
Abstract
Background
Epilepsy is genetically complex neurological disorder affecting millions of people of different age groups varying in its type and severity. Copy number variants (CNVs) are key players in the genetic etiology of numerous neurodevelopmental disorders and prior findings also revealed that chromosomal aberrations are more susceptible against the pathogenesis of epilepsy. Novel technologies, such as array comparative genomic hybridization (array-CGH), may help to uncover the pathogenic CNVs in patients with epilepsy.
Results
This study was carried out by high density whole genome array-CGH analysis with blood DNA samples from a cohort of 22 epilepsy patients to search for CNVs associated with epilepsy. Pathogenic rearrangements which include 6p12.1 microduplications in 5 patients covering a total region of 99.9kb and 7q32.3 microdeletions in 3 patients covering a total region of 63.9kb were detected. Two genes BMP5 and PODXL were located in the predicted duplicated and deleted regions respectively. Furthermore, these CNV findings were confirmed by qPCR.
Conclusion
We have described, for the first time, several novel CNVs/genes implicated in epilepsy in the Saudi population. These findings enable us to better describe the genetic variations in epilepsy, and could provide a foundation for understanding the critical regions of the genome which might be involved in the development of epilepsy.
Similar content being viewed by others
Background
Epilepsy is one of the most common neurological disorder in humans with 1% prevalence and a lifetime incidence of up to 3%; characterized by recurrent and unprovoked seizures because of an abnormal electrical activity in central nervous system (CNS) [1]. More than 50 distinct epilepsy syndromes have been recognized with a broad range of clinical features; roughly it can be divided into idiopathic or symptomatic epilepsies. Metabolic disorders, infections, stroke, head trauma or brain tumors may cause symptomatic epilepsy whereas idiopathic seizure occur mainly because of genetic contribution [2]. Since long, it is observed that idiopathic epilepsy has a genetic component and its genetic etiology can be determined in a small fraction of cases. Development of genome wide technologies such as array-CGH and single-nucleotide polymorphism genotyping enabled the detection of submicroscopic microdeletions and microduplications also known as CNVs [3, 4]. CNVs being an important component of human genetic variation are key players in the genetic etiology of numerous neurodevelopmental disorders. A number of studies have recently highlighted the role of CNVs in the etiology of various disorders including autism [5, 6], intellectual disability (ID) [7] and schizophrenia [8, 9]. Recent studies on six genomic regions showed recurrent microdeletions on chromosomes 15q13.3, 16p13.11 and 15q11.2 and were identified as necessary genetic factors influencing idiopathic generalized epilepsy (IGE) [10–12]. Databases of normal and pathogenic genome variations are available on the web and are extremely valuable tools for interpreting CNVs identified in patients (Database of Genomic Variants: DGV; Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources: DECIPHER). Recent studies showed that 267 different genomic loci have been associated with well described microdeletion/microduplication syndromes (MMSs) [13]; these informations are constantly updated. In this study, we hypothesized that epilepsy could be caused by CNVs and that genes within those CNVs would be novel candidate genes for epilepsy. We selected a cohort of 22 patients with different types of epilepsies and performed high density whole genome array-CGH which showed novel CNVs/genes deletion and duplication that might be the contributory factors in the genetic etiology of epilepsy.
Results and discussion
Array-CGH results showed gains as well as losses in different genomic regions of 19 epilepsy patients from a cohort of 22 epilepsy patients. But, we reported the results of only 8 patients satisfying the cut off value of duplications and deletions (0.8 for duplication and -1.0 for deletion). Microduplication of 6p12.1 was observed in five patients including two from the same family and microdeletion of 7q32.3 in three patients was also found. These CNV findings were confirmed by qPCR.
Detection of 6p12.1 microduplications
Whole genome 2x 400K oligonucleotide based microarray analysis showed 99.9kb duplication at cytoband 6p12.1. Figure 2 presented 5 horizontal red lines of 5 patients with duplicated regions; each red line is marked with each patients ID along with the duplicated region of that specific patient. Out of these 5 patients, 2 were members of the same family whereas the rest of 3 were sporadic cases. No deletion or duplication was observed in father: 05, normal daughter: 1192 and normal son: 1191 (Figure 1). In affected mother: 1190, a microduplication of 51.6kb (55722611-55774293) and in affected daughter: 1193, a microduplication of 55.8kb (55718462-55774293) was observed. Rest of 3 sporadic cases also showed duplications of different sizes such as 241 showed a duplication of 95.6kb (55678636-55774293); 494 showed a duplication of 82.5kb (55691753-55774293) and 440 showed a duplication of 51.7kb (55726917-55778628). The gene that was observed in the duplicated region of all the patients is BMP5 (Table 1 and Figure 2); determined by UCSC Genome Browser (http://genome.ucsc.edu/) and Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home). BMP5 is a member of BMP family which belongs to transforming growth factor-β (TGFβ) superfamily. It is expressed in hippocampus, cerebellum and striatum [14, 15]. Studies also showed its expression in developing rat superior cervical ganglion corresponding to initial extension of primary dendrites and in early postnatal period during maximal dendritic growth. Immunofluorescence analysis of cultured superior cervical ganglion cells detected Bmp5 all over cell body of glial [16]. It was also observed that cell proliferation was reduced by BMP5 through cell death into the progenitor cells specifically in ventral forebrain. Insertion of BMP5 in developing chicken prosencephalon causes loss of ventral forebrain secondary to immense cell death localized to that region. Experimental embryos showed a loss of basal telencephalon due to implantation of beads soaked in recombinant BMP5 or BMP4 into neural tube of chicken forebrain that resulted in holoprosencephaly (a single cerebral hemisphere), cyclopia (a single midline eye) and loss of ventral midline structures [17]. Noradrenergic (NA) neurons are critical modulators of brain functions and have been implicated in common CNS disorders [18]. Locus coeruleus (LC), the major NA nucleus providing the main source of noradrenaline in the brain is formed in rostral hindbrain during embryogenesis. LC development requires either Bmp5 or Bmp7 and one is able to compensate the loss of the other. Also, the position of mid-hindbrain organizer determines the size of LC and propose that Bmp5/7 play an important role in mediating this organizer function [19]. Studies also uncovered novel roles of BMP signaling during development of heart, allantois, branchial arches, somites and forebrain development. Bmp5 do not appear to be involved in establishing pattern in these tissues but necessary for proliferation and maintenance of specific cell populations [20]. Observed duplications in all patients with reference to controls were confirmed by qPCR which showed a significant fold increase in all patients compared to controls Figure 3).

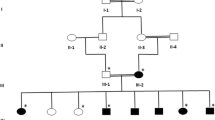
Pedigree of the epilepsy family; 05: normal father; 1190: affected mother; 1191: normal son: 1192: normal daughter: 1193: affected daughter.

Summary of genome wide gains in five patients. Whole genome 2x 400K oligonucleotide based microarray analysis showing 99.9kb duplication at cytoband 6p12.1. Five horizontal red lines showing duplicated regions of five patients; each red line is marked with each patients ID along with the duplicated region: 1193; 55718462-55774293, 241; 55678636-55774293, 1190; 55722611-55774293, 494; 55691753-55774293 and 440; 55726917-55778628. The gene predicted in the duplicated region is BMP5.

Confirmation of CNVs by qPCR. (A) The results showed BMP5 copy number amplifications in two patients; mother (1190) and the proband (1193) but not in healthy individuals. (B) BMP5 copy number amplifications were observed in three patients (241, 440 and 494) but not in healthy individuals.
Detection of 7q32.3 microdeletions
Whole genome 2x 400K oligonucleotide based microarray analysis showed 63.9kb microdeletion at cytoband 7q32.3. Figure 4 showed 3 horizontal green lines of 3 patients with deleted regions; each green line is marked with each patients ID along with the deleted region of that specific patient. In patient 1090, a microdeletion of 63.9kb (130860344-130924315); in 1186, a microdeletion of 59.5kb (130864738-130924315) and in 495, a deletion of 56.6kb (130864738-130921416) was observed. The gene that was observed in the deleted regions of all the patients was PODXL (Table 1 and Figure 4); determined by UCSC Genome Browser (http://genome.ucsc.edu/) and Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home). Podocalyxin (PODXL) is type-1 membrane mucin protein belongs to CD34 family expressed abundantly in kidney epithelial cells (podocytes) [21]; its expression has also reported in the brain with maximum level in cortical plate, hippocampus, cerebellum and basal forebrain nuclei [22, 23]. In humans, it is also linked with malignant progression of brain astrocytic tumors [24]. Studies also showed that ventricles of Podxl−/− mice were enlarged in coronal and horizontal brain MR images which were also confirmed through histological analysis of paraffin embedded brain [25]. Reason of this could be either an enhanced formation or reduced disposal of cerebrospinal fluid (CSF) or it might be due to a combination of both mechanisms. Enhanced volume of CSF might be the result of a new steady state equilibrium of choroid plexuses ion membrane transporters; Na+-K+ATPase, K+ channels and Na+-2Cl−-K+ co-transporters expressed in apical membrane and Cl−-HCO3 exchangers, a variety of Na+ coupled HCO3− transporters and K+-Cl− cotransporters expressed in the basolateral membrane [26]. Further analysis is required to check the involvement of these ion transporters toward ventricles enlargement in the absence of PODXL. Numerous human pathological disorders are linked with enlarged ventricles such as trauma, autism, bardet-biedl syndrome and alzheimer disease [27–29]. However, an earlier and possibly best human pathological condition studied was schizophrenia [30]. Finding of enlarged ventricles in the patients having first schizophrenia episode has raised the question of whether this neurodegenerative disease exhibited at the time of symptom onset or it is considered as a neurodevelopmental process which produces abnormal brain volumes at an early age [31]. Observed deletions in all the patients with reference to controls were confirmed by qPCR which showed a significant fold decrease in all the patients compared to controls (Figure 5).

Summary of genome wide losses in three patients. Whole genome 2x 400K oligonucleotide based microarray analysis showing 63.9kb deletion at cytoband 7q32.3. Three horizontal green lines showing deleted regions of three patients; each green line is marked with each patients ID along with the deleted region: 1090; 130860344-130924315, 1186; 130864738-130924315 and 495; 130864738-130921416. The gene which is observed in the deleted region is PODXL.

Confirmation of CNVs by qPCR. The results showed PODXL copy number deletions in three patients (1090, 495 and 1186) but not in healthy individuals.
Conclusion
CNVs are major contributory factors in developing the neurological disorders and also contribute in the genetic etiology of epilepsy. In this study, we described, for the first time, some of the novel CNVs/genes implicated in epilepsy in the Saudi population. Our findings present a better description of genetic variations in epilepsy, and could provide a foundation for the understanding of the critical regions of the genome which might be involved in the development of epilepsy. More comprehensive descriptions of patients and identification of possible causative CNVs and genes in the CNV regions will be required to identify new syndromes and improve the diagnoses of epilepsy.
Methods
Patients and families
22 patients were selected with primary diagnosis of different types of epilepsies including epilepsy (Epi), epilepsy plus mental retardation (Epi+MR) and juvenile myoclonic epilepsy (JME) shown in the Table 1. Details of all patients are as follows; Case 1190 and 1193: 1193 is a 4 years old female proband; her seizures were started at the age of 3 years. Her 33 years old mother 1190 had a consanguineous marriage and also suffering with seizures that were started at the age of 17 years. 05: father; 1192: sister and 1191: brother of the proband were normal and no CNV was found in any of these normal members of the family. One patient of the same family was died at the age of 5 years due to seizures (Figure 1). Case 440: She is 39 years old female patient of JME and her seizures were started the age of 16 years. Case 241: He is 14 years old male patient of JME and his seizures were started the age of 15 years. Case 1090: she is 15 years old female patient of epilepsy, her parents had a consanguineous marriage and both are normal. Blood samples of her parents, one normal sister and one normal brother was not available. Her seizures were started when she was 8 years old. Case 1186: He is 27 years old male patient of JME and his seizures were started the age of 16 years. Case 495: She is 13 years old female patient of epilepsy plus mental retarded. Her seizures were started at the age of 6 years. Case 494: He is 7 years old male patient of epilepsy plus mental retarded. His seizures were started the age of 3 years.
DNA preparation
Blood samples from all the patients were taken at King Abdulaziz University Hospital with their informed consent after the approval of ethical committee. DNA was extracted by using QIAamp DNA blood mini kit (http://www.Qiagen.com) and was quantified by NanoDrop ND-10000 Spectrophotometer.
Array-CGH analysis
Array-CGH analysis was done by Agilent sure print G3 Hmn CGH 2x 400K arrays (Agilent Technologies, Santa Clara, USA) following manufacturer protocol. Briefly, 500ng patient’s DNA and also reference DNA of the same sex (Promega, Madison, USA) were digested with RsaI (Promega, Madison, USA) and AluI (Promega, Madison, USA) for 2hrs at 37ºC. Furthermore, these samples were labeled by random primers through Agilent labeling kit (Agilent Technologies, Santa Clara, USA) following manufacturer guidelines. Patient’s DNA was labeled with Cy5-dUTP whereas reference DNA was labelled with Cy3-dUTP. Labeled products were purified by Microcon YM-30 filter units (Millipore, Billerica, USA). Patient and reference DNAs were mixed with Cot-1 DNA (Invitrogen, USA) blocking agent and hybridization buffer following by Agilent instructions. After denaturation at 95ºC and pre-annealing at 37ºC, hybridization was done at 65ºC for 40hrs. After the two washing steps, array was analyzed through Agilent Scanner (G2505C) and Feature Extraction Software (V.1.5.1.0). Data was analyzed by using Partek Genomics Suite Software, Cytogenomics Software (V.2.0.6.0; Agilent Technologies, USA) and publicly available DGV.
Validation of CNVs by real-time quantitative PCR
The microdeletions and microduplications were confirmed by real-time quantitative PCR (qPCR). Targeted sequences were chosen for qPCR by using Primer-3 Software (V.0.4.0). Two pair of primers were selected for each of genomic region of two target genes; bone morphogenetic protein-5 (BMP5) and podocalyxin-like protein (PODXL); an endogenous gene β2-microglobulin (B2M) and control samples. PCR was done in 10μl reaction volume comprising of 05μl SYBR-Green qPCR master mix (KAPA Biosystems, USA), 10 pmol forward and reverse primers and 20ng genomic DNA. Reaction cycling conditions were as follows; denaturation at 95°C for 10minutes, annealing at 60°C for 1minute, holding stage at 60°C for 30seconds followed by 45 cycles at 60°C for 15seconds. Each run of 96 well plate included control samples, target genes, reference gene and non-template control for each gene. Plate was analyzed with StepOnePlus™ Real-Time PCR Systems (StepOnePlus™ Real-Time PCR, Applied Biosystems, Canada) raw data was acquired using Light Cycler 480 Software. Ct is the threshold cycle defined as the mean cycle at which the fluorescence curve reaches an arbitrary threshold; ΔCt is the difference between the Ct of the target gene and that of reference gene; ΔΔCt is ΔCt value of patients obtained by dividing the patient ΔCt value with ΔCt value of control sample. T-test with significant P-value <0.05 was used to find out statistical significance of predicted copy number alterations.
Abbreviations
- Array-CGH:
-
Array comparative genomic hybridization
- CNVs:
-
Copy number variations
- qPCR:
-
Real-time quantitative PCR
- CNS:
-
Central nervous system
- BMP5:
-
Bone morphogenetic protein-5
- PODXL:
-
Podocalyxin-like protein
- B2M:
-
β2-microglobulin
- IGE:
-
Idiopathic generalized epilepsy
- JME:
-
Juvenile myoclonic epilepsy
- Epi+MR:
-
Epilepsy plus mental retardation
- DGV:
-
Database of Genomic Variant.
References
Hauser WA, Annegers JF, Rocca WA: Descriptive epidemiology of epilepsy: contributions of population-based studies from Rochester, Minnesota. Mayo Clinic proceedings. 1996, 71 (6): 576-586. 10.4065/71.6.576.
Davidsson J, Collin A, Olsson ME, Lundgren J, Soller M: Deletion of the SCN gene cluster on 2q24.4 is associated with severe epilepsy: an array-based genotype-phenotype correlation and a comprehensive review of previously published cases. Epilepsy research. 2008, 81 (1): 69-79. 10.1016/j.eplepsyres.2008.04.018.
Koolen DA, Pfundt R, de Leeuw N, Hehir-Kwa JY, Nillesen WM, Neefs I, Scheltinga I, Sistermans E, Smeets D, Brunner HG, et al: Genomic microarrays in mental retardation: a practical workflow for diagnostic applications. Human mutation. 2009, 30 (3): 283-292. 10.1002/humu.20883.
Gurnett CA, Hedera P: New ideas in epilepsy genetics: novel epilepsy genes, copy number alterations, and gene regulation. Archives of neurology. 2007, 64 (3): 324-328. 10.1001/archneur.64.3.324.
Christian SL, Brune CW, Sudi J, Kumar RA, Liu S, Karamohamed S, Badner JA, Matsui S, Conroy J, McQuaid D, et al: Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biological psychiatry. 2008, 63 (12): 1111-1117. 10.1016/j.biopsych.2008.01.009.
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al: Structural variation of chromosomes in autism spectrum disorder. American journal of human genetics. 2008, 82 (2): 477-488. 10.1016/j.ajhg.2007.12.009.
Friedman JM, Baross A, Delaney AD, Ally A, Arbour L, Armstrong L, Asano J, Bailey DK, Barber S, Birch P, et al: Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. American journal of human genetics. 2006, 79 (3): 500-513. 10.1086/507471.
Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, et al: Large recurrent microdeletions associated with schizophrenia. Nature. 2008, 455 (7210): 232-236. 10.1038/nature07229.
Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, et al: Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008, 320 (5875): 539-543. 10.1126/science.1155174.
de Kovel CG, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, Kluck C, Muhle H, von Spiczak S, Ostertag P, et al: Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain : a journal of neurology. 2010, 133 (Pt 1): 23-32.
Dibbens LM, Mullen S, Helbig I, Mefford HC, Bayly MA, Bellows S, Leu C, Trucks H, Obermeier T, Wittig M, et al: Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Human molecular genetics. 2009, 18 (19): 3626-3631. 10.1093/hmg/ddp311.
Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, et al: 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nature genetics. 2009, 41 (2): 160-162. 10.1038/ng.292.
Weise A, Mrasek K, Klein E, Mulatinho M, Llerena JC, Hardekopf D, Pekova S, Bhatt S, Kosyakova N, Liehr T: Microdeletion and microduplication syndromes. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2012, 60 (5): 346-358. 10.1369/0022155412440001.
Charytoniuk DA, Traiffort E, Pinard E, Issertial O, Seylaz J, Ruat M: Distribution of bone morphogenetic protein and bone morphogenetic protein receptor transcripts in the rodent nervous system and up-regulation of bone morphogenetic protein receptor type II in hippocampal dentate gyrus in a rat model of global cerebral ischemia. Neuroscience. 2000, 100 (1): 33-43. 10.1016/S0306-4522(00)00246-3.
Harvey BK, Hoffer BJ, Wang Y: Stroke and TGF-beta proteins: glial cell line-derived neurotrophic factor and bone morphogenetic protein. Pharmacology & therapeutics. 2005, 105 (2): 113-125. 10.1016/j.pharmthera.2004.09.003.
Beck HN, Drahushuk K, Jacoby DB, Higgins D, Lein PJ: Bone morphogenetic protein-5 (BMP-5) promotes dendritic growth in cultured sympathetic neurons. BMC neuroscience. 2001, 2: 12-10.1186/1471-2202-2-12.
Golden JA, Bracilovic A, McFadden KA, Beesley JS, Rubenstein JL, Grinspan JB: Ectopic bone morphogenetic proteins 5 and 4 in the chicken forebrain lead to cyclopia and holoprosencephaly. Proceedings of the National Academy of Sciences of the United States of America. 1999, 96 (5): 2439-2444. 10.1073/pnas.96.5.2439.
Berridge CW, Waterhouse BD: The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain research Brain research reviews. 2003, 42 (1): 33-84. 10.1016/S0165-0173(03)00143-7.
Tilleman H, Hakim V, Novikov O, Liser K, Nashelsky L, Di Salvio M, Krauthammer M, Scheffner O, Maor I, Mayseless O, et al: Bmp5/7 in concert with the mid-hindbrain organizer control development of noradrenergic locus coeruleus neurons. Molecular and cellular neurosciences. 2010, 45 (1): 1-11. 10.1016/j.mcn.2010.05.003.
Solloway MJ, Robertson EJ: Early embryonic lethality in Bmp5;Bmp7 double mutant mice suggests functional redundancy within the 60A subgroup. Development. 1999, 126 (8): 1753-1768.
Kerjaschki D, Sharkey DJ, Farquhar MG: Identification and characterization of podocalyxin--the major sialoprotein of the renal glomerular epithelial cell. The Journal of cell biology. 1984, 98 (4): 1591-1596. 10.1083/jcb.98.4.1591.
Vitureira N, McNagny K, Soriano E, Burgaya F: Pattern of expression of the podocalyxin gene in the mouse brain during development. Gene expression patterns : GEP. 2005, 5 (3): 349-354. 10.1016/j.modgep.2004.10.002.
Lin WL, Pang VF, Liu CH, Chen JY, Shen KF, Lin YY, Yu CY, Hsu YH, Jou TS: Pleomorphic extra-renal manifestation of the glomerular podocyte marker podocalyxin in tissues of normal beagle dogs. Histochemistry and cell biology. 2007, 127 (4): 399-414. 10.1007/s00418-006-0252-8.
Hayatsu N, Kaneko MK, Mishima K, Nishikawa R, Matsutani M, Price JE, Kato Y: Podocalyxin expression in malignant astrocytic tumors. Biochemical and biophysical research communications. 2008, 374 (2): 394-398. 10.1016/j.bbrc.2008.07.049.
Nowakowski A, Alonso-Martin S, Gonzalez-Manchon C, Larrucea S, Fernandez D, Vilar M, Cerdan S, Ayuso MS, Parrilla R: Ventricular enlargement associated with the panneural ablation of the podocalyxin gene. Molecular and cellular neurosciences. 2010, 43 (1): 90-97. 10.1016/j.mcn.2009.09.011.
Brown PD, Davies SL, Speake T, Millar ID: Molecular mechanisms of cerebrospinal fluid production. Neuroscience. 2004, 129 (4): 957-970.
Hardan AY, Minshew NJ, Mallikarjuhn M, Keshavan MS: Brain volume in autism. Journal of child neurology. 2001, 16 (6): 421-424. 10.1177/088307380101600607.
Davis RE, Swiderski RE, Rahmouni K, Nishimura DY, Mullins RF, Agassandian K, Philp AR, Searby CC, Andrews MP, Thompson S, et al: A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proceedings of the National Academy of Sciences of the United States of America. 2007, 104 (49): 19422-19427. 10.1073/pnas.0708571104.
Nestor SM, Rupsingh R, Borrie M, Smith M, Accomazzi V, Wells JL, Fogarty J, Bartha R, Alzheimer's Disease Neuroimaging I: Ventricular enlargement as a possible measure of Alzheimer's disease progression validated using the Alzheimer's disease neuroimaging initiative database. Brain : a journal of neurology. 2008, 131 (Pt 9): 2443-2454.
Johnstone EC, Crow TJ, Frith CD, Husband J, Kreel L: Cerebral ventricular size and cognitive impairment in chronic schizophrenia. Lancet. 1976, 2 (7992): 924-926.
Steen RG, Mull C, McClure R, Hamer RM, Lieberman JA: Brain volume in first-episode schizophrenia: systematic review and meta-analysis of magnetic resonance imaging studies. The British journal of psychiatry : the journal of mental science. 2006, 188: 510-518. 10.1192/bjp.188.6.510.
Acknowledgements
This project was supported by NSTIP strategic technologies program in the Kingdom of Saudi Arabia- Project No. (12-BIO3059-03). The authors also, acknowledge with thanks Science and Technology Unit and Deanship of Scientific Research (DSR), under Grant no. (HiCi/1432/6-1) King Abdulaziz University for their technical and financial support. This article processing and publishing charges is paid by CEGMR. HSJ is supported by KACST project code "8-MED 120-3".
This article has been published as part of BMC Genomics Volume 16 Supplement 1, 2015: Selected articles from the 2nd International Genomic Medical Conference (IGMC 2013): Genomics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/16/S1
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no conflicts of interest.
Authors' contributions
MIN, MF and AGC designed the study. MF and MIN performed the experiments, analyzed the data and wrote the manuscript, MMA performed microarray. AGC, TAK, MMJ, MHQ, HSJ contributed in writing and editing the manuscript. All authors read and approved the final manuscript.
Muhammad Imran Naseer, Muhammad Faheem contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Naseer, M.I., Faheem, M., Chaudhary, A.G. et al. Genome wide analysis of novel copy number variations duplications/deletions of different epileptic patients in Saudi Arabia. BMC Genomics 16 (Suppl 1), S10 (2015). https://doi.org/10.1186/1471-2164-16-S1-S10
Published:
DOI: https://doi.org/10.1186/1471-2164-16-S1-S10