
Abstract
Background
Among functional elements of a metazoan gene, enhancers are particularly difficult to find and annotate. Pioneering experiments in Drosophila have demonstrated the value of enhancer "trapping" using an invertebrate to address this functional genomics problem.
Results
We modulated a Sleeping Beauty transposon-based transgenesis cassette to establish an enhancer trapping technique for use in a vertebrate model system, zebrafish Danio rerio. We established 9 lines of zebrafish with distinct tissue- or organ-specific GFP expression patterns from 90 founders that produced GFP-expressing progeny. We have molecularly characterized these lines and show that in each line, a specific GFP expression pattern is due to a single transposition event. Many of the insertions are into introns of zebrafish genes predicted in the current genome assembly. We have identified both previously characterized as well as novel expression patterns from this screen. For example, the ET7 line harbors a transposon insertion near the mkp3 locus and expresses GFP in the midbrain-hindbrain boundary, forebrain and the ventricle, matching a subset of the known FGF8-dependent mkp3 expression domain. The ET2 line, in contrast, expresses GFP specifically in caudal primary motoneurons due to an insertion into the poly(ADP-ribose) glycohydrolase (PARG) locus. This surprising expression pattern was confirmed using in situ hybridization techniques for the endogenous PARG mRNA, indicating the enhancer trap has replicated this unexpected and highly localized PARG expression with good fidelity. Finally, we show that it is possible to excise a Sleeping Beauty transposon from a genomic location in the zebrafish germline.
Conclusions
This genomics tool offers the opportunity for large-scale biological approaches combining both expression and genomic-level sequence analysis using as a template an entire vertebrate genome.
Similar content being viewed by others


Background
Human, mouse and rat genomes likely have less than 40 000 genes each [1–4]. This is only two to three times as many genes as in Caenorhabditis elegans and Drosophila melanogaster, and only six times as many as Saccharomyces cerevisiae [5–7]. The increased complexity of vertebrates therefore can not be simply accounted for by a larger gene number. A part of the increased complexity is thought to be accomplished by alternative splicing, RNA editing and the use of protein modifications to generate a variety of protein products from a single gene, but everything starts with increased complexity at the level of transcriptional regulation. While promoters are relatively simple and short in yeast, their complexity increases in multicellular organisms, making regulatory sequences ever harder to identify. In humans, enhancer elements can be located over a megabase away from the transcriptional start site [8]. Furthermore, current gene prediction programs used to annotate genomes often fail to correctly identify the 5' start site of a transcription unit, making in silico analysis of the regulatory sequences even more complex. To further complicate the matter, enhancer sequences diverge in evolution, co-evolving with their respective transcription factors, and often do not work across large evolutionary distances – worm to fly, for example [9]. This makes information from non-vertebrate model systems sometimes inapplicable to vertebrate sequences.
Enhancer detection ("trapping") using insertion site context vectors was popularized as a genomics tool in Drosophila. The first fly enhancer trap vectors were based on the P element transposon and often used the transposase's own promoter fused to the beta-galactosidase reporter gene for enhancer detection [10–12]. Several of the enhancer trap lines were shown to express the Lac Z reporter in cells corresponding to the expression patterns of nearby genes, validating the approach [12, 13]. In other work, promoters such as engrailed, fushi tarazu and Hsp70 were successfully developed for enhancer trapping in the fruitfly [14–16]. Further modifications to the system included the implementation of a bipartite system with a Gal4 transactivator [17], green fluorescent protein (GFP) [18], and even a GFP-Lac Z fusion protein [19] as reporters. In addition to the P element, other transposons such as hobo and piggyBac with insertion site preferences distinct from those of the P element have been used in Drosophila [20, 21]. The availability of a variety of transposons, promoters and reporters for enhancer trapping in the fruitfly enabled researchers to obtain enhancer trap insertions into a considerable fraction of Drosophila genes (reviewed by [22]) and allows an investigator to choose vectors most suitable for the problem at hand.
The ability to excise from a genomic location has been instrumental to the utility of P element based vectors. For mutation-causing insertions, reversion of the mutant phenotype by P element excision proves that a given insertion causes the mutation. Since the mutagenicity of Drosophila enhancer trap transposons is not significantly higher than the average 15% rate obtained with regular P elements, most insertions do not result in a mutation [22]. In these instances, the P element's ability to induce genomic deletions by "imprecise excision" can be used to obtain a mutation in the neighboring gene(s) [23].
The success of enhancer trapping in Drosophila prompted application of this approach in the mouse [24–27]. As was the case in Drosophila, the lac Z reporter was shown to be expressed in part of the target gene's expression domain [28]. Despite the considerable success of these early experiments, enhancer detection as an experimental approach in mouse was not explored further, giving way to different versions of gene traps (for a review, see [29]).
We believe the success of enhancer trapping in Drosophila can be largely attributed to the advantages of this experimental system over mouse. In Drosophila, large numbers of transgenic organisms can be readily generated and screened for gene expression patterns. It is far less practical in the mouse. This is partly due to the availability of efficient and precise transgene delivery tools in the fruitfly: the native P element, hobo and piggyBac transposons. In contrast, early mouse experiments were carried out by non-facilitated DNA transgenesis. This approach is less efficient and prone to induce deletions and other genome rearrangements in the recipient locus, as noted in the first published mouse enhancer trap locus [25, 30]. The compact nature of the Drosophila genome also contributed to the success of enhancer trapping, making the path from an enhancer trap insertion to the identification of the affected gene straightforward, especially once the Drosophila genome was sequenced.
The zebrafish Danio rerio is a vertebrate model system that provides many of the advantages found in invertebrates. A few hundred transparent, externally developing embryos can be obtained from a single pair of fish per week. The zebrafish genome is about two-fold smaller than the mouse genome, and its sequencing and annotation are nearing completion. Finally, transposon tools for efficient and precise transgene delivery into the zebrafish genome are available. We focused our research on the Sleeping Beauty (SB) transposon system [31, 32]. While not as efficient as the highest titer retrovirus used in zebrafish [33, 34], the Sleeping Beauty transposon system offers advantages in expression as well as ease of construction and testing of diverse vectors that can be done using basic molecular biology tools. Furthermore, the SB system offers the possibility of transposase-induced excision out of the genome to induce local deletions or to revert possible mutant phenotypes.
In this report, we investigated the potential of the SB transposon system for enhancer detection in zebrafish. Our results indicate that zebrafish enhancer trap lines with diverse GFP expression patterns can be readily generated using the SB system. Most of the obtained lines harbor a single transposon insertion event, facilitating the rapid identification of transposon insertion sites responsible for specific GFP expression patterns. We show that two enhancer trap lines exhibit GFP expression patterns matching the expression patterns of the target genes, and that both expected and novel gene expression patterns can be identified using this genomics tool. We conclude that enhancer trapping using the Sleeping Beauty transposon system is a viable experimental approach using as template a vertebrate genome.
Results
The Sleeping Beauty transposon can detect enhancers in cis
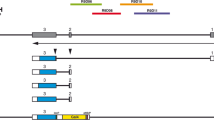
We have previously established multiple zebrafish lines using SB transposons with ubiquitous and tissue-specific promoters driving reporter expression [32]. Surprisingly, we did not observe any dependency of the expression pattern on the genomic context of the transposon insertion. Multiple studies describing insertion-site dependent transgene expression in vertebrates have suggested that many of those events are due to the transgene falling under control of nearby enhancers [35–40]. For enhancer detection approaches it is imperative that the reporter gene be sensitive to neighboring transcriptional regulatory elements. At least three explanations can be put forward to explain the absence of expression patterns in our previous work in zebrafish. First, the Sleeping Beauty transposons are flanked by relatively large inverted repeats. These repeats might function as silencer elements and not allow for transcriptional regulation across them. Second, the promoter we used (Xenopus laevis EF1α, [41]) may not be subject to transcriptional regulation by tissue-specific enhancers. Third, the expression level from the selected promoter may be too high to be effectively modulated, as enhancer traps usually contain attenuated promoters. To test these hypotheses, we decided to produce an artificial enhancer trapping event by cloning a tissue-specific promoter / enhancer just outside the inverted repeats and test for an increase in tissue-specific expression in injected embryos (Figure 1). We started with the transposon used in our previous work, pT2/S1 EF1α-GM2, which contains a shortened version of the Xenopus laevis EF1α promoter driving the GM2 version of GFP in a pT2 transposon vector [32]. We took advantage of the observation that relatively few pT2/S1 EF1α-GM2 injected embryos express GFP in the eye. We added a lens-specific Xenopus laevis γ1-Crystallin promoter [42] to the pT2/S1 EF1α-GM2 vector, as we had previously shown that this promoter specifically expresses in the lens of injected (F0) and transgenic (F1) zebrafish [32]. Embryos injected with pT2/S1 EF1α-GM2 or γ Cry1/pT2/S1 EF1α-GM2 were scored for any GFP fluorescence and for eye-specific GFP fluorescence at 3 dpf (Figure 1). The addition of γ Cry1 to pT2/S1 EF1α-GM2 caused a modest (two-fold) increase in injected embryos showing any GFP fluorescence. In contrast, the increase in eye-specific GFP expression was ten-fold (Figure 1). We concluded that at least in this assay, the Sleeping Beauty inverted repeat sequences do not block transcriptional regulation and that the EF1α promoter can be subject to transcriptional regulation from external, tissue-specific regulatory sequence elements.

Artificial enhancer trapping with a Sleeping Beauty transposon. Comparison of GFP expression in embryos injected with pT2/S1 EF1α or γCry1/pT2/S1 EF1α. Plasmids are diagramed as cartoons on the top of the picture. The SB transposon's inverted repeats are shown as boxes with open triangles, and the GFP open reading frame is depicted as a grey arrow. The gamma-Crystallin promoter/enhancer is shown as a hatched box. DNA-injected embryos which survived to 3 dpf were counted and scored for GFP fluorescence anywhere in the embryo (any GFP) and for fluorescence in the eye (eye GFP), even if there was additional fluorescence elsewhere. The average percentage of embryos positive for particular GFP fluorescence in three independent experiments is shown ± standard deviation.
Promoter truncations and pilot screens
We next tested the hypothesis that the absence of expression patterns in our previous work was due to the fact that S1 EF1α is expressed too strongly in transgenic animals. Since most successful enhancer traps in Drosophila and mice were based on truncated or weak promoters, we decided to attenuate the S1 EF1α promoter by removing sequences upstream of Bst 1107I (S2 EF1α) and Eco RI (S3 EF1α) restriction enzyme sites (Figure 2). We then co-injected the corresponding transposon constructs with SB10 transposase mRNA to assess germline transmission, expression and enhancer trapping rates. In pilot experiments, progeny from over 20 fish were screened with each construct. While overall germline transmission and expression rates were comparable (Figure 2), there was a difference in the expression patterns of the two constructs in transgenic embryos. Most of the transgenic animals generated with pT2/S3 EF1α-GM2 exhibited weak GFP expression, and we could not detect any expression patterns (data not shown). In contrast, when pT2/S2 EF1α-GM2 was used, most of the GFP-positive fish exhibited fairly strong, ubiquitous expression. Closer analysis indicated that many of these "ubiquitous" expression patterns were rather unique, with GFP expression often noted to be particularly strong in some tissues, consistent with a tissue-specific expression pattern superimposed on a ubiquitous expression pattern (data not shown). In most cases, GFP expression segregated as a single integration event, indicating that a tissue-specific expression pattern was not likely being masked by a ubiquitous expression pattern from a different insertion event. A similar phenomenon has been observed with Drosophila enhancer traps [11]. We speculate that in those instances, the GFP expression cassette may have fallen under the control of multiple enhancers – some tissue-specific, some ubiquitous. Alternatively, the ubiquitous expression may stem from the ubiquitous activity of the EF1α promoter used in the screen, with tissue-specific enhancers only elevating the expression levels in certain tissues, but not restricting it. We did not consider such expression patterns valuable and did not establish any fish lines with such GFP expression. Importantly, one of the founders in the pT2/S2 EF1α-GM2 pilot screen yielded three kinds of GFP expression in its progeny. Some were ubiquitously GFP positive, some showed a hatching gland-specific expression profile, and some exhibited both. When three F1 fish with both ubiquitous and hatching gland specific expression were raised and outcrossed, the two expression patterns exhibited independent segregation: 24% of the embryos were GFP negative, 26% expressed GFP ubiquitously, 25% had hatching gland-specific GFP expression, and 25% had both hatching gland-specific and ubiquitous expression (n = 245). Independent segregation indicates that the two transposon insertions causing the two expression patterns are unlinked. Two independent integration events were confirmed by Southern hybridization and inverse PCR (data not shown). The hatching gland-specific GFP expressing embryos were used to establish our first enhancer trap line, ET1 (Figure 3A). We concluded from our pilot screens that pT2/S2 EF1α-GM2 demonstrated the desired properties for potential use in enhancer trapping studies.

EF1α promoter truncations and endogenous enhancer trap screening. A diagram of the S1 EF1α promoter [32, 41]. Restriction enzyme sites are shown on top as single letters. S is Sph I, N is Nhe I, B is Bst 1107I and R is Eco RI. G/C, G and C rich box. Sp1, Sp1-like site. TATA, TATA box. Numbering below is relative to the first T of the TATA box. The table below the diagram shows the results of the pilot and scale-up (*) screens. Transgenesis and expression rates are shown, non-expressing transposon insertions were not scored. Transgenesis and expression rate from scale-up screen (#) is an underestimate since many founders were screened by incross and crosses from doubly transgenic founders were scored as a single transmission event (see text).

Enhancer trap lines exhibit a variety of unique GFP expression patterns. (A). Lateral view of GFP expression in Enhancer Trap line 1 (ET1) at 38 hours post fertilization (hpf). (B) ET3 at 5–6 somite stage. (C) ET3 at 36 hpf. (D) ET4 at 26 hpf. (E) ET5 at 30 hpf. (F) ET5 at 48 hpf. (G) ET6 at 26 hpf. (H) ET7 at 32 hpf. (I) Ventral view of ET7 at 5 dpf. (J) Lateral view of ET8 at 26 hpf. (K) Dorsal view of ET9 at 28 hpf. (L) Lateral view of ET9 at 30 hpf. In all panels, anterior is to the left. See text for details.
Germline excision of a Sleeping Beauty transposon insertion
We have previously demonstrated the excision of a Sleeping Beauty transposon from the genome in somatic tissues of transposase-injected zebrafish embryos [43]. We tested if such an excision event could be inherited by examining transposon excision in the germline. Embryos homozygous for the ET1 insertion were injected with SB10 transposase mRNA, and while some were used for a somatic excision assay the rest were raised to test for germline transmission of an excision event. A PCR reaction on genomic DNA from transposase-injected embryos with primers flanking ET1 insertion point produced two bands. A large band corresponded in size to the transposon insertion allele, and a small band corresponded to a transposon-less allele (data not shown). Both cannonical Sleeping Beauty transposon footprints (ATGTCAT and ATGACAT, [44, 45]) were obtained upon cloning and sequencing of the smaller band, indicating a transposase-mediated excision and DNA repair. 26 fish were screened for germline transmission (see Materials and Methods), and one was shown to transmit the expected excision footprint. We conclude that the Sleeping Beauty transposon can be excised from a genomic location in the zebrafish germline.
pT2/S2 EF1α-GM2 scale-up screening: 10% of GFP-expressing integrations yield tissue-specific patterns
One tissue-specific expression pattern was recovered from our pilot screen. We sought to recover more patterns and to test if enhancer detection in zebrafish is amenable to scale-up. To that end, we co-injected 3248 zebrafish embryos with the pT2/S2 EF1α-GM2 and SB10 transposase mRNA mix. 2102 embryos survived to day 3 for scoring, of which 848 were mosaic GFP positive and were selected to be raised. 330 survived to adulthood and were screened for germline transmission of GFP expression, primarily by sibling incrossing. This approach provided a lower estimate of the transgenesis and expression rate because it does not distinguish instances were both parents are transgenic. In this screen, at least 80 of the founder fish produced GFP-expressing progeny resulting in a minimum estimate of a 24% transgenesis rate. The actual transgenesis rate is closer to 30% because most of the fish were screened by incross, and if a pair produced GFP-expressing progeny, only one parent was counted as a transmitter. Eight of the GFP-expressing fish displayed distinct GFP expression patterns (Figure 3). Together with the pilot screen, 9 tissue-specific expression lines were obtained from 90 transgenic founder fish (10%) using the pT2/S2 EF1α-GM2 transposon.
Recovered expression patterns label a diverse array of tissues during embryogenesis
GFP expression in ET1 can be first observed in the polster region at 7–8 somite stage (not shown). The expression is very pronounced between 20 and 40 hours post-fertilization (hpf), when it marks the hatching gland (Figure 3A). Expression disappears as the hatching gland is resorbed. Line ET3 represents a pattern with the earliest onset of expression. Anterior localization of GFP in the diencephalon is detected by 5–6 somite stage in this line (Figure 3B). Extremely bright anterior expression persists in the ventral diencephalon (Figure 3C) and by 6 days post-fertilizations (dpf) is restricted slightly more posterior in the midline. The onset of expression for ET4 is 18 hpf with a bilateral expression pattern in cranial sensory ganglia that remains strong until 2 dpf and is undetectable by 5 dpf. This anterior expression in ET4 seems to label the lateral line ganglia both anterior and posterior to the otic vesicle (Figure 3D), however, no expression is detected in the lateral line in the trunk. In ET5 a single bilateral patch of strong GFP expression in the hyoid arch is observed by 24 hpf (Figure 3E), that by 48 hpf marks a more anterior location in the embryo (Figure 3F). Expression in this line is greatly diminished by 3 dpf and is undetectable by 5 dpf. Strong GFP expression is observed in ET6 by 26 hpf as a bilateral expression pattern consisting of two distinct patches in a subset of cranial sensory ganglia/placodes (Figure 3G). The expression weakens by 2 dpf and is undetectable by 3 dpf. GFP expression in ET7 begins weakly in the midbrain-hindbrain boundary (MHB) at 12–14 somites with the most pronounced expression in the anterior side of the MHB detected by 26 hpf (Figure 3H). Robust expression in the heart is first detected at around 32 hpf and remains ventricle-specific through 5 dpf (Figure 3I), even though expression in the brain is no longer restricted to the MHB. GFP expression in ET8 is already localized by 10–12 somites and remains strong in the telencephalon, and posterior side of the MHB through 26 hpf (Figure 3J). By 3 dpf the localized anterior expression is undetectable over autofluorescence, however, caudal expression appears to be enhanced in the dorsal neural tube. The onset of expression in ET9 occurs around 22 hpf and is difficult to detect by 2 dpf. At 28–30 hpf (Figure 3K,3L), three distinct expression domains are apparent in the telencephalon, diencephalon and hindbrain of ET9.
The ET2 line expresses GFP specifically in the motoneurons
We analyzed the ET2 line in detail because of the highly specific expression of GFP in these fish. GFP expression was first observed at the 16 somite stage, when 2 bilateral cells in the spinal cord of the 10 anterior somites become GFP positive (Figure 4). At later stages, multiple cells per somite become GFP positive, either due to continued expression of GFP mRNA or due to segregation of GFP to daughter cells. GFP expression follows the wave of somitogenesis, with the posterior-most somites lagging in GFP expression. At about 24 hours, ventrally-projecting axons become visible by GFP fluorescence. Later yet a pattern of nodes appears along the axons (Figure 4). Based on the position of neuronal cell body and the axonal trajectory, we conclude that caudal primary motoneurons express GFP in this line [46]. To our knowledge, this is the first gene to be specifically expressed only in this subpopulation of motoneurons. We therefore sought to identify the locus tagged by this transposon insertion.

The ET2 transgenic fish line expresses GFP in caudal primary motoneurons. GFP expression in ET2 was visualized in motoneurons using a bandpass GFP filter set at various stages of embryonic development. In all panels anterior is to the left. (A) The onset of GFP expression in ET2 line at 16 somite stage. (B) 26 somite stage. (C) 24 hpf. (D, E) 36 hpf. Axonal trajectories are visible at 24 and 36 hpf.
Southern analysis indicated that there is a single transposon insertion in this line, and it is linked to GFP expression (Figure 5). Inverse PCR identified a transposase-mediated insertion into a TA dinucleotide at position 256083 on contig ctg9701 (zebrafish genome assembly Zv3). The insertion occurred into a Genescan-predicted gene. Further analysis indicated that the Genescan-predicted gene actually consists of parts of at least two different genes, myoferlin and poly(ADP-ribose) glycohydrolase (PARG). The insertion located in the PARG part of the predicted transcript, 649 nucleotides from an exon just upstream of the PARG catalytic site. To confirm that the transposon insertion into the PARG gene induced GFP expression in primary motoneurons, we prepared genomic DNA from both GFP positive and GFP negative embryos from an independent outcross, and we conducted a PCR with NeuroIns-F1 and NeuroIns-R1 primers specific to the flanking sequences. In GFP negative embryos, a 0.5 kb band corresponding to wild type locus is noted. In GFP positive embryos, the same band is seen in addition to a larger band of approximately 2.4 kb, corresponding to a locus with transposon insertion (Figure 5). Since the inverse PCR and confirming PCR was performed on DNA from different batches of embryos, we can exclude the possibility of DNA contamination or fish husbandry error and conclude that the enhancer trap transposon insertion into the PARG gene causes GFP expression in caudal primary motoneurons.

Identification of the transposition event in the ET2 line. (A) The pT2/S2 EF1α transposon insertion into zebrafish genome is shown; restriction enzyme sites and primers used for molecular analysis are indicated. Transposon IR/DR's are shown as solid boxes with open triangles, and the GFP open reading frame is shown as a grey arrow. Genomic DNA is shown as a dotted line. N is Nsi I, E is Eco RV. (B) Southern blot on ET2 line outcross embryos. DNA from GFP positive (lanes 1 and 2) and GFP negative (lanes 3 and 4) embryos was digested with Nsi I (lanes 1 and 3) or Eco RV (lanes 2 and 4) and probed with a GFP-specific probe. (C) Linkage of the transposon insertion event to GFP expression. Primers flanking the transposon insertion event (arrows) were used to conduct PCR on DNA from GFP positive (lane 2) and GFP negative (lane 3) embryos from an ET2 outcross different from the one used in (B). Lane 1, λ Eco 47III Marker (Fermentas Inc).
GFP expression in ET2 line matches the expression of the endogenous PARG gene
Poly(ADP-ribosyl)ation is a protein modification that is extensively studied at the biochemical level and is associated with changes in DNA replication, recombination, repair and transcription [47], for a review, see [48]. Recently poly(ADP-ribosyl)ation was demonstrated to have a role in long term memory in the sea slug Aplysia [49]. Most organisms have multiple genes for poly (ADP-ribose) polymerases but only a single known gene for poly (ADP-ribose) glycohydrolase [48]. PARG activity is noted to be expressed in many cell lines, among them neuronal [50–53], but the tissue specificity of PARG expression during embryogenesis has not been reported for any organism. To test if the pT2/S2 EF1α-GM2 enhancer trap recapitulates the expression pattern of the endogenous PARG gene, we conducted whole mount in situ hybridization on ET2 outcross embryos to compare PARG and GFP reporter expression (Figure 6). In situ hybridization visualizes axonal cell bodies, the position of which appears indistinguishable with both PARG and GFP probes. We therefore conclude that GFP mRNA expression in this enhancer trap line faithfully recapitulates the expression of the zebrafish PARG gene during embryogenesis.

GFP expression in ET2 line embryos is indistinguishable from endogenous PARG gene expression. 23 hpf embryos collected from a heterozygous outcross were photographed for GFP fluorescence and sibling embryos were fixed for in situ hybridization. (A) In situ hybridization with PARG antisense probe. (B) In situ with GFP antisense probe. (C) Visualization of GFP expression in living embryos using a bandpass GFP filter set. (D) The same embryo as in (C) photographed using a bandpass GFP filter set with a low level of bright field illumination to visualize GFP expression in relative position to the somites.
Molecular analysis of other enhancer trap lines identifies target genes
We characterized insertion events in other enhancer trap lines. GFP positive F1 or F2 fish were outcrossed, and embryos were sorted into GFP positive and GFP negative pools. Genomic DNA was prepared from each pool, and Southern analysis was conducted to assess transposon copy number and linkage to the GFP expression pattern. In all lines except ET1 (see above), a single GFP expression-linked transposon insertion event was detected by Southern hybridization. We then conducted inverse PCR analysis on the DNA from GFP positive embryos to identify the insertion locus. For verification, DNA from embryos from an independent outcross was prepared and PCR was run with primers flanking the insertion site to link GFP expression and transposon insertion at a particular locus. Verified enhancer trap loci are presented in Table 1. Notably, seven of the insertions have occurred into introns of Genescan-predicted genes. Four of the tagged genes show significant homologies to previously identified genes: PARG (see above), MAPK upstream kinase-binding inhibitory protein (MBIP) [54], a member of cytochrome P450 superfamily and Nidogen [55]. The other three tagged predicted genes do not have significant homologies to previously identified genes. In the two lines which have insertions into intergenic regions, transposons have integrated less than 25 kb from the nearest predicted transcript.
ET7 line has a transposon insertion near mkp3 locus and matches mkp3 expression pattern
The transposon insertion in ET7 line has occurred into a predicted novel gene (Table 1). Closer investigation of the target locus revealed the presence of a previously characterized zebrafish mkp3 gene within 30 kb of the insertion site. Our attempts to amplify the predicted novel candidate gene from maternal and post-somitogenesis zebrafish cDNA libraries using 2 different primer pairs failed, while mkp3 cDNA was readily amplified in parallel PCR reactions (data not shown). This suggests that the novel target gene may be a false prediction by Genescan. The mkp3 gene encodes a dual specificity phosphatase which was cloned as a member of fgf8 synexpression group and is a negative feedback regulator of FGF8 signaling. mkp3 is expressed in the midbrain-hindbrain boundary, forebrain, tailbud, branchial arches, developing ear, pectoral fin buds and other tissues [56, 57]. GFP expression in ET7 line closely mimics mkp3 mRNA expression pattern in 23 hour embryo (Figure 7). The only significant difference is that GFP expression is stronger in somites and not as bright in the tailbud, even though the tailbud expression becomes brighter at later stages of development (data not shown). We did not observe GFP expression in the pectoral fin buds, even though we reproduced mkp3 expression in the fin buds just after after 24 hpf by in situ (data not shown, [56, 57]. An intriguing possibility is that mkp3 expression in pectoral fin buds may be controlled by a different enhancer, one we do not detect in this transgenic line. Additionally, ET7 expresses GFP in the heart after 24 hpf, and the expression clearly localizes to the ventricle at 5 dpf (Figure 3I). Expression of mkp3 in the heart after 24 hpf was not reported, and we did not conduct in situ hybridization on late pharyngula stage embryos to test for it. However, fgf8 is expressed in the ventricle of the zebrafish heart at 36 hpf [58]. Taken together, this suggests that GFP expression in ET7 line mimics a subset of the complete expression pattern of the zebrafish mkp3 gene.

GFP expression in ET7 line matches mkp3 mRNA expression. (A) GFP fluorescence photograph of an ET7 embryo at 23 hpf. (B) In situ hybridization on 23 hpf wild type embryo using mkp3 antisense RNA probe.
Discussion
In this paper, we describe the first use of enhancer trapping, or enhancer detection, as an experimental approach in zebrafish. We show that Sleeping Beauty transposons can trap enhancers by testing an artificial enhancer trapping event in vivo. This approach is likely to also be useful in the construction and testing of other trap vectors: gene (5' exon) and polyA (3' exon) and other related constructs. We then constructed two further truncations to the S1 EF1α promoter in the transgenesis cassette [32] and found one to be particularly suitable for enhancer trapping. Ten percent (9 of 90) of GFP-expressing transgenic fish generated lines with unique GFP expression patterns. All reagents described in this paper, including the enhancer trap fish strains, are readily available upon request http://beckmancenter.ahc.umn.edu.
Many of the obtained enhancer trap lines express GFP in the nervous system. This was previously observed with both mouse and Drosophila enhancer trap vectors and was speculated to stem from the transcriptional complexity of neural tissue [11, 28]. Several of our lines also exhibit some level of GFP expression in the eye. At least two explanations can be put forward to explain this observation. First, many genes are expressed in the developing eye. Thus, the eye expression that we see may reflect expression of the tagged genes in the eye. Second, optical properties of the tissues in the eye may permit detection of GFP expression that is lower that what would be required for detection in other tissues.
The ET2 line harbors a transposon insertion into the zebrafish gene for poly(ADP-ribose) glycohydrolase (PARG). We demonstrate that both PARG and GFP in ET2 line are expressed in caudal primary motoneurons of 23 hour old embryos. Thus, GFP expression in the ET2 line mimics that of an endogenous gene (PARG), indicating that transgene expression is under control of an endogenous enhancer. A very intriguing question is what the actual trapped enhancer sequence is, how far away from the genomic enhancer the trap can insert and still detect it, and weather artificial enhancer trap approach (Figure 1) can be used to answer these questions.
The ET7 line has a transposon insertion into a predicted novel gene 30 kb downstream of the zebrafish mkp3 locus. GFP expression in that line closely resembles part of the mkp3 expression domain, suggesting that the enhancer trap transposon in that line is under control of a subset of mkp3 enhancer elements.
Zebrafish enhancer trap lines will be valuable in future developmental genetics studies, be it classical mutagenesis or morpholino "knockdown" screening [59]. GFP expression can be used as a sensitive marker for certain tissue or cell types. For example, the ET1 line expresses GFP in the hatching gland. The expression of the hgg1 gene is specific to the polster and hatching gland depends on nodal signaling and is absent in one-eyed-pinhead mutants [60]. We phenocopied the one-eyed-pinhead mutation by morpholino injection in ET1 homozygotes and observed a complete loss of hatching gland-specific GFP expression (data not shown). While the ET1 line expresses GFP in an organ that can be readily observed using regular light microscopy techniques, other lines visualize tissues that are not nearly as easily morphologically accessible. In particular, the ET2 line visualizes the position of primary motoneuron cell bodies and axonal trajectory. Morpholinos against known genes or new members of the zebrafish secretome [61] can be screened for effects on neuronal cell body position or axonal pathfinding in the developing embryos by injection into ET2 line embryos. The ET7 line may provide a fluorescent readout of FGF8 signaling, thus facilitating the identification of genes involved in that signaling pathway.
A further utility offered by the transposon system is the possibility to revert a mutant phenotype or to generate localized deletions by transposon excision [23]. We successfully excised the transposon in the germline of the ET1 line, resulting in the expected transposon footprint. It has been shown that excision of the Sleeping Beauty transposon from a plasmid results in local deletions with fairly high frequency which is dependent on the cell type or tissue used [45]. Furthermore, the frequency of imprecise excision of Sleeping Beauty transposons significantly increases in cells with a compromised DNA repair pathway [62, 63]. It remains to be determined how frequently the excision of a Sleeping Beauty transposon from a genomic location in zebrafish germ line is accompanied by a deletion of flanking genomic DNA, and it should be possible to compromise the embryo's DNA repair machinery to induce such deletions at a high frequency.
Our experiments indicate that enhancer detection using Sleeping Beauty transposons is an easily scalable and efficient experimental technique in zebrafish. Obtaining fish with different GFP expression patterns is not the rate limiting step in this process. Preliminary molecular analysis of the insertion site is also straightforward using inverse PCR techniques. Identification of candidate genes should benefit from the progress in zebrafish genome sequencing and annotation. The main bottleneck step is the detailed biological analysis of GFP and the corresponding candidate gene expression profile.
In Drosophila, the generation of transposase-expressing lines of flies made enhancer detection and P-element mutagenesis in general a mainstream approach. Even without a similar gain in efficiency, transposase expressing fish lines would make enhancer trapping as well as related gene- and poly(A)-trap methodologies even more accessible for high-throughput functional analysis of the vertebrate genome.
Methods
Plasmid construction
pT2/S1 EF1α-GFP (pDB358) was previously published [32]. To make γ Cry/pT2/S1 EF1α-GFP (pDB375), a Bam HI-Hind III fragment from Cry1-GFP3 [42] containing part of the X. laevis γ-Cry1 promoter was cloned into the Ecl136II site of pDB358. To produce pT2/S2 EF1α-GFP (pDB371), a part of the EF1α promoter was deleted from pDB358 by ligation of the Bst 1107I-Nco I and Nhe I-Nco I fragments of pDB358. Similarly, the Eco RI-Nco I and Nhe I-Nco I fragments of pDB358 were ligated to produce pT2/S3 EF1α-GFP (pDB372).
Inverse PCR, PCR and primer sequences
For inverse PCR experiments, zebrafish genomic DNA was digested and ligated as described [64]. 1 and 2.5 microliters of the ligation reaction were used for the first PCR reaction with RP1/LP1 or RP1/GFP-R1 primers in total volume of 25 μl. 1 μl of the first PCR reaction was used as a template for the second (nested) PCR reaction with primer pairs RP2/LP2 or RP2/GFP-R2, respectively. Expand Hi Fi PCR system (Roche) was used for all PCR reactions. A MJ Research PTC-100 PCR machine was used for PCR with the following program : 92°C 4 min., 92°C 10 sec., 60°C 30 sec., 68°C 6 min., 30 cycles. Starting at cycle 11, 20 sec. per cycle was added to the extension time. The same PCR reaction with an annealing temperature 55°C was used for amplification with primers flanking transposon insertion sites, and for amplification of partial PARG cDNA from a maternal cDNA library. Primer sequences are: LP1 GTGTCATGCACAAAGTAGATGTCC [32]; LP2 ACTGACTTGCCAAAACTATTGTTTG; nRP1 CTAGGATTAAATGTCAGGAATTGTG; RP2 GTGAGTTTAAATGTATTTGGCTAAG; GFP-R1 TTCGGGCATGGCACTCTTG; GFP-R2 TATGATCTGGGTATCTCGCAA; NeuroC1-F1 CGTAAAGATGCCTTGTTCAGAA; NeuroC1-R1 ATTCCGTGACTCTCCTGAAATA; NeuroIns-F1 GGCTTGCATACATGACTAATG; NeuroIns-R1 GAAGACTGAAGTCCTCAAACT; HG1-1 ACATTGAGCCACTAAGCATTG; HG-2 TGTGTGCACTTAAGGGGCGA. Mkp3-F1 AGTGTTGCATTCTCCAGGATA; Mkp3-R1 TGACACAGAACTTCCCTGAAC; EF1a-F2 TTCCTGCAGGTCGACTCT; GFP-R0 GTGTAATCCCAGCAGCTG. Information about other primers is available from the authors upon request.
In situ hybridization
A partial sequence for the zebrafish poly(ADP-ribose) glycohydrolase cDNA was amplified using primers neuroC1-F1 and neuroC1-R1 and cloned using a Topo TA cloning kit (Invitrogen) to make pDB376. To make antisense RNA probe, pDB376 was digested with Spe I and transcribed with T7 RNA polymerase (Promega) and DIG labeling kit (Roche). GFP probe was made by amplifying GFP with 46 base pairs of EF1α promoter from pT2/S1 EF1α-GM2 using primers EF1a-F2 and GFP-R0, and cloning it into Topo TA cloning kit resulting in pSS100. pSS100 was linearized with Spe I and transcribed with T7 RNA polymerase using DIG labeling kit. To make mkp3 antisense probe, mkp3 cDNA was amplified from maternal cDNA library with primers Mkp3-F1 and Mkp3-R1 and cloned into Topo TA cloning kit to produce pDB528. The plasmid was linearized with Spe I and transcribed with T7 polymerase using DIG labeling kit.
Screening for germline transmission of Sleeping Beauty transposons
Embryos injected with SB10 transposase mRNA and transposon DNA mix were raised as described [32, 64]. In pilot screens, adult fish were outcrossed to brass for ease of husbandry. All collected embryos were screened for GFP expression at 1 day post fertilization (dpf) and 3 dpf. We set an arbitrary 200 embryo cutoff for screening, meaning that if less that 200 embryos were obtained from a founder, an additional cross was set up and to obtain additional embryos for screening. Analysis of transgenesis data from pilot screens indicated that 10% of transgenic lines would have been missed if cutoff was set at 100 embryos, and this less stringent coverage protocol was used in scale up screen. Also, we decided to limit screening to 1 dpf since none of the transgenics would have been missed in the pilot screens without the 3 dpf screening.
Transposon excision in the germline
Homozygous ET1 embryos were injected with SB10 transposase mRNA, raised and screened for loss of hatching gland specific GFP expression, or for a change in the GFP expression pattern. Twenty six fish were screened (R0, for Remobilization), and 2 gave GFP negative embryos, with an additional 2 giving ubiquitously GFP positive embryos, suggesting that germline remobilization events may have occurred in as many as 15% of transposase injected embryos. Ubiquitous GFP positive embryos (one in each of the two R0) did not survive. Of the two R0's that gave GFP negative embryos, one gave mosaic hatching gland expression in the next generation. PCR with transposon specific and flanking primers did not show any changes in the locus. The second R0 produced 19 embryos that were GFP negative from the total of 671 embryos obtainted. An R1 adult was outcrossed, embryo DNA was prepared, and PCR with primers HG1-1 and HG1-2 was conducted. The resulting PCR fragment was cloned using PCR 4 Topo cloning kit (Invitrogen). Plasmids were sequenced using M13 Forward primer, and one clone with a transposon footprint was identified. To confirm that it was not due to PCR contamination, a second clutch of embryos was obtained, the procedure was repeated, and the same footprint was obtained (data not shown).
References
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, Szustakowki J, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, International Human Genome Sequencing C: Initial sequencing and analysis of the human genome.[see comment][erratum appears in Nature 2001 Aug 2;412(6846):565 Note: Szustakowki J [corrected to Szustakowski J]]. Nature. 2001, 409 (6822): 860-921. 10.1038/35057062.
Venter JC, Adams MD, Myers EWLiPW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X: The sequence of the human genome.[see comment][erratum appears in Science 2001 Jun 5;292(5523):1838]. Science. 2001, 291 (5507): 1304-1351. 10.1126/science.1058040.
Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES, Mouse Genome Sequencing C: Initial sequencing and comparative analysis of the mouse genome.[see comment]. Nature. 2002, 420 (6915): 520-562. 10.1038/nature01262.
Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, Okwuonu G, Hines S, Lewis L, DeRamo C, Delgado O, Dugan-Rocha S, Miner G, Morgan M, Hawes A, Gill R, Celera , Holt RA, Adams MD, Amanatides PG, Baden-Tillson H, Barnstead M, Chin S, Evans CA, Ferriera S, Fosler C, Glodek A, Gu Z, Jennings D, Kraft CL, Nguyen T, Pfannkoch CM, Sitter C, Sutton GG, Venter JC, Woodage T, Smith D, Lee HM, Gustafson E, Cahill P, Kana A, Doucette-Stamm L, Weinstock K, Fechtel K, Weiss RB, Dunn DM, Green ED, Blakesley RW, Bouffard GG, De Jong PJ, Osoegawa K, Zhu B, Marra M, Schein J, Bosdet I, Fjell C, Jones S, Krzywinski M, Mathewson C, Siddiqui A, Wye N, McPherson J, Zhao S, Fraser CM, Shetty J, Shatsman S, Geer K, Chen Y, Abramzon S, Nierman WC, Havlak PH, Chen R, Durbin KJ, Egan A, Ren Y, Song XZ, Li B, Liu Y, Qin X, Cawley S, Cooney AJ, D'Souza LM, Martin K, Wu JQ, Gonzalez-Garay ML, Jackson AR, Kalafus KJ, McLeod MP, Milosavljevic A, Virk D, Volkov A, Wheeler DA, Zhang Z, Bailey JA, Eichler EE, Tuzun E, Birney E, Mongin E, Ureta-Vidal A, Woodwark C, Zdobnov E, Bork P, Suyama M, Torrents D, Alexandersson M, Trask BJ, Young JM, Huang H, Wang H, Xing H, Daniels S, Gietzen D, Schmidt J, Stevens K, Vitt U, Wingrove J, Camara F, Mar Alba M, Abril JF, Guigo R, Smit A, Dubchak I, Rubin EM, Couronne O, Poliakov A, Hubner N, Ganten D, Goesele C, Hummel O, Kreitler T, Lee YA, Monti J, Schulz H, Zimdahl H, Himmelbauer H, Lehrach H, Jacob HJ, Bromberg S, Gullings-Handley J, Jensen-Seaman MI, Kwitek AE, Lazar J, Pasko D, Tonellato PJ, Twigger S, Ponting CP, Duarte JM, Rice S, Goodstadt L, Beatson SA, Emes RD, Winter EE, Webber C, Brandt P, Nyakatura G, Adetobi M, Chiaromonte F, Elnitski L, Eswara P, Hardison RC, Hou M, Kolbe D, Makova K, Miller W, Nekrutenko A, Riemer C, Schwartz S, Taylor J, Yang S, Zhang Y, Lindpaintner K, Andrews TD, Caccamo M, Clamp M, Clarke L, Curwen V, Durbin R, Eyras E, Searle SM, Cooper GM, Batzoglou S, Brudno M, Sidow A, Stone EA, Payseur BA, Bourque G, Lopez-Otin C, Puente XS, Chakrabarti K, Chatterji S, Dewey C, Pachter L, Bray N, Yap VB, Caspi A, Tesler G, Pevzner PA, Haussler D, Roskin KM, Baertsch R, Clawson H, Furey TS, Hinrichs AS, Karolchik D, Kent WJ, Rosenbloom KR, Trumbower H, Weirauch M, Cooper DN, Stenson PD, Ma B, Brent M, Arumugam M, Shteynberg D, Copley RR, Taylor MS, Riethman H, Mudunuri U, Peterson J, Guyer M, Felsenfeld A, Old S, Mockrin S, Collins F, Rat Genome Sequencing Project C: Genome sequence of the Brown Norway rat yields insights into mammalian evolution.[see comment]. Nature. 2004, 428 (6982): 493-521. 10.1038/nature02426.
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG: Life with 6000 genes. Science. 1996, 274 (546): 563-547. 10.1126/science.274.5287.546.
Genome sequence of the nematode C. elegans: a platform for investigating biology. The C. elegans Sequencing Consortium. Science. 1998, 282 (5396): 2012-2018. 10.1126/science.282.5396.2012.
Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YH, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, Gabor GL, Abril JF, Agbayani A, An HJ, Andrews-Pfannkoch C, Baldwin D, Ballew RM, Basu A, Baxendale J, Bayraktaroglu L, Beasley EM, Beeson KY, Benos PV, Berman BP, Bhandari D, Bolshakov S, Borkova D, Botchan MR, Bouck J, Brokstein P, Brottier P, Burtis KC, Busam DA, Butler H, Cadieu E, Center A, Chandra I, Cherry JM, Cawley S, Dahlke C, Davenport LB, Davies P, de Pablos B, Delcher A, Deng Z, Mays AD, Dew I, Dietz SM, Dodson K, Doup LE, Downes M, Dugan-Rocha S, Dunkov BC, Dunn P, Durbin KJ, Evangelista CC, Ferraz C, Ferriera S, Fleischmann W, Fosler C, Gabrielian AE, Garg NS, Gelbart WM, Glasser K, Glodek A, Gong F, Gorrell JH, Gu Z, Guan P, Harris M, Harris NL, Harvey D, Heiman TJ, Hernandez JR, Houck J, Hostin D, Houston KA, Howland TJ, Wei MH, Ibegwam C, Jalali M, Kalush F, Karpen GH, Ke Z, Kennison JA, Ketchum KA, Kimmel BE, Kodira CD, Kraft C, Kravitz S, Kulp D, Lai Z, Lasko P, Lei Y, Levitsky AA, Li J, Li Z, Liang Y, Lin X, Liu X, Mattei B, McIntosh TC, McLeod MP, McPherson D, Merkulov G, Milshina NV, Mobarry C, Morris J, Moshrefi A, Mount SM, Moy M, Murphy B, Murphy L, Muzny DM, Nelson DL, Nelson DR, Nelson KA, Nixon K, Nusskern DR, Pacleb JM, Palazzolo M, Pittman GS, Pan S, Pollard J, Puri V, Reese MG, Reinert K, Remington K, Saunders RD, Scheeler F, Shen H, Shue BC, Siden-Kiamos I, Simpson M, Skupski MP, Smith T, Spier E, Spradling AC, Stapleton M, Strong R, Sun E, Svirskas R, Tector C, Turner R, Venter E, Wang AH, Wang X, Wang ZY, Wassarman DA, Weinstock GM, Weissenbach J, Williams SM, Woodage T, Worley KC, Wu D, Yang S, Yao QA, Ye J, Yeh RF, Zaveri JS, Zhan M, Zhang G, Zhao Q, Zheng L, Zheng XH, Zhong FN, Zhong W, Zhou X, Zhu S, Zhu X, Smith HO, Gibbs RA, Myers EW, Rubin GM, Venter JC: The genome sequence of Drosophila melanogaster. Science. 2000, 287 (5461): 2185-2195. 10.1126/science.287.5461.2185.
Nobrega MA, Ovcharenko I, Afzal V, Rubin EM: Scanning human gene deserts for long-range enhancers. Science. 2003, 302 (5644): 413-10.1126/science.1088328.
Ruvinsky I, Ruvkun G: Functional tests of enhancer conservation between distantly related species. Development. 2003, 130 (21): 5133-5142. 10.1242/dev.00711.
O'Kane CJ, Gehring WJ: Detection in situ of genomic regulatory elements in Drosophila. Proc Natl Acad Sci U S A. 1987, 84 (24): 9123-9127.
Bellen HJ, O'Kane CJ, Wilson C, Grossniklaus U, Pearson RK, Gehring WJ: P-element-mediated enhancer detection: a versatile method to study development in Drosophila. Genes Dev. 1989, 3 (9): 1288-1300.
Bier E, Vaessin H, Shepherd S, Lee K, McCall K, Barbel S, Ackerman L, Carretto R, Uemura T, Grell E: Searching for pattern and mutation in the Drosophila genome with a P-lacZ vector. Genes Dev . 1989, 3 (9): 1273-1287.
Wilson C, Pearson RK, Bellen HJ, O'Kane CJ, Grossniklaus U, Gehring WJ: P-element-mediated enhancer detection: an efficient method for isolating and characterizing developmentally regulated genes in Drosophila. Genes Dev. 1989, 3 (9): 1301-1313.
Hama C, Ali Z, Kornberg TB: Region-specific recombination and expression are directed by portions of the Drosophila engrailed promoter. Genes Dev. 1990, 4 (7): 1079-1093.
Kassis JA, Noll E, VanSickle EP, Odenwald WF, Perrimon N: Altering the insertional specificity of a Drosophila transposable element. Proc Natl Acad Sci U S A. 1992, 89 (5): 1919-1923.
Mlodzik M, Hiromi Y: Enhancer Trap Method in Drosophila: Its Application to Neurobiology. Methods in Neurosciences. 1992, 9: 397-414.
Brand AH, Perrimon N: Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development . 1993, 118 (2): 401-415.
Phelps CB, Brand AH: Ectopic gene expression in Drosophila using GAL4 system. Methods. 1998, 14 (4): 367-379. 10.1006/meth.1998.0592.
Timmons L, Becker J, Barthmaier P, Fyrberg C, Shearn A, Fyrberg E: Green fluorescent protein/beta-galactosidase double reporters for visualizing Drosophila gene expression patterns. Dev Genet. 1997, 20 (4): 338-347. 10.1002/(SICI)1520-6408(1997)20:4<338::AID-DVG5>3.0.CO;2-8.
Smith D, Wohlgemuth J, Calvi BR, Franklin I, Gelbart WM: hobo enhancer trapping mutagenesis in Drosophila reveals an insertion specificity different from P elements. Genetics. 1993, 135 (4): 1063-1076.
Horn C, Offen N, Nystedt S, Hacker U, Wimmer EA: piggyBac-based insertional mutagenesis and enhancer detection as a tool for functional insect genomics. Genetics. 2003, 163 (2): 647-661.
Bellen HJ: Ten years of enhancer detection: lessons from the fly. Plant Cell. 1999, 11 (12): 2271-2281. 10.1105/tpc.11.12.2271.
Robertson HM, Preston CR, Phillis RW, Johnson-Schlitz DM, Benz WK, Engels WR: A stable genomic source of P element transposase in Drosophila melanogaster. Genetics. 1988, 118 (3): 461-470.
Allen ND, Cran DG, Barton SC, Hettle S, Reik W, Surani MA: Transgenes as probes for active chromosomal domains in mouse development. Nature. 1988, 333 (6176): 852-855. 10.1038/333852a0.
Kothary R, Clapoff S, Brown A, Campbell R, Peterson A, Rossant J: A transgene containing lacZ inserted into the dystonia locus is expressed in neural tube. Nature. 1988, 335 (6189): 435-437. 10.1038/335435a0.
Gossler A, Joyner AL, Rossant J, Skarnes WC: Mouse embryonic stem cells and reporter constructs to detect developmentally regulated genes. Science. 1989, 244 (4903): 463-465.
Bonnerot C, Grimber G, Briand P, Nicolas JF: Patterns of expression of position-dependent integrated transgenes in mouse embryo. Proc Natl Acad Sci U S A. 1990, 87 (16): 6331-6335.
Korn R, Schoor M, Neuhaus H, Henseling U, Soininen R, Zachgo J, Gossler A: Enhancer trap integrations in mouse embryonic stem cells give rise to staining patterns in chimaeric embryos with a high frequency and detect endogenous genes. Mech Dev. 1992, 39 (1–2): 95-109. 10.1016/0925-4773(92)90029-J.
Stanford WL, Cohn JB, Cordes SP: Gene-trap mutagenesis: past, present and beyond. Nat Rev Genet. 2001, 2 (10): 756-768. 10.1038/35093548.
Brown A, Copeland NG, Gilbert DJ, Jenkins NA, Rossant J, Kothary R: The genomic structure of an insertional mutation in the dystonia musculorum locus. Genomics. 1994, 20 (3): 371-376. 10.1006/geno.1994.1190.
Ivics Z, Hackett PB, Plasterk RH, Izsvak Z: Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997, 91 (4): 501-510. 10.1016/S0092-8674(00)80436-5.
Davidson AE, Balciunas D, Mohn D, Shaffer J, Hermanson S, Sivasubbu S, Cliff MP, Hackett PB, Ekker SC: Efficient gene delivery and gene expression in zebrafish using the Sleeping Beauty transposon. Developmental Biology. 2003, 263 (2): 191-202. 10.1016/j.ydbio.2003.07.013.
Gaiano N, Amsterdam A, Kawakami K, Allende M, Becker T, Hopkins N: Insertional mutagenesis and rapid cloning of essential genes in zebrafish. Nature. 1996, 383 (6603): 829-832. 10.1038/383829a0.
Chen W, Burgess S, Golling G, Amsterdam A, Hopkins N: High-throughput selection of retrovirus producer cell lines leads to markedly improved efficiency of germ line-transmissible insertions in zebra fish.[erratum appears in J Virol 2002 May;76(10):5300]. Journal of Virology. 2002, 76 (5): 2192-2198. 10.1128/jvi.76.5.2192-2198.2002.
Jaenisch R, Jahner D, Nobis P, Simon I, Lohler J, Harbers K, Grotkopp D: Chromosomal position and activation of retroviral genomes inserted into the germ line of mice. Cell. 1981, 24 (2): 519-529. 10.1016/0092-8674(81)90343-3.
Lacy E, Roberts S, Evans EP, Burtenshaw MD, Costantini FD: A foreign beta-globin gene in transgenic mice: integration at abnormal chromosomal positions and expression in inappropriate tissues. Cell . 1983, 34 (2): 343-358. 10.1016/0092-8674(83)90369-0.
Bayer TA, Campos-Ortega JA: A transgene containing lacZ is expressed in primary sensory neurons in zebrafish. Development . 1992, 115 (2): 421-426.
Lin S, Yang S, Hopkins N: lacZ expression in germline transgenic zebrafish can be detected in living embryos. Developmental Biology . 1994, 161 (1): 77-83. 10.1006/dbio.1994.1009.
Field HA, Ober EA, Roeser T, Stainier DY: Formation of the digestive system in zebrafish. I. Liver morphogenesis. Dev Biol . 2003, 253 (2): 279-290. 10.1016/S0012-1606(02)00017-9.
Grabher C, Henrich T, Sasado T, Arenz A, Wittbrodt J, Furutani-Seiki M: Transposon-mediated enhancer trapping in medaka. Gene . 2003, 322: 57-66. 10.1016/j.gene.2003.09.009.
Johnson AD, Krieg PA: A Xenopus laevis gene encoding EF-1 alpha S, the somatic form of elongation factor 1 alpha: sequence, structure, and identification of regulatory elements required for embryonic transcription. Dev Genet. 1995, 17 (3): 280-290.
Offield MF, Hirsch N, Grainger RM: The development of Xenopus tropicalis transgenic lines and their use in studying lens developmental timing in living embryos. Development. 2000, 127 (9): 1789-1797.
Dupuy AJ, Clark K, Carlson CM, Fritz S, Davidson AE, Markley KM, Finley K, Fletcher CF, Ekker SC, Hackett PB, Horn S, Largaespada DA: Mammalian germ-line transgenesis by transposition. Proc Natl Acad Sci U S A. 2002, 99 (7): 4495-4499. 10.1073/pnas.062630599.
Plasterk RH, Izsvak Z, Ivics Z: Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet. 1999, 15 (8): 326-332. 10.1016/S0168-9525(99)01777-1.
Liu G, Aronovich EL, Cui Z, Whitley CB, Hackett PB: Excision of Sleeping Beauty transposons: parameters and applications to gene therapy. J Gene Med. 2004, 6 (5): 574-583. 10.1002/jgm.486.
Lewis KE, Eisen JS: Paraxial mesoderm specifies zebrafish primary motoneuron subtype identity. Development. 2004, 131 (4): 891-902. 10.1242/dev.00981.
Chambon P, Weill JD, Mandel P: Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963, 11: 39-43.
Masutani M, Nakagama H, Sugimura T: Poly(ADP-ribose) and carcinogenesis. Genes, Chromosomes & Cancer. 2003, 38 (4): 339-348. 10.1002/gcc.10250.
Cohen-Armon M, Visochek L, Katzoff A, Levitan D, Susswein AJ, Klein R, Valburn M, Schwartz JH: Long-term memory requires poluADP-ribosylation. Science. 2004, 304 (5678): 1820-1822. 10.1126/science.1096775.
Ying W, Swanson RA: The poly(ADP-ribose) glycohydrolase inhibitor gallotannin blocks oxidative astrocyte death. Neuroreport . 2000, 11 (7): 1385-1388.
Ohashi S, Kanai M, Hanai S, Uchiumi F, Maruta H, Tanuma S, Miwa M: Subcellular localization of poly(ADP-ribose) glycohydrolase in mammalian cells. Biochem Biophys Res Commun. 2003, 307 (4): 915-921. 10.1016/S0006-291X(03)01272-5.
Sevigny MB, Silva JM, Lan WC, Alano CC, Swanson RA: Expression and activity of poly(ADP-ribose) glycohydrolase in cultured astrocytes, neurons, and C6 glioma cells. Brain Res Mol Brain Res. 2003, 117 (2): 213-220. 10.1016/S0169-328X(03)00325-5.
Uchiumi F, Ikeda D, Tanuma S: Changes in the activities and gene expressions of poly(ADP-ribose) glycohydrolases during the differentiation of human promyelocytic leukemia cell line HL-60. Biochim Biophys Acta. 2004, 1676 (1): 1-11. 10.1016/j.bbaexp.2003.10.001.
Fukuyama K, Yoshida M, Yamashita A, Deyama T, Baba M, Suzuki A, Mohri H, Ikezawa Z, Nakajima H, Hirai S, Ohno S: MAPK upstream kinase (MUK)-binding inhibitory protein, a negative regulator of MUK/dual leucine zipper-bearing kinase/leucine zipper protein kinase. J Biol Chem . 2000, 275 (28): 21247-21254. 10.1074/jbc.M001488200.
Kohfeldt E, Sasaki T, Gohring W, Timpl R: Nidogen-2: a new basement membrane protein with diverse binding properties. J Mol Biol. 1998, 282 (1): 99-109. 10.1006/jmbi.1998.2004.
Kawakami Y, Rodriguez-Leon J, Koth CM, Buscher D, Itoh T, Raya A, Ng JK, Esteban CR, Takahashi S, Henrique D, Schwarz MF, Asahara H, Izpisua Belmonte JC: MKP3 mediates the cellular response to FGF8 signalling in the vertebrate limb. Nat Cell Biol. 2003, 5 (6): 513-519. 10.1038/ncb989.
Tsang M, Maegawa S, Kiang A, Habas R, Weinberg E, Dawid IB: A role for MKP3 in axial patterning of the zebrafish embryo. Development. 2004, 131 (12): 2769-2779. 10.1242/dev.01157.
Reifers F, Walsh EC, Leger S, Stainier DY, Brand M: Induction and differentiation of the zebrafish heart requires fibroblast growth factor 8 (fgf8/acerebellar). Development. 2000, 127 (2): 225-235.
Nasevicius A, Ekker SC: Effective targeted gene 'knockdown' in zebrafish.[see comment]. Nature Genetics. 2000, 26 (2): 216-220. 10.1038/79951.
Gritsman K, Zhang J, Cheng S, Heckscher E, Talbot WS, Schier AF: The EGF-CFC protein one-eyed pinhead is essential for nodal signaling. Cell. 1999, 97 (1): 121-132. 10.1016/S0092-8674(00)80720-5.
Klee EW, Ekker SC, Ellis LB: Target selection for Danio rerio functional genomics. Genesis. 2001, 30 (3): 123-125. 10.1002/gene.1045.
Yant SR, Kay MA: Nonhomologous-end-joining factors regulate DNA repair fidelity during Sleeping Beauty element transposition in mammalian cells. Molecular & Cellular Biology. 2003, 23 (23): 8505-8518. 10.1128/MCB.23.23.8505-8518.2003.
Izsvak Z, Stuwe EE, Fiedler D, Katzer A, Jeggo PA, Ivics Z: Healing the wounds inflicted by sleeping beauty transposition by double-strand break repair in mammalian somatic cells. Molecular Cell. 2004, 13 (2): 279-290. 10.1016/S1097-2765(03)00524-0.
Hermanson S, Davidson AE, Sivasubbu S, Balciunas D, Ekker SC: Sleeping Beauty Transposon for Efficient Gene Delivery. Methods in Cell Biology. 2004, 77: 351-364.
Acknowledgements
We thank Paul Phelps, Sandra Leo, Amanda Mahoney and Tessa Hodapp for help with fish screening, and Aubrey Nielsen, Rachel Bowers, Dan Carlson and Pat Cliff for fish maintenance and Perry Hackett for critical reading of the manuscript. We thank all members of the Arnold and Mabel Beckman Center for Transposon Research for valuable discussions. This research was supported by the Arnold and Mabel Beckman Foundation and the National Institutes of Health (DA14546).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' Contributions
The experiments described in this paper were planned, conducted and analyzed as a joint effort between the authors. In particular, DB, AD, SH and ZW contributed to fish screening, line establishment and to scientific descriptions of these lines, DB and SS to molecular analysis, AD and DB to GFP expression and in situ analysis. DB designed and built the transposons used in this study and was responsible for drafting the manuscript for publication. SE conceived and supervised the study and edited the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Balciunas, D., Davidson, A.E., Sivasubbu, S. et al. Enhancer trapping in zebrafish using the Sleeping Beauty transposon. BMC Genomics 5, 62 (2004). https://doi.org/10.1186/1471-2164-5-62
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-5-62