. DNA sequencing and some informatics support was provided by the U.S. Department of Energy Joint Genome Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
This study was conceived by SH, JLB and AM. The laboratory work was done by SH, except for shotgun sequencing of the BAC clones and sequence assembly, which was conducted by JVK. In silico analyses were performed by SH. The manuscript was drafted by SH and read and revised by all other authors.
Electronic supplementary material
12864_2007_1030_MOESM1_ESM.jpeg
{kind=link}
Additional file 1: Vista plot of HoxAa cluster based on LAGAN alignment with reference sequence Astatotilapia burtoni. (JPEG 1 MB)
12864_2007_1030_MOESM2_ESM.jpeg
{kind=link}
Additional file 2: Vista plot of HoxAb cluster based on LAGAN alignment with reference sequence Astatotilapia burtoni. (JPEG 719 KB)
12864_2007_1030_MOESM3_ESM.jpeg
{kind=link}
Additional file 3: Vista plot of HoxBa cluster based on LAGAN alignment with reference sequence Gasterosteus aculeatus. (JPEG 836 KB)
12864_2007_1030_MOESM4_ESM.jpeg
{kind=link}
Additional file 4: Vista plot of HoxBb cluster based on LAGAN alignment with reference sequence Astatotilapia burtoni. (JPEG 683 KB)
12864_2007_1030_MOESM5_ESM.jpeg
{kind=link}
Additional file 5: Vista plot of HoxCa cluster based on LAGAN alignment with reference sequence Astatotilapia burtoni. (JPEG 1 MB)
12864_2007_1030_MOESM6_ESM.jpeg
{kind=link}
Additional file 6: Vista plot of HoxDa cluster based on LAGAN alignment with reference sequence Astatotilapia burtoni. (JPEG 901 KB)
12864_2007_1030_MOESM7_ESM.jpeg
{kind=link}
Additional file 7: Vista plot of HoxDb cluster based on LAGAN alignment with reference sequence Tetraodon nigroviridis. (JPEG 550 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hoegg, S., Boore, J.L., Kuehl, J.V. et al. Comparative phylogenomic analyses of teleost fish Hox gene clusters: lessons from the cichlid fish Astatotilapia burtoni. BMC Genomics 8, 317 (2007). https://doi.org/10.1186/1471-2164-8-317
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-8-317