
Abstract
Background
Availability of DNA sequence information is vital for pursuing structural, functional and comparative genomics studies in plastids. Traditionally, the first step in mining the valuable information within a chloroplast genome requires sequencing a chloroplast plasmid library or BAC clones. These activities involve complicated preparatory procedures like chloroplast DNA isolation or identification of the appropriate BAC clones to be sequenced. Rolling circle amplification (RCA) is being used currently to amplify the chloroplast genome from purified chloroplast DNA and the resulting products are sheared and cloned prior to sequencing. Herein we present a universal high-throughput, rapid PCR-based technique to amplify, sequence and assemble plastid genome sequence from diverse species in a short time and at reasonable cost from total plant DNA, using the large inverted repeat region from strawberry and peach as proof of concept. The method exploits the highly conserved coding regions or intergenic regions of plastid genes. Using an informatics approach, chloroplast DNA sequence information from 5 available eudicot plastomes was aligned to identify the most conserved regions. Cognate primer pairs were then designed to generate ~1 – 1.2 kb overlapping amplicons from the inverted repeat region in 14 diverse genera.
Results
100% coverage of the inverted repeat region was obtained from Arabidopsis, tobacco, orange, strawberry, peach, lettuce, tomato and Amaranthus. Over 80% coverage was obtained from distant species, including Ginkgo, loblolly pine and Equisetum. Sequence from the inverted repeat region of strawberry and peach plastome was obtained, annotated and analyzed. Additionally, a polymorphic region identified from gel electrophoresis was sequenced from tomato and Amaranthus. Sequence analysis revealed large deletions in these species relative to tobacco plastome thus exhibiting the utility of this method for structural and comparative genomics studies.
Conclusion
This simple, inexpensive method now allows immediate access to plastid sequence, increasing experimental throughput and serving generally as a universal platform for plastid genome characterization. The method applies well to whole genome studies and speeds assessment of variability across species, making it a useful tool in plastid structural genomics.
Similar content being viewed by others


Background
Chloroplast DNA (cpDNA) represents an extranuclear capsule of genetic information, encoding essential structural and enzymatic proteins of the organelle. This satellite genome contains a wealth of information that has been shaped by speciation, rendering it a rich resource to trace evolutionary relationships between photosynthetic taxa [1]. Genetic manipulation of the chloroplast genome can transform the chloroplast into a bioreactor, allowing large-scale production of proteins vital to agriculture or pharmacology [2, 3]. Maternal inheritance of plastid genome in most species ensures gene containment in genetically modified plants making it an attractive alternative for the integration of foreign genes [4, 5].
Genomic and phylogenetic studies and efficient genetic modification begin with the base material of plastid DNA sequence. Despite their relatively small size, few plastid genomes have been fully sequenced, thus limiting comparative genomics studies across the species. Complete sequence coverage has been resolved for only 13 species representing model and crop plants, namely, Arabidopsis thaliana [6], Atropa belladonna [7], Medicago truncatula (GenBank: NC_003119), Oryza sativa [8], Spinacea oleracea [9], Nicotiana tabacum [10], Triticum aestivum [11], Lotus corniculatus [12], Zea mays [13], Panax ginseng [14], Cucumis sativus (GenBank: NC_007144), Glycine max [15] and Saccharum officinarum [16]. Comparison of a small set of representative coding or intergenic sequences derived from a large number of species has been used to perform phylogenetic studies [17], but there are many unresolved phylogenetic questions [18]. Use of complete genome data is another emerging approach in plant phylogenetics [19]. There are at least three federally-sponsored, multi-institution endeavors underway to sequence about 200 plastid genomes from plants [20–22].
There are a number of challenges to rapid access to chloroplast DNA sequence from a given species. Typical sequencing efforts begin with construction of chloroplast plasmid or other genomic libraries. Construction of such resources requires isolation of pure plastid DNA, which may be troublesome in some species. Even shotgun sequencing to appreciable coverage is considerably expensive and time consuming. From the standpoint of method and cost, the generation of plastid sequence data is considerable and a potential hindrance to productive data mining and engineering efforts. One recent report describes a sophisticated methodology using FACS (fluorescence-assisted cell sorting) and RCA (rolling circle amplification) for sequencing a plastid genome [23]. The degree of technological sophistication is inversely proportional to its wider applicability due to the prohibitive costs associated with it, slowing chloroplast genomics studies. These large-scale studies are expensive and prohibit testing of additional related species or ecotypes that may be informative. Additionally, smaller research programs do not necessarily have access to these tools and techniques. Most importantly some rare and/or difficult-to-obtain taxa that are not amenable to large-scale chloroplast DNA extraction can not be analyzed by the existing methodology. While downstream processing of sequence information is highly streamlined due to the presence of freely available tools on the World Wide Web, such as DOGMA [24, 25], the lack of cost-effective innovation in rapid sequence acquisition has restricted plastid informatics studies.
To address these issues, we exploited the fact that chloroplast genomes are extremely well conserved in size, gene arrangement, and coding sequence, at least within major subgroups of the plant kingdom [26, 27]. We formulated the hypothesis that conserved islands of cpDNA sequence could serve as universal anchors to generate overlapping PCR products comprised of conserved coding regions, and adjacent polymorphic intergenic regions. The resulting amplicons could then be sequenced, assembled, annotated and analyzed. This approach exploits the high degree of sequence conservation and general synteny within discrete portions of chloroplast genome. In this report this powerful technique has been applied to the large inverted repeat (IR) region of strawberry (Fragaria × ananassa) and peach (Prunus persica). The entire ~30 kb region was amplified from total DNA, sequenced, annotated and submitted to public databases in several days for a fraction of the cost of traditional or other recently published approaches [28]. To further validate this application, corresponding regions were amplified from a series of other eudicots and a monocot of agricultural importance as well as two gymnosperms (Pinus and the distant vascular plant Gingko) and a pteridophyte (Equisetum). This universal method represents a rapid, inexpensive means to obtain complete coverage of many higher plant plastid genome regions, and even substantial coverage from distant genera. The sequence information generated form this method can hasten phylogenetic and genomics studies and also help in identification of regulatory elements necessary for design of transformation vectors for the manipulation of chloroplast genomes of new species.
Results
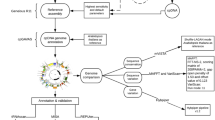
In order to expand ongoing Rosaceae genomics studies [29], the original goal of this work was to use existing informatics resources to devise a PCR-based strategy to obtain plastid DNA sequence for cultivated strawberry and peach. This information would assist in identifying indel (insertion/deletion) polymorphisms or SNPs (single nucleotide polymorphisms) that could serve as an additional tool for phylogenetic analysis [30] and also allow the design of vectors useful for strawberry plastid engineering. A schematic explanation of the technique is shown in Figure 1. If a given primer pair fails to generate an amplicon in initial PCR trials, the forward primer can then be paired with the reverse primer from the next primer pair to obtain coverage of that region. Figure 2 demonstrates proof-of-concept, as universal primer set derived from the IR of five sequenced eudicots is sufficient to amplify the corresponding ~30 Kb region in commercial strawberry. The 27 primer pairs generate amplicons spanning this region. The corresponding PCR products were sequenced, and the sequence was immediately deposited to public databases. Here we proceeded from computational analyses to finished strawberry and peach IR sequence in one week for <$500.

Schematic representation of the ASAP approach. Three rounds of PCR allow for 100% coverage of a given region. F and R suffix to the numbers represent forward and reverse universal primers.

ASAP profile from Fragaria × ananassa. A. Round 1 touchdown PCR. B. Round 2 touchdown PCR and C. Round 3 touchdown extension PCR.
Since the method proved useful in strawberry and peach, its applicability across plant species was assessed. Total genomic DNA was derived from 13 diverse plant species and subjected to the ASAP protocol using the primers listed in Table 1 and the conditions stated in Table 2. The ASAP primer set effectively generated expected amplicons from all eudicot species tested. Expected results were obtained in Nicotiana and Arabidopsis. Complete coverage was obtained with the first round of PCR and the amplicon sizes were consistent with predictions (Table 1). Comparison of agarose gel electrophoresis profiles from the IR region revealed clearly discernible amplified fragment length polymorphisms (AFLPs) in the regions amplified with primer pairs 11, 17 and 27 (Figure 3). The profiles for these two species are in complete agreement with the calculated sizes.

Composite ASAP PCR profiles from 8 plant species. At– Arabidopsis thaliana, Nt– Nicotiana tabacum, Cs– Citrus sinensis, Pp– Prunus persica, Le– Lycopersicon esculentum, Ah– Amaranthus hypochondriacus, Ch– Coleus hybrida and Ls– Lactuca sativa. Horizontal lines across each species indicate 1 kb size. Vertical columns indicate the amplicons generated from a given primer pair in the 8 plant species.
The maize plastid genome lacks the ycf 2 open reading frame in the IR region, therefore primer pairs 3 to 9 failed to produce any amplicons, as anticipated (Figure 4A). Similarly, primer pairs 11, 26 and 27 did not produce any amplicons. Using bl2Seq program the maize IR region was compared with the associated primers and no significant sequence similarity was found between them. Interestingly, one primer each in primer pairs 1 and 10 had very low sequence similarity and yet the amplicons were obtained. Using three rounds of PCR (Table 2) 100% coverage was obtained even in the monocot plastome (Figure 4C), indicating the applicability of eudicot-based primer designs to this taxonomic group. Absence of amplicons from primer pair 26 and 27 could be due to the fact that the primers annealed in the spacer region which could be unique to the eudicots. The results of the reactions are presented in both Table 3 and Figure 3. Table 3 presents the conditions required to produce the amplified regions from individual species, whether the products were obtained from PCR round 1, round 2 or round 3. Figure 3 shows the complete array of amplified products corresponding to the amplification conditions presented in Table 2.

Zea mays ASAP PCR profiles. A. Round 1 and 2 touchdown PCR. B. Round 3 touchdown extension PCR and C. Composite profile from A and B.
Fragaria and Prunus (Rosaceae)
Complete coverage of the plastid IR region from Fragaria was obtained after proceeding through three rounds of ASAP PCR (Table 2) with the 27 pairs of primers (Figure 2). These amplicons were generated using Pfu Turbo DNA polymerase (Stratagene Inc., Carlsbad, CA) in order to minimize potential errors generated during PCR reactions. These amplicons were directly sequenced in a 96-well format. The sequence was assembled and annotated as described in Methods.
Interestingly, in Prunus complete coverage was obtained with Round 2 PCR. Sequence comparison with Fragaria revealed that Prunus and Fragaria share considerable sequence similarity in the IR region as expected being from the same phylogenetic group. This is another demonstration of the utility of this technique where two members of the same taxonomic group were sequenced and compared in a very short time frame and in a cost-effective manner.
Other eudicots and identification of a variable region
The ASAP protocol was attempted in other eudicot species for which plastid genome sequence has not been reported. In Citrus and Lycopersicon complete coverage was obtained after Round 1 PCR and for the remaining species almost 99 – 100% coverage was obtained using Round 2 PCR conditions. Electrophoresis profiles revealed highly discernible AFLPs amongst different plant species. The most consistently variable region was represented by primer pair 11. In tobacco this amplicon represents sequences for orf 87/ycf 15, orf 92, orf 115, trn L and orf 79. Gel electrophoresis profiles of amplicons generated from this primer pair revealed a great range of variability across all species tested (Figure 5A). AFLPs were discernable by gel electrophoresis between two solanaceous species, Nicotiana and Lycopersicon. Sequencing and alignment of this region from two members of Solanaceae, tobacco and tomato revealed a 95% – 98% sequence similarity in the aligning sequences. Tomato had two deletions in the region coding for orf 92 and ycf 15 in tobacco, which reconciles the smaller amplicon size. On the other extreme is the representative member of Caryophyllaceae, Amaranthus, where sequencing and subsequent alignments with tobacco revealed absence of ORFs between ycf 2 and orf 92 – trn L-CAA region (Figure 5B). Thus the ASAP method provides the advantage of analyzing a large region from a number of species and identifying a highly variable region at the same time.

A. PCR amplicons generated from primer pair 11 in 9 plant species. Fa– Fragaria × ananassa/Rosaceae, Pp– Prunus persica/Rosaceae, At– Arabidopsis thaliana/Crucifereae, Cs– Citrus sinensis/Rutaceae, Ch– Coleus hybrida/Lamiaceae, Nt– Nicotiana tabacum/Solanaceae, Le– Lycopersicon esculentum/Solanaceae, Ls– Lactuca sativa/Asteraceae, Ah– Amaranthus hypochondriacus/Caryophyllaceae. B. Schematic representation of the polymorphism between the amplicons generated from primer pair 11 in tobacco, tomato and amaranthus.
Distant species
To test the limits of this methodology, the same 27 pairs of primers were used against total DNA from Pisum sativum, Ginkgo biloba, Pinus taeda and Equisetum hyemale. These species represent a unique member of Fabaceae (Pisum – largest deletion resulting in removal of the rRNA cluster; has only one IR), an ancient and contemporary gymnosperm, and a pteridophyte. The primer pairs designed for eudicot plastid genomes were able to amplify the regions corresponding to primer pairs 14 to 25 in Pisum. In tobacco these primer pairs amplify the 98494 – 110052 nt region of the IR that includes the rrn operon. The bl2Seq program was used to determine the sequence similarity between the 27 primer pairs and the Pisum chloroplast genome sequence (Kindly provided by John Gray, John Innes Institute, UK). The observed amplicon patterns are consistent with what is anticipated from the sequence data. Primer pairs that failed to generate an amplicon do not share a significant sequence similarity with the Pisum plastid genome sequence (Figure 6). In the two gymnosperms, similar amplicon patterns were generated from the rrn operon region. Again in the pteridophyte only the primer pairs corresponding to the rrn operon produced an amplicon. The Equisetum chloroplast genome does possess ycf 2 gene but comparative sequence analysis with higher plant ycf 2 revealed no significant sequence similarity. Interestingly the amino acid sequence similarity was almost 94% (data not shown).

Composite ASAP PCR profiles from 4 unique plant species. Ps – Pisum sativum, Gb – Gingko biloba, Pt – Pinus taeda, Eh – Equisetum hyemale.
Discussion
Chloroplast sequencing efforts of model photosynthetic organisms have provided a wealth of information detailing structural features and plant phylogeny, as well as a basis for manipulation of the plastid genome in the interest of bioengineering. Current molecular phylogenetic studies are carried out using large or complete plastid genome sequences or small coding or intergenic sequences and the Amplification, Sequencing, and Annotation of Plastomes (ASAP) method caters to both approaches. Rapid generation of plastid sequence information is necessary as it can facilitate better design of plastid transformation vectors [15]. One of the factors in successful engineering of cotton and carrot plastomes was the use of species-specific flanking sequences in the transformation vectors [31, 32].
The simple yet powerful ASAP technique described herein expands the capacity for any laboratory to dissect the chloroplast genome at the informatics level with a basic set of available resources. The obvious limitation of ASAP method is that some plastid genomes have undergone extensive rearrangements [33]. Therefore, this method complements other strategies [28] for obtaining plastid genome sequences and provides a convenient platform for plastomes that share a considerable level of synteny. A long range PCR based approach was used earlier to sequence the Amborella trichopoda plastid genome [34], requiring nebulization of large amplicons and cloning prior to sequencing. In contrast, the ASAP method performs the dual task of generating small amplicons for direct sequencing and the electrophoresis profiles provide structural genomics information. Direct sequencing of the PCR products limits incorrect base calls resulting from amplification or sequencing errors, as large pools of products are sequenced and even early PCR errors will not be scored incorrectly. In a worst-case scenario a base call at a given position will be unclear, requiring re-amplification and re-sequencing of the amplicon. This approach allows the implementation of non-proofreading, highly processive polymerases that limit costs yet generate substantial quantities of template for downstream analyses.
ASAP also allows description and characterization of the frequent islands of high sequence identity present within the coding regions of sequenced genomes. Using local identities found within the cpDNA sequences of five sequenced eudicot plant species, primer pairs were designed to produce overlapping amplicons that bracket sequences of plastid genes and the rich sequence variability resident to their adjacent intergenic regions. The ASAP method was herein shown to generate representative regions of 10 diverse plant species to almost 100% coverage. Even the rrn region was also amplified from four divergent genomes studied, such as Pisum, Equisetum, Pinus and Ginkgo, with expected efficiency. The success of the method suggests it an excellent first step in the analysis of any novel plastid genome. The ASAP method may be the only practical approach for some rare and/or difficult-to-obtain taxa that are not amenable to chloroplast DNA extraction.
One caveat of this technique is that plastid sequences are not confined to the chloroplast. Plastid DNA sequences are represented in both mitochondrial and nuclear genomes, and may serve as templates for the amplicons generated with the 27 primer pairs. The high copy number of plastomes in green leaf derived total DNA and long primers (26 bp each) used in this approach should preferentially amplify chloroplast sequences. Additionally, nuclear integrated plastid sequences are continuously shuffled and eliminated [35], making them less likely to be incorrectly amplified via this approach. However, with the design of specific primers this technique could be extended to plastid genomes of distant phylogenetic groups or mitochondrial genomes in species where high level of synteny is present.
Conclusion
The ASAP method represents a rapid means to generate a large amount of plastid genome information from simple PCR steps to facilitate bioinformatic dissection and functional genomic studies. The products generated spotlight AFLPs that serve as low-resolution beacons to report regions of high diversity, such as the hypervariable Region 11 (Figure 5). These regions may be particularly meaningful for phylogenetic analyses. In this capacity the ASAP methods may facilitate studies of hypervariable specific regions of the plastid genome, helping to quickly identify areas of likely importance, bypassing the necessity to sequence an entire genome as a first step. The ASAP method eliminates cloning steps, thus negating the need to identify and trim plasmid sequences before assembly. Another powerful facet of the technique is the potential to generate plastid fragment fingerprint for any species. AFLPs produced may be immediately comparable to those produced by other genomes, revealing deviations in otherwise conserved sequence, thus informing of structural-genomic variation. Even the absence of products in related species may be extremely informative.
To summarize, the ASAP method has the following advantages: The method is ideal for laboratories or programs with limited resources interested in obtaining chloroplast DNA sequence information for their particular research interest. ASAP method may be the only practical approach to obtain chloroplast DNA sequence from rare or small plant samples. This method provides a fingerprint of a given chloroplast region, which can be readily compared amongst different genera and give information of structural variability even without the sequence information. Another practical application of this approach is the use of PCR amplicons generated by ASAP method for construction of chloroplast genome microarrays from a given plant species.
Specific to this report, the method was used to amplify and sequence the large IR region from octoploid strawberry and peach. This ~30 kbp region from strawberry was amplified in 33 PCR reactions and then sequenced. The PCR fragments were generated in three thermalcycler runs over two days, and the entire process, from leaf to data on the server, was performed in under a week for under $500. With this minimal time and capital investment roughly 25% of the chloroplast genome has been deciphered; bidirectionally and with complete coverage. The entire process is now being scaled up to sequence an entire plastid genome within the context of a 96-well plate. In this system 1.2 to 1.5 kb amplicons may be produced and sequenced from this standard format. The common format also lends itself to robotic manipulation, and it is exciting to speculate that a single set of 96 primer pairs may be matched to a high-throughput robotic amplification and sequencing system to generate plastid DNA sequence at the rate of a plastid genome per day. It is our hope that this methodology will hasten study of chloroplast sequences, especially those from unusual organisms or those not considered worthy of large investment.
Methods
Primer design
The cpDNA sequences of five eudicot plant species namely Nicotiana tabacum (NC_001879), Arabidopsis thaliana (NC_000932), Atropa belladonna (NC_004561), Spinacea oleracea (NC_002202) and Panax ginseng (NC_006290) were aligned using ClustalW [36]. Tobacco chloroplast genome sequence was used as a reference and its coding regions were delineated in the aligned sequences. Highly conserved, putative primer sites were derived by hand parsing the aligned sequences of the IR B region. Primer candidates satisfied several criteria. A candidate primer must be resident to the coding region or conserved intergenic region and primer pairs must be spaced by ~1.0 to 1.2 kb. The primers must share 95% sequence identity among the representative plastid genomes. For universal, large-format application in simultaneous PCR reactions the primers should maintain approximately 50% GC content and a Tm of approximately 50°C. Table 1 lists the primer sequences, annealing sites, respective position in tobacco, Arabidopsis and maize plastid genomes and the expected amplicon sizes. This set of primers, used to amplify the large IR region in this manuscript, will be supplied by the authors upon request.
DNA preparation and primary optimization
Total plant DNA was isolated from fresh leaf tissue using the Qiagen DNeasy Kit (Qiagen Inc., Valencia, CA) according to manufacturer's instructions, except that the homogenized plant material was mixed in Buffer AP1 for 10 min on a platform vortexer for 10 min and the sample was centrifuged for 10 min at 1000 rpm to remove any debris at this stage. The supernatant was then incubated at 65°C for 10 min and from this point on the manufacturer's protocol was followed. DNA was isolated from two plant species namely Arabidopsis thaliana (ecotype Col-0) and tobacco (Nicotiana tabacum). These two species served as a positive control and a basis for detection of computationally predicted AFLPs. Upon validation of the technique with known genomes, amplification was performed on several crops of agricultural importance namely, strawberry (Fragaria × ananassa; cv. Strawberry Festival), Sweet orange (Citrus sinensis), lettuce (Lactuca sativa), peach (Prunus persica; cv. UF Sun), Tomato (Lycopersicon esculentum), coleus (Coleus × hybrida), Amaranthus (Amaranthus hypochondricus). Maize (Zea mays; W22) was tested as a model monocot. Since it has been sequenced, amplification discrepancies related to insertion/deletion and/or rearrangement can be anticipated and circumvented. The technique was also performed on diverse plant species to test the range of the approach. These species included Pisum sativum L. (Little Marvel), which does not have an IR region and has only one copy of the rRNA genes, an ancient gymnosperm (Ginkgo biloba), a contemporary gymnosperm loblolly pine (Pinus taeda), along with horsetail, (Equisetum hyemale), a pteridophyte which represents an ancient plastid genome that would test and define the limit of the application of the eudicot species-based primer designs reported here.
Before performing reactions with the 27 primer pairs that define the large IR, it was important to establish the amount of total DNA to use in each reaction. Variability in amplification may be introduced from several sources, namely the relative amount of chloroplast DNA to total DNA ratio and the tendency for inhibitory compounds to co-purify with DNA templates in total DNA isolation [37]. For instance, the relative cpDNA: total DNA ratio will be significantly different between Arabidopsis (haploid genome size ~140 Mbp) and Pinus (haploid genome size ~21658 Mbp), two organisms with massively different nuclear genome sizes. DNA preparation from some species, such as strawberry, may introduce phenolics or polysaccharides that could inhibit efficient amplification during PCR.
To accommodate both of these variables an initial reaction set is performed using the most conserved primers, those representing the 16 S rrn locus (primer set 19; Table 1). These primers predictably generate a fragment in all species tested, yet the yield varies considerably based on the amount of template used in the reaction. For instance, while coleus was best amplified with 1 ng per reaction, Equisetum required 10 ng per reaction (data not shown). The fidelity of the reaction is extremely dependent upon total isolated DNA concentration and the inherent cpDNA: nuclear DNA ratio in total DNA. A pilot experiment testing for the production of PCR product over 4–5 orders of magnitude must be performed to optimize conditions for subsequent reactions.
PCR conditions for plastome amplification
Touchdown PCR was utilized to generate PCR amplicons [38]. The first set of reactions was performed using Round I conditions, conditions conducive to amplification in the species for which the primers were designed (Figure 1; Table 2). This reaction would routinely produce amplicons in 24 out of 27 reactions. Reactions that failed to produce a product were reconstituted from fresh reagents and template, and PCR is performed using Reaction II conditions (Table 2). Typically this amplification was sufficient to obtain complete coverage of the large IR in 7 of the 10 species studied (Table 3). Amplicons generated from this approach will be made feely available upon request.
If Round II conditions failed to produce a PCR product it could be assumed that sequence differences in the primer landing site are present or those sites are deleted or rearranged. In this case PCR is performed using each fragment-specific primer and primers from adjacent fragments that successfully amplified in Round I and/or Round II. The process is outlined in Figure 1.
Sequencing was performed directly from PCR products in a 96-well format at the University of Florida ICBR Core Facility using ET Terminator (Amersham Inc, Schaumburg, IL) as reported earlier [29]. A 5 μl aliquot of PCR product from a 50 μl reaction was analyzed by gel electrophoresis to verify purity and concentration. PCR products were treated with ExoSAP to remove primers and nucleotides. Each amplicon was sequenced bidirectionally using the primers used in initial amplification. Sequencing primers were added at 10 pmol/μl in a 10 μl final reaction volume. Sequences with Phred score of >20 were used for assembly. Both strands were sequenced for each fragment thus providing a 2X coverage and, 4X coverage in the overlapping regions.
Assembly, annotation and dissemination of the sequences from Fragaria and Prunus
Sequences generated from a primer pair were first aligned using Blast 2 sequences (bl2seq) available at NCBI website [39, 40]. Individual amplicons derived from this process were further assembled using CAP3 [41, 42]. The sequence was then annotated using DOGMA [24, 25]. Chloroplast genome sequences from the IR B region of Fragaria and Prunus were submitted to the GenBank under accession numbers FACPINVREP (Fa) and PPCPINVREP (Pp).
Abbreviations
- ASAP:
-
Amplification, sequencing, annotation of plastomes
- ORF:
-
Open reading frame
- AFLP:
-
Amplified fragment length polymorphism
- SNP:
-
Single nucleotide polymorphism
- PCR:
-
Polymerase chain reaction
- FACS:
-
Fluorescence-assisted cell sorting
- RCA:
-
Rolling circle amplification
- DOGMA:
-
Dual organeller genome annotator
References
Palmer JD: Chloroplast DNA and molecular phylogeny. Bioessays. 1985, 2: 263-267. 10.1002/bies.950020607.
Maliga P: Plastid transformation in higher plants. Annu Rev Plant Biol. 2004, 55: 289-313. 10.1146/annurev.arplant.55.031903.141633.
Grevich JJ, Daniell H: Chloroplast genetic engineering: Recent advances and future perspectives. Critical Reviews in Plant Sciences. 2005, 24: 83-107. 10.1080/07352680590935387.
Daniell H, Kumar S, Dufourmantel N: Breakthrough in chloroplast genetic engineering of agronomically important crops. Trends Biotechnol. 2005, 23: 238-245. 10.1016/j.tibtech.2005.03.008.
Dhingra A, Daniell H: Arabidopsis Protocols. Edited by: Salinas J and Sanchez-Serrano JJ. 2005, Totowa, New Jersey, Humana Press, 323: 525- Chloroplast genetic engineering via organogenesis or somatic embryogenesis, Second, Methods in Molecular Biology.
Sato S, Nakamura Y, Kaneko T, Asamizu E, Tabata S: Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res. 1999, 6: 283-290. 10.1093/dnares/6.5.283.
Schmitz-Linneweber C, Regel R, Du TG, Hupfer H, Herrmann RG, Maier RM: The plastid chromosome of Atropa belladonna and its comparison with that of Nicotiana tabacum: the role of RNA editing in generating divergence in the process of plant speciation. Mol Biol Evol. 2002, 19: 1602-1612.
Hiratsuka J, Shimada H, Whittier R, Ishibashi T, Sakamoto M, Mori M, Kondo C, Honji Y, Sun CR, Meng BY: The complete sequence of the rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol Gen Genet. 1989, 217: 185-194.
Schmitz-Linneweber C, Maier RM, Alcaraz JP, Cottet A, Herrmann RG, Mache R: The plastid chromosome of spinach (Spinacea oleracea): complete nucleotide sequence and gene organization. Plant Mol Biol. 2001, 45: 307-315. 10.1023/A:1006478403810.
Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K, Ohto C, Torazawa K, Meng BY, Sugita M, Deno H, Kamogashira T, Yamada K, Kusuda J, Takaiwa F, Kato A, Tohdoh N, Shimada H, Sugiura M: The complete nucleotide sequence of tobacco chloroplast genome:its gene organization and expression. EMBO J. 1986, 5: 2043-2049.
Ogihara Y, Isono K, Kojima T, Endo A, Hanaoka M, Shiina T, Terachi T, Utsugi S, Murata M, Mori N, Takumi S, Ikeo K, Gojobori T, Murai R, Murai K, Matsuoka Y, Ohnishi Y, Tajiri H, Tsunewaki K: Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 2002, 266: 740-746. 10.1007/s00438-001-0606-9.
Kato T, Kaneko T, Sato S, Nakamura Y, Tabata S: Complete structure of the chloroplast genome of a legume, Lotus japonicus. 2000, 7: 323-330.
Maier RM, Neckermann K, Igloi GL, Kossel H: Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J Mol Biol. 1995, 251: 614-628. 10.1006/jmbi.1995.0460.
Kim KJ, Lee HL: Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11: 247-261. 10.1093/dnares/11.4.247.
Saski C, Lee SB, Daniell H, Wood TC, Tomkins J, Kim HG, Jansen RK: Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. 2005, 59: 309-322.
Asano T, Tsudzuki T, Takahashi S, Shimada H, Kadowaki K: Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: a comparative analysis of four monocot chloroplast genomes. DNA Res. 2004, 11: 93-99. 10.1093/dnares/11.2.93.
Soltis DE, Albert VA, Savolainen V, Hilu K, Qiu YL, Chase MW, Farris JS, Stefanovic S, Rice DW, Palmer JD, Soltis PS: Genome-scale data, angiosperm relationships, and 'ending incongruence': a cautionary tale in phylogenetics. Trends in Plant Science. 2004, 9: 477-483. 10.1016/j.tplants.2004.08.008.
Wolf PG, Rowe CA, Sinclair RB, Hasebe M: Complete nucleotide sequence of the chloroplast genome from a leptosporangiate fern, Adiantum capillus-veneris L. DNA Research. 2003, 10: 59-65. 10.1093/dnares/10.2.59.
Martin W, Deusch O, Stawski N, Grunheit N, Goremykin V: Chloroplast genome phylogenetics: why we need independent approaches to plant molecular evolution. Trends Plant Sci. 2005, 10: 203-209. 10.1016/j.tplants.2005.03.007.
Project DT: . [http://www.flmnh.ufl.edu/deeptime/]
Project CGS: . [http://www.genome.clemson.edu/projects/Chloroplast/]
project C: . [http://evogen.jgi.doe.gov/second_levels/chloroplasts/jansen_project_home/chlorosite.html]
Wolf PG, Karol KG, Mandoli DF, Kuehl J, Arumuganathan K, Ellis MW, Mishler BD, Kelch DG, Olmstead RG, Boore JL: The first complete chloroplast genome sequence of a lycophyte, Huperzia lucidula (Lycopodiaceae). Gene. 2005, 350: 117-128. 10.1016/j.gene.2005.01.018.
Website D: . [http://bugmaster.jgi-psf.org/dogma/]
Wyman SK, Jansen RK, Boore JL: Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004, 20: 3252-3255. 10.1093/bioinformatics/bth352.
Palmer JD: Comparative organization of chloroplast genomes. Annu Rev Genet. 1985, 19: 325-354. 10.1146/annurev.ge.19.120185.001545.
Odintsova MS, Iurina NP: Plastidic genome of higher plants and algae: structure and function. Mol Biol (Mosk). 2003, 37: 768-783.
Jansen RK, Raubeson LA, Boore JL, dePamphilis CW, Chumley TW, Haberle RC, Wyman SK, Alverson AJ, Peery R, Herman SJ: Methods for obtaining and analyzing whole chloroplast genome sequences. Methods in Enzymology. 2005, , Academic Press, 348-384. 10.1016/S0076-6879(05)95020-9. Volume 395
Folta KM, Staton M, Stewart PJ, Jung S, Bies DH, Jesudurai C, Main D: Expressed sequence tags (ESTs) and simple sequence repeat (SSR) markers from octoploid strawberry (Fragaria x ananassa). BMC Plant Biol. 2005, 5: 12-10.1186/1471-2229-5-12.
Lin J, Davis TM: S1 analysis of long PCR heteroduplexes: detection of chloroplast indel polymorphisms in Fragaria. Theoretical and Applied Genetics. 2000, 101: 415-420. 10.1007/s001220051498.
Kumar S, Dhingra A, Daniell H: Plastid-expressed betaine aldehyde dehydrogenase gene in carrot cultured cells, roots, and leaves confers enhanced salt tolerance. Plant Physiology. 2004, 136: 2843-2854. 10.1104/pp.104.045187.
Kumar S, Dhingra A, Daniell H: Stable transformation of the cotton plastid genome and maternal inheritance of transgenes. Plant Molecular Biology. 2004, 56: 203-216. 10.1007/s11103-004-2907-y.
Cosner ME, Jansen RK, Palmer JD, Downie SR: The highly rearranged chloroplast genome of Trachelium caeruleum (Campanulaceae): multiple inversions, inverted repeat expansion and contraction, transposition, insertions/deletions, and several repeat families. Curr Genet. 1997, 31: 419-429. 10.1007/s002940050225.
Goremykin VV, Hirsch-Ernst KI, Wolfl S, Hellwig FH: Analysis of the Amborella trichopoda chloroplast genome sequence suggests that Amborella is not a basal angiosperm. Mol Biol Evol. 2003, 20: 1499-1505. 10.1093/molbev/msg159.
Matsuo M, Ito Y, Yamauchi R, Obokata J: The rice nuclear genome continuously integrates, shuffles, and eliminates the chloroplast genome to cause chloroplast-nuclear DNA flux. Plant Cell. 2005, 17: 665-675. 10.1105/tpc.104.027706.
website CW: . [http://www.ebi.ac.uk/clustalw/]
Demeke T, Adams RP: The effects of plant polysaccharides and buffer additives on PCR. Biotechniques. 1992, 12: 332-334.
Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS: Touchdown PCR to circumvent spurious priming during gene amplification. Nucleic Acids Research. 1991, 19: 4008-4008.
Tatusova TA, Madden TL: BLAST 2 SEQUENCES, a new tool for comparing protein and nucleotide sequences. Fems Microbiology Letters. 1999, 174: 247-250. 10.1016/S0378-1097(99)00149-4.
Huang XQ, Madan A: CAP3: A DNA sequence assembly program. Genome Research. 1999, 9: 868-877. 10.1101/gr.9.9.868.
Huang XQ, Madan A: . [http://genome.cs.mtu.edu/cap/cap3.html]
Acknowledgements
The authors would like to thank Professor Pam Soltis and Professor Doug Soltis, University of Florida, for critical review of the work and for their helpful comments and suggestions. The authors would also like to thank Jeremy Ramdial and Gene Peir for their technical support, Dr. Angel Alpuche-Solis, IPICYT, Mexico for Amaranthus and Lactuca sativa DNA; Professor John Davis, University of Florida for Pinus taeda DNA; Professor Harry Klee, University of Florida for tomato DNA; Professor Karen Koch, University of Florida for maize DNA; Professor Ben Elmo Whitty, University of Florida for tobacco plants and Professor John Gray, John Innes Institute, UK for pea chloroplast genome sequence. The authors acknowledge Denise Tombolato and Philip Stewart for critical reading and evaluation of this manuscript. This work was performed with support from NSF grant #0416877 (KMF) and funding from IFAS at the University of Florida.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AD conceived of the ASAP methodology, designed primers, prepared DNA, performed all PCR/sequencing reactions, and assembled/reported all sequences. AD also prepared all figures and tables for the manuscript. KMF assisted in shaping the ASAP concept and prepared the first draft of the manuscript. Both authors contributed to the development of the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Dhingra, A., Folta, K.M. ASAP: Amplification, sequencing & annotation of plastomes. BMC Genomics 6, 176 (2005). https://doi.org/10.1186/1471-2164-6-176
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-6-176