
Abstract
Background
Small G proteins, which are essential regulators of multiple cellular functions, are activated by guanine nucleotide exchange factors (GEFs) that stimulate the exchange of the tightly bound GDP nucleotide by GTP. The catalytic domain responsible for nucleotide exchange is in general associated with non-catalytic domains that define the spatio-temporal conditions of activation. In the case of small G proteins of the Arf subfamily, which are major regulators of membrane trafficking, GEFs form a heterogeneous family whose only common characteristic is the well-characterized Sec7 catalytic domain. In contrast, the function of non-catalytic domains and how they regulate/cooperate with the catalytic domain is essentially unknown.
Results
Based on Sec7-containing sequences from fully-annotated eukaryotic genomes, including our annotation of these sequences from Paramecium, we have investigated the domain architecture of large ArfGEFs of the BIG and GBF subfamilies, which are involved in Golgi traffic. Multiple sequence alignments combined with the analysis of predicted secondary structures, non-structured regions and splicing patterns, identifies five novel non-catalytic structural domains which are common to both subfamilies, revealing that they share a conserved modular organization. We also report a novel ArfGEF subfamily with a domain organization so far unique to alveolates, which we name TBS (TB C-S ec7).
Conclusion
Our analysis unifies the BIG and GBF subfamilies into a higher order subfamily, which, together with their being the only subfamilies common to all eukaryotes, suggests that they descend from a common ancestor from which species-specific ArfGEFs have subsequently evolved. Our identification of a conserved modular architecture provides a background for future functional investigation of non-catalytic domains.
Similar content being viewed by others

Background
Guanine Nucleotide Exchange Factors (GEFs) are obligatory components of signaling cascades regulated by small GTP-binding proteins (called small G proteins hereafter). Their biochemical activity is to stimulate the dissociation of the tightly bound GDP nucleotide from the small G protein in response to cellular signals. Thereby, they favor the binding of the more abundant cellular GTP, organizing the active conformation of the small G protein which can recruit its effectors (reviewed in [1]). Each small G protein family features its own ensemble of GEFs characterized by a conserved catalytic domain responsible for nucleotide exchange, which is generally combined with non-catalytic domains that define the spatio-temporal conditions of activation. In the case of small G proteins of the Arf family, which are major regulators in membrane trafficking (reviewed in [2]), the exchange domain is a conserved module of ~200 amino acids called the Sec7 domain [3]. Its biochemical (reviewed in [4]) and structural [5, 6] mechanisms have been investigated in detail. Remarkably, the Sec7 domain is the only domain that is conserved in all ArfGEFs (reviewed in [7, 8]) and it is to some extent interchangeable between species [9]. In contrast, little is known about the functions of the other domains, which are likely to determine intracellular localization of ArfGEFs and their responsiveness to specific signals.
As for most small G proteins, Arf family members are outnumbered by ArfGEFs in many species. In humans for instance, 5 Arf proteins have been identified, and there are at least 13 proteins carrying a Sec7 domain, of which most have been characterized as bona fide ArfGEFs (reviewed in [7, 8]). Thus an individual Arf protein may be activated by more than one GEF, emphasizing that essential aspects in building up the Arf responses may be encoded by the modular architecture of their GEFs. Sequence similarity in the non-catalytic regions forms the basis for the classification of ArfGEFs into subfamilies. 8 subfamilies are currently identified in eukaryotes with sizes ranging from small (~40–80 kD including CYH, EFA6 and FBS), to medium (~100–150 kD, including BRAG/LONER, SYT1, SYT2) and large (~170–200 kD) ArfGEFs (reviewed in [7, 8]). Large ArfGEFs comprise two subfamilies which we will refer to as the BIG and GBF subfamilies after the name of their human representatives. The GBF subfamily includes human GBF1 [10], Arabidopsis GNOM [11] and Saccharomyces Gea1 and Gea2 [12], the BIG subfamily human BIG1 and BIG2 [13, 14] and yeast Sec7p [15]. An additional subfamily called RalF is found in Rickettsie and Legionella bacteria, likely acting on an host Arf pathway [16]. Analysis of the CYH and EFA6 subfamilies, present only in multicellular animals, and that of the large ArfGEFs, found in all eukaryotes, have yielded most of the functional data currently available. CYH and EFA6 are active on Arf6 at the plasma membrane where they may function in the crosstalk of membrane traffic, cytoskeleton dynamics and signalling in endosomal pathways (reviewed in [17]). Most members of the BIG and GBF subfamilies characterized so far function in vesicular trafficking at the Golgi [12, 14, 18], except for BIG2, which also localizes on recycling endosomes [19], and GNOM which acts in the endosomal recycling pathway [11].
The domain architecture of non-catalytic regions of ArfGEFs, hence their contribution to specific aspects of the build-up of the Arf response, is essentially not established except for those ArfGEFs with domains found in other classes of cellular regulators. The known domains include membrane-interacting PH domains in the CYH (reviewed in [20]), EFA6 [21] and possibly BRAG/LONER[22] subfamilies, and a putative F-box in the FBS subfamily [23], a protein-protein interaction domain that has been involved in the recruitment of substrates to the SCF ubiquitination machinery. Coiled-coil structures have also been predicted in the N-terminus of the CYH subfamily and in the C-terminus of the EFA6 subfamily. In CYH, they are involved in dimerization [3], recruitment of partners [24] and Golgi targeting [25], and in actin remodeling functions in the case of EFA6 [21]. On the other hand, although the functions of BIG and GBF subfamilies have been the subject of many investigations, their architecture is barely described, making it difficult to associate biochemical activities with their molecular structure.
Here we investigate the domain architecture in the BIG and GBF subfamilies, including all sequences from fully annotated eukaryotic genomes and our novel annotation of Sec7-containing proteins from the Paramecium tetraurelia alveolate. Sequence comparisons combined with secondary structures and splicing patterns analysis identifies five novel domains that are conserved between BIG and GBF subfamilies, thus unifying them as a higher order subfamily with a probable common ancestor. Our analysis of Sec7-domain containing sequences from Paramecium also introduces a novel subfamily of ArfGEFs unique to alveolates, which we call TBS (TB C-S ec7).
Results and discussion
A conserved domain architecture in BIG and GBF subfamilies
The BIG and GBF subfamilies are the only ArfGEFs subfamilies common to all eukaryotes [8] and the sole ArfGEFs present in plants [26] (Figure 1). They are therefore possible representatives of ancestral ArfGEF functions and may provide a model to understand the nature and implementation of activities associated with the exchange function carried by the conserved Sec7 domain. However, domain 'hunting' in BIG and GBF subfamilies was complicated by the facts that the Sec7 domain is their only domain that could be identified from known domain repertoires, and that their poorly characterized non-catalytic regions were not found outside these ArfGEF subfamilies. Alternatively, we based our search of candidate structural domains in BIGs and GBFs on the bioinformatics analysis of their own sequences, taking advantage of the growing number of sequences from fully annotated genomes from mammals, insects, plants, nematode, and fungi, to which we included our annotation of Sec7-containing proteins from the newly sequenced genome of Paramecium.

Venn diagram of the nine Sec7-containing subfamilies sorted according to the species where each subfamily has been found. The TBS subfamily was identified in this study. The BIG and GBF subfamilies are merged in a higher order subfamily (GBG), and are the only subfamily common to all eukaryotes.
Multiple alignments of 42 sequences (listed in Table 1) revealed that the BIG and GBF subfamilies share an unexpected conserved architecture (schematized in Figure 2). Two homology domains are located in N-terminus of the Sec7 domain – the DCB (~150 aa) and HUS (H omology U pstream of S ec7, ~170 aa) domains – and three in its C-terminus -the HDS1 (H omology D ownstream of S ec7, ~130 aa), HDS2 (~160 aa) and HDS3 (~120 aa) domains (Figure 3,4,5,6,7). In Arabidopsis GNOM, the DCB domain is included in an N-terminal region of ~250 residues involved in dimerization and possibly binding to cyclophilin5 and called the D imerization/C yclophilin B inding region [27], after which the new domain was named (Figure 3). All domains are predicted to have a high content of α-helices that co-align in the multiple sequence alignments, reinforcing the prediction of sequence similarities and suggesting that these domains form folded structural units that may share common functional features. Except for the N-terminal DCB domain which is also found in the yeast protein Ysl2p [28], all of them are unique to these two ArfGEFs subfamilies within the detection limits of the BLAST search. The HUS domain features a remarkably conserved N(Y/F)DC(D/N) motif, which we call the HUS box, which is predicted to locate in a loop where it may be available for functional interactions (Figure 4). The N- and C-terminal ends of BIGs and GBFs are more variable, including an unusual enrichment in Asp/Glu or Pro residues in some members. A specific feature of BIG members is that their C-terminus is in general less variable than that of GBFs, and is predicted with a significant amount of secondary structures. In contrast to the predicted structural domains, the intervening regions are highly variable in length and do not yield aligned sequences. Analysis of their amino-acid composition reveals a paucity of hydrophobic residues which is predicted to associate with an essentially unfolded conformation, suggesting that they act as linkers to tether the functional domains together.

The common domain architecture of the BIG and GBF subfamilies. From N- to C-terminus : DCB , HUS, Sec7, HDS1, HDS2, HDS3. Linker regions of variable length and sequence are shown in grey, with alternate splicing sites in human GBF1, BIG1 and BIG2 in black, white and grey diamond shapes respectively. Interactions reported in the litterature are indicated in boxes of width corresponding to the mapped regions, except for myosin IXb interaction which was studied only with full-length BIG1. Arrows indicate predicted Protein kinase A-anchoring motifs. 1 [45]; 2 [27]; 3 [46]; 4 [47]; 5 [48]; 6 [49]; 7 [50]; 8 [51].

The conserved domains of the BIG/GBF subfamily: DCB domain. Multiple sequence alignement of the conserved domains from BIG and GBF representative sequences showing secondary structure predictions that co-align in all sequences. Colour coding is red for invariant residues, yellow for a sequence similarity score threshold of 0.15 using the BLOSUM62 matrix. The gap in helix 4 is due to an insert in the drosophila Q9V696 sequence, and may be resulting from a sequence annotation error.

The conserved domains of the BIG/GBF subfamily: HUS domain. See Figure 3 legend for alignment details. The highly conserved HUS motif is boxed in blue. The gap in helix 5 domain is due to an insert in the Arabidopsis 3g43300 sequence, and may be resulting from a sequence annotation error.

The conserved domains of the BIG/GBF subfamily: HDS1 domain. See Figure 3 legend for alignment details.

The conserved domains of the BIG/GBF subfamily: HDS2 domain. See Figure 3 legend for alignment details.

The conserved domains of the BIG/GBF subfamily: HDS3 domain. See Figure 3 legend for alignment details.
To further investigate the predicted organization of BIGs and GBFs in 6 conserved helical domains connected by variable linkers, splicing patterns of human BIGs and GBFs were analyzed in the large number of cDNAs and ESTs in the databases that correspond to GBF/BIG transcripts. This revealed the use of alternate splice donor and acceptor sites predicted to yield proteins with insertions and deletions ranging from 1 to 38 residues, and a number of splice variants arising from exon skipping (Table 2). Strikingly, all observed sequence variations occur in regions identified as linkers between conserved domains (Figure 2). Together with our domain analysis, this suggests that splicing at non-canonical exon/intron boundaries is only tolerated in regions of the protein where the impact upon folding of domains with essential function would be minimal.
Evolution of BIGs and GFBs from a common ancestor
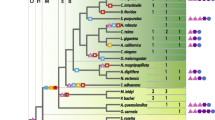
Combined, our analysis reveals that the BIG and GBF subfamilies share the same overall domain organization, and are likely to descend from a common ancestor gene that duplicated first to form the BIG and GBF groups, and again within these groups to yield species-specific BIG and GBF members. These two subfamilies can therefore be unified as a higher order ArfGEF subfamily (called below GBG for G BF/B IG G EFs), from which unrooted phylogenetic trees can be built (Figure 8). Unlike previous phylogenetic analysis which compared ArfGEFs based on their Sec7 domains after diverging non-catalytic regions have been trimmed [8], our trees were established from the simultaneous alignment of all 6 conserved domains (DCB, HUS, Sec7, HDS1, HDS2 and HDS3), excluding variable linkers. The same tree topology was obtained with both neighbor-joining and maximum likelihood methods, and was retained using any one of the new conserved domains alone (data not shown). Bootstrap analysis strongly supported this topology for most branches. Only a few small branches located at the base of the groups were found in less than 60% of the trials in one of the two methods, but this never occurred with both methods simultaneously.

Unrooted neighbour-joining phylogenetic tree of the BIG/GBF subfamily. Colour coding for the main groups is green for plants, marine blue for fungi, orange and red for animals, cyan for protists. Branches found in less than 60% bootstrap trials by either the neighbor-joining or the maximum likelihood method are in dotted lines. Species abbreviations are as in Table 1.
The tree topology strongly suggests that in most organisms, GBG members sort in separate branches, corresponding to their classification in BIGs and GBFs. Remarkably, our annotation of Sec7-containing proteins in the genome of Paramecium reveals the first departure from this distribution, as all GBGs in this species are located in a single branch, which is closer to the BIGs. This unexpected tree topology may indicate that alveolates diverged from animals/fungi and plants before the duplication of an ancestral GBG into the BIG and GBF, and that GBGs in that organism are representative of this ancestral gene. Alternatively, duplication may have been followed by loss of GBF genes. Current knowledge of the phylogenetic branching of alveolates relative to the plants and animal/fungi branches does not permit resolution between those two possibilities.
GBGs in plants: refining the functional evolution of BIGs and GBFs
In fungi and mammals, BIGs and GBFs are represented by only one or two members, whose functions in vesicular trafficking at the Golgi within each group appear largely overlapping [12, 14]. In contrast, plants encode a large number of GBGs in both the BIG and GBF branches but lack other ArfGEFs (Figures 1, 8). In Arabidopsis, none of the GBGs map to duplicated chromosomes where identical functions may be encoded [26, 29]. In addition, comparative analysis with the rice genome nearing completion identifies at least five branches each represented by one rice and one or two Arabidopsis homologs (Figure 8). This correspondence between two highly divergent plant species indicates that GBGs diversified early during plant evolution, probably reflecting functional specialization along with the establishment of plant multicellularity. While GNOM has a plant-specific function in recycling plasma-membrane proteins needed for cell-cell communication and cell polarity establishment [11], possibly closer to the function of EFA6 or CYH subfamilies in metazoans, other plant GBGs are expected to fulfill the presumed ancient function of regulating Golgi trafficking exemplified by mammalian and yeast GBGs. Comparison of orthologous pairs in plants further reveals that they have different sensitivities to Brefeldin A (a widely used fungal inhibitor of Golgi traffic) as predicted from the sequences of the binding site of the drug carried by the Sec7 domain [6]. This observation clearly illustrates that differences in outcome following BrefeldinA treatment may not reflect differences in underlying molecular mechanisms, but instead simply reflect neutral sequence differences at the Sec7 domains between species. In particular, not all BIGs may be BFA-sensitive or GBFs BFA-resistant, unlike suggested by their original nomenclature.
A novel ArfGEF subfamily in alveolates
A remarkable evolutionary feature of ArfGEFs is that while GBGs seem to be ubiquitous to all eukaryotes, fungi and animals kingdoms evolved their own ArfGEFs subfamilies unrelated to those of the other kingdoms. We thus addressed the question of whether Paramecium, which has a large number of GBGs (at least five, of which four are present as pairs as the result of recent duplications) but appears to lack the specialization into the BIG and GBF subgroups, has the same ArfGEF distribution as plants or features a second ArfGEF subfamily. We thus searched the newly sequenced genome from Paramecium tetraurelia and the available alveolate genomes from Cryptosporidium parvum and Tetrahymena thermophila for additional Sec7-containing proteins. This identified a novel putative ArfGEF subfamily characterized by the association of the Sec7 domain with a TBC (Tre/Bub2/Cdc16) domain (Figure 9), which was found only in the protists kingdom. The TBC domain is predicted to carry a GAP (GTPase activating protein) activity towards small G proteins of the Rab family [30], suggesting a potential crosstalk between Rab and Arf pathways. Such a relationship between these two small G proteins families, which are major regulators of membrane traffic, would not be unprecedented, as for example the SYT1 ArfGEF gene was identified in yeast by its genetic interactions with Rab proteins in the exocytic pathway [31]. Interestingly, alveolates have specialized exocytic pathways based on a membrane organelle lying beneath the plasma membrane, the trichocyst, where this unique ArfGEF family may potentially function.

TBS: a novel ArfGEF subfamily in alveolates. Top: Domain structure of the TBS subfamily. Below: Sequences of the TBC domain from Paramecium TBS aligned with TBC domains from known RabGAPs. Secondary structures are from the crystal structure of yeast GYP1 [30].
Conclusion
A conserved scenario for the activation of Arf proteins by their GEFs?
The identification of a conserved modular architecture in all GBG subfamily members suggests that the mechanistic basis for their activation of Arf is likely to follow a similar scenario. Candidate functions for the conserved domains include oligomerization, the collection of input signals, membrane localization, regulation of the exchange activity, scaffolding of Arf proteins to their downstream effectors, not excluding signaling to partners outside the Arf pathways. Dimerization has been reported in the BIG subgroup for BIG1, which forms heterodimers with the highly homologous BIG2 ArfGEF [14], and in the GBF subgroup for GNOM, which forms homodimers [27]. The conservation of the DCB domain in GBGs, which is responsible for the dimerization of GNOM, suggests that such a dimerization function may be general to this domain in GBGs. Another unresolved issue is the conservation of the cellular partners effecting the functions associated with the conserved domains. Our identification of an almost invariant motif in the HUS domain argues in favor of this domain interacting with a conserved partner. However, the ancient divergence into the BIG and GBF groups and their subsequent divergence into species-specific members suggest that specialized requirements are likely to have evolved in most organisms, possibly yielding less conserved partners outside the Sec7 and HUS domains. Finally, whereas in plants all ArfGEFs are predicted to function according to the scheme defined by the conserved domains, other species have additional ArfGEF subfamilies with a modular architecture unrelated to that of the GBG subfamily. It is not known to what extent the GBG's scenario for Arf activation will also apply to non-GBGs ArfGEFs, acting alone or in association with protein partners. In the case of the GBGs, our definition of the structural homology domains as reported here should now provide a robust background for future investigations of their interactions and functions.
Methods
Protein sequence databases were searched with amino acid sequences from human BIG1, human GBF1 and Arabidopsis GNOM using the BLAST algorithm [32]. Paramecium tetraurelia genes were identified with the BLAST algorithm using genome sequence data from Genoscope [33] and manually annotated using Artemis [34]. Tetrahymena sequences were retrieved from the Tetrahymena thermophila genome sequencing project server [35]. Arabidopsis sequences were retrieved from the Arabidopsis Genome Initiative database [36], rice sequences from the TIGR Rice annotation project [37]. Splice variants for hGBF1, hBIG1 and hBIG2 were identified from information provided under Aceview in the December (03) release for their respective listings at the NCBI [38]. Multiple sequence alignments were performed using ClustalW [39] with default alignment parameters or T-coffee [40, 41]. Reliability of the alignments was evaluated according to the T-coffee score, and ranged from average to good for all predicted domains. Average sequence identities were respectively 24 % (DCB domain), 26 % (HUS domain), 44% (Sec7 domain), 26% (HDS1 domain), 28% (HDS2 domain) and 21% (HDS3 domain). Aligned sequences were displayed with ESPript [42] using a similarity global score of 0.15 calculated using the BLOSUM62 matrix. Unrooted phylogenetic trees were generated using the neighbor-joining algorithm of ClustalW excluding gapped regions, and with a maximum likelihood method using the PHYML package [43]. Phylogenetic trees for individual domains was performed on the subset of sequences used in Figure 3. The reliability of the trees was assessed by a bootstrap analysis (1000 replicates). Trees were drawn with TreeView version 1.6.6. Secondary structure predictions on aligned sequences were carried out with the PHD program along with the ClustalW multiple alignment [39]. Non-structured linkers poor in hydrophobic residues were predicted with the PONDR algorithm [44].
Abbreviations
- GEF:
-
Guanine nucleotide exchange factor. CYH: cytohesins/ARNO
- EFA:
-
Exchange Factor for Arf6
- FBS:
-
F-Box/Sec7
- TBS:
-
TBC/Sec7
- GBF:
-
Golgi-associated BFA-resistant guanine nucleotide exchange Factor
- BIG:
-
BFA-Inhibited Guanine nucleotide exchange factor
- GBG:
-
GBF/BIG Gefs
- SYT1:
-
S uppressor of ypt. DCB: Dimerization/Cyclophilin Binding
- HUS:
-
Homology Upstream of Sec7
- HDS:
-
Homology Downstream of Sec7
- TBC:
-
Tre/Bub2/Cdc16
- SCF:
-
Skp1/Cull1/F box.
References
Cherfils J, Chardin P: GEFs: structural basis for their activation of small GTP-binding proteins. Trends Biochem Sci. 1999, 24: 306-311. 10.1016/S0968-0004(99)01429-2.
Chavrier P, Goud B: The role of ARF and Rab GTPases in membrane transport. Curr Opin Cell Biol. 1999, 11: 466-475. 10.1016/S0955-0674(99)80067-2.
Chardin P, Paris S, Antonny B, Robineau S, Beraud-Dufour S, Jackson CL, Chabre M: A human exchange factor for ARF contains Sec7- and pleckstrin-homology domains. Nature. 1996, 384: 481-484. 10.1038/384481a0.
Pasqualato S, Renault L, Cherfils J: The GDP/GTP cycle of Arf proteins. Structural and biochemical aspects. The ARF Book Richard A Kahn, editor Kluwer Academic Publishers. 2004, 23-48.
Goldberg J: Structural basis for activation of ARF GTPase: mechanisms of guanine nucleotide exchange and GTP-myristoyl switching. Cell. 1998, 95: 237-248. 10.1016/S0092-8674(00)81754-7.
Renault L, Guibert B, Cherfils J: Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature. 2003, 426: 525-530. 10.1038/nature02197.
Jackson CL, Casanova JE: Turning on ARF: the Sec7 family of guanine-nucleotide-exchange factors. Trends Cell Biol. 2000, 10: 60-67. 10.1016/S0962-8924(99)01699-2.
Cox R, Mason-Gamer RJ, Jackson CL, Segev N: Phylogenetic analysis of Sec7-domain-containing Arf nucleotide exchangers. Mol Biol Cell. 2004, 15: 1487-1505. 10.1091/mbc.E03-06-0443.
Peyroche A, Paris S, Jackson CL: Nucleotide exchange on ARF mediated by yeast Gea1 protein. Nature. 1996, 384: 479-481. 10.1038/384479a0.
Claude A, Zhao BP, Kuziemsky CE, Dahan S, Berger SJ, Yan JP, Armold AD, Sullivan EM, Melancon P: GBF1: A novel Golgi-associated BFA-resistant guanine nucleotide exchange factor that displays specificity for ADP-ribosylation factor 5. J Cell Biol. 1999, 146: 71-84.
Geldner N, Anders N, Wolters H, Keicher J, Kornberger W, Muller P, Delbarre A, Ueda T, Nakano A, Jürgens G: The Arabidopsis GNOM ARF-GEF mediates endosomal recycling, auxin transport, and auxin-dependent plant growth. Cell. 2003, 112: 219-230. 10.1016/S0092-8674(03)00003-5.
Peyroche A, Courbeyrette R, Rambourg A, Jackson CL: The ARF exchange factors Gea1p and Gea2p regulate Golgi structure and function in yeast. J Cell Sci. 2001, 114: 2241-2253.
Mansour SJ, Skaug J, Zhao XH, Giordano J, Scherer SW, Melançon P: p200 ARF-GEP1: a Golgi-localized guanine nucleotide exchange protein whose Sec7 domain is targeted by the drug brefeldin A. Proc Natl Acad Sci U S A. 1999, 96: 7968-7973. 10.1073/pnas.96.14.7968.
Yamaji R, Adamik R, Takeda K, Togawa A, Pacheco-Rodriguez G, Ferrans VJ, Moss J, Vaughan M: Identification and localization of two brefeldin A-inhibited guanine nucleotide-exchange proteins for ADP-ribosylation factors in a macromolecular complex. Proc Natl Acad Sci U S A. 2000, 97: 2567-2572. 10.1073/pnas.97.6.2567.
Achstetter T, Franzusoff A, Field C, Schekman R: SEC7 encodes an unusual high molecular weight protein required fir membrane traffic from the yeast Golgi apparatus. J Biol Chem. 1988, 263: 11711-11717.
Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR: A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science. 2002, 295: 679-682. 10.1126/science.1067025.
Donaldson JG: Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J Biol Chem. 2003, 278: 41573-41576. 10.1074/jbc.R300026200.
Zhao X, Lasell TK, Melançon P: Localization of large ADP-ribosylation factor-guanine nucleotide exchange factors to different Golgi compartments: evidence for distinct functions in protein traffic. Mol Biol Cell. 2002, 13: 119-133. 10.1091/mbc.01-08-0420.
Shin HW, Morinaga N, Noda M, Nakayama K: BIG2, a guanine nucleotide exchange factor for ADP-ribosylation factors: its localization to recycling endosomes and implication in the endosome integrity. Mol Biol Cell. 2004, 15: 5283-5294. 10.1091/mbc.E04-05-0388.
Cullen PJ, Chardin P: Membrane targeting: what a difference a G makes. Curr Biol. 2000, 10: R876-8. 10.1016/S0960-9822(00)00818-6.
Derrien V, Couillault C, Franco M, Martineau S, Montcourrier P, Houlgatte R, Chavrier P: A conserved C-terminal domain of EFA6-family ARF6-guanine nucleotide exchange factors induces lengthening of microvilli-like membrane protrusions. J Cell Sci. 2002, 115: 2867-2879.
Chen EH, Pryce BA, Tzeng JA, Gonzalez GA, Olson EN: Control of myoblast fusion by a guanine nucleotide exchange factor, loner, and its effector ARF6. Cell. 2003, 114: 751-762. 10.1016/S0092-8674(03)00720-7.
Ilyin GP, Rialland M, Pigeon C, Guguen-Guillouzo C: cDNA cloning and expression analysis of new members of the mammalian F-box protein family. Genomics. 2000, 67: 40-47. 10.1006/geno.2000.6211.
Mansour M, Lee SY, Pohajdak B: The N-terminal coiled coil domain of the cytohesin/ARNO family of guanine nucleotide exchange factors interacts with the scaffolding protein CASP. J Biol Chem. 2002, 277: 32302-32309. 10.1074/jbc.M202898200.
Lee SY, Pohajdak B: N-terminal targeting of guanine nucleotide exchange factors (GEF) for ADP ribosylation factors (ARF) to the Golgi. J Cell Sci. 2000, 113 ( Pt 11): 1883-1889.
Jürgens G, Geldner N: Protein secretion in plants: from the trans-Golgi network to the outer space. Traffic. 2002, 3: 605-613. 10.1034/j.1600-0854.2002.30902.x.
Grebe M, Gadea J, Steinmann T, Kientz M, Rahfeld JU, Salchert K, Koncz C, Jürgens G: A conserved domain of the arabidopsis GNOM protein mediates subunit interaction and cyclophilin 5 binding. Plant Cell. 2000, 12: 343-356. 10.1105/tpc.12.3.343.
Jochum A, Jackson D, Schwarz H, Pipkorn R, Singer-Kruger B: Yeast Ysl2p, homologous to Sec7 domain guanine nucleotide exchange factors, functions in endocytosis and maintenance of vacuole integrity and interacts with the Arf-Like small GTPase Arl1p. Mol Cell Biol. 2002, 22: 4914-4928. 10.1128/MCB.22.13.4914-4928.2002.
Initiative AG: Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000, 408: 796-815. 10.1038/35048692.
Rak A, Fedorov R, Alexandrov K, Albert S, Goody RS, Gallwitz D, Scheidig AJ: Crystal structure of the GAP domain of Gyp1p: first insights into interaction with Ypt/Rab proteins. Embo J. 2000, 19: 5105-5113. 10.1093/emboj/19.19.5105.
Jones S, Jedd G, Kahn RA, Franzusoff A, Bartolini F, Segev N: Genetic interactions in yeast between Ypt GTPases and Arf guanine nucleotide exchangers. Genetics. 1999, 152: 1543-1556.
Bork Group's WU-BLAST2 Search Service at EMBL. [http://dove.embl-heidelberg.de/Blast2/]
Paramecium Genomics. [http://paramecium.cgm.cnrs-gif.fr/]
The Sanger Institute: Informatics Software: Artemis. [http://www.sanger.ac.uk/Software/Artemis/]
The TIGR Tetrahymena thermophila Genome Project. [http://www.tigr.org/tdb/e2k1/ttg/]
TAIR BLAST. [http://www.arabidopsis.org/Blast/]
TIGR Rice Genome Annotation. [http://www.tigr.org/tdb/e2k1/osa1/]
AceView. [http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/]
Pole Bioinformatique Lyonnais. [http://pbil.univ-lyon1.fr/]
Notredame C, Higgins DG, Heringa J: T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000, 302: 205-217. 10.1006/jmbi.2000.4042.
Tcoffee. [http://igs-server.cnrs-mrs.fr/Tcoffee/tcoffee_cgi/index.cgi]
Easy execution of ESPript 2.x / ENDscript 1.x. [http://ribosome.toulouse.inra.fr/ESPript/cgi-bin/ESPript.cgi]
PHYML: fast, accurate estimation of large PHYlogenies by Maximum Likelihood. [http://atgc.lirmm.fr/phyml/]
Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z: Intrinsic disorder and protein function. Biochemistry. 2002, 41: 6573-6582. 10.1021/bi012159+.
Padilla PI, Chang MJ, Pacheco-Rodriguez G, Adamik R, Moss J, Vaughan M: Interaction of FK506-binding protein 13 with brefeldin A-inhibited guanine nucleotide-exchange protein 1 (BIG1): effects of FK506. Proc Natl Acad Sci U S A. 2003, 100: 2322-2327. 10.1073/pnas.2628047100.
Li H, Adamik R, Pacheco-Rodriguez G, Moss J, Vaughan M: Protein kinase A-anchoring (AKAP) domains in brefeldin A-inhibited guanine nucleotide-exchange protein 2 (BIG2). Proc Natl Acad Sci U S A. 2003, 100: 1627-1632. 10.1073/pnas.0337678100.
Chantalat S, Park SK, Hua Z, Liu K, Gobin R, Peyroche A, Rambourg A, Graham TR, Jackson CL: The Arf activator Gea2p and the P-type ATPase Drs2p interact at the Golgi in Saccharomyces cerevisiae. J Cell Sci. 2004, 117: 711-722. 10.1242/jcs.00896.
Chantalat S, Courbeyrette R, Senic-Matuglia F, Jackson CL, Goud B, Peyroche A: A novel Golgi membrane protein is a partner of the ARF exchange factors Gea1p and Gea2p. Mol Biol Cell. 2003, 14: 2357-2371. 10.1091/mbc.E02-10-0693.
Charych EI, Yu W, Miralles CP, Serwanski DR, Li X, Rubio M, De Blas AL: The brefeldin A-inhibited GDP/GTP exchange factor 2, a protein involved in vesicular trafficking, interacts with the beta subunits of the GABA receptors. J Neurochem. 2004, 90: 173-189. 10.1111/j.1471-4159.2004.02481.x.
Garcia-Mata R, Sztul E: The membrane-tethering protein p115 interacts with GBF1, an ARF guanine-nucleotide-exchange factor. EMBO Rep. 2003, 4: 320-325. 10.1038/sj.embor.embor762.
Saeki N, Tokuo H, Ikebe M: BIG1 is a binding partner of myosin IXB and regulates its Rho gap activity. J Biol Chem. 2005
Acknowledgements
This work was supported by a Human Frontiers in Science Program grant to G.J., P.M. and J.Ch. We thank Genoscope for access to Paramecium whole genome shotgun primary data and Linda Sperling (CNRS, Gif-sur-Yvette) for help with annotations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
B.M. and V.B. carried out sequence and phylogenetic analysis. A.J. participated in the domain analysis. J.Co. annotated Paramecium sequences. D.S. and P.M. performed splicing pattern analysis. N.K. and G.J. analyzed the distribution of large ArfGEFs in plants. J.Ch. conceived and coordinated the study and wrote the manuscript.
Barbara Mouratou, Valerie Biou contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Mouratou, B., Biou, V., Joubert, A. et al. The domain architecture of large guanine nucleotide exchange factors for the small GTP-binding protein Arf. BMC Genomics 6, 20 (2005). https://doi.org/10.1186/1471-2164-6-20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-6-20