
Abstract
Background
With the development of space science, it is important to analyze the relationship between the space environment and genome variations that might cause phenotypic changes in microbes. Klebsiella pneumoniae is commonly found on the human body and is resistant to multiple drugs. To study space-environment-induced genome variations and drug resistance changes, K. pneumoniae was carried into outer space by the Shenzhou VIII spacecraft.
Results
The K. pneumoniae strain LCT-KP289 was selected after spaceflight based on its phenotypic differences compared to the ground-control strain. Analysis of genomic structural variations revealed one inversion, 25 deletions, fifty-nine insertions, two translocations and six translocations with inversions. In addition, 155 and 400 unique genes were observed in LCT-KP214 and LCT-KP289, respectively, including the gene encoding dihydroxyacetone kinase, which generates the ATP and NADH required for microbial growth. Furthermore, a large number of mutant genes were related to transport and metabolism. Phylogenetic analysis revealed that most genes in these two strains had a dN/dS value greater than 1, indicating that the strain diversity increased after spaceflight. Analysis of drug-resistance phenotypes revealed that the K. pneumoniae strain LCT-KP289 was resistant to sulfamethoxazole, whereas the control strain, LCT-KP214, was not; both strains were resistant to benzylpenicillin, ampicillin, lincomycin, vancomycin, chloramphenicol and streptomycin. The sulfamethoxazole resistance may be associated with sequences in Scaffold7 in LCT-KP289, which were not observed in LCT-K214; this scaffold contained the gene sul1. In the strain LCT-KP289, we also observed a drug-resistance integron containing emrE (confers multidrug resistance) and ant (confers resistance to spectinomycin, streptomycin, tobramycin, kanamycin, sisomicin, dibekacin, and gentamicin). The gene ampC (confers resistance to penicillin, cephalosporin-ii and cephalosporin-i) was present near the integron. In addition, 30 and 26 drug-resistance genes were observed in LCT-KP289 and LCT-KP214, respectively.
Conclusions
Comparison of a K. pneumoniae strain obtained after spaceflight with the ground-control strain revealed genome variations and phenotypic changes and elucidated the genomic basis of the acquired drug resistance. These data pave the way for future studies on the effects of spaceflight.
Similar content being viewed by others


Background
With the rapid development of space technology, space capsules are frequently launched to explore the universe; therefore, it is important to understand space biology. Space environmental physics has elucidated the existence of properties including electromagnetic radiation, microgravity, high vacuum and strong magnetic field in the space environment; however, it is important to understand the impact of these factors on organisms. Paul et al. focused on plants, which are significant components of biological systems, and discussed the adaption and growth tropism of plants in the microgravity environment in a space shuttle [1]. Gridhani et al. examined proton-induced perturbations in gene expression, cell cycle and cell division as well as the differences between the effects of protons and high-energy proton radiation [2, 3]. Gao et al. observed that bacterial metabolism was significantly altered in the space environment [4]; furthermore, exposure to the space environment might cause genetic damage [5]. Tixador et al. studied the growth and antibiotic resistance of Escherichia coli during the mission of the space shuttle Discovery [6]. However, mutations caused by the space environment have not been examined at the genomic level. Klebsiella pneumoniae is an important Gram-negative, opportunistic pathogen that causes severe diseases such as septicemia, pneumonia, urinary tract infections, and soft-tissue infections [7]. Many clinical strains of K. pneumoniae are highly resistant to antibiotics, which poses a major threat to global public health. Over the past decade, the physiology, biochemistry, and regulation of K. pneumoniae pathways have been extensively studied [8–11]. However, the effect of spaceflight on K. pneumoniae has not been examined at the genomic level. K. pneumoniae is well-suited for such studies because of its characteristics.
In 2011, the Shenzhou VIII spacecraft carried K. pneumoniae strains into outer space for approximately 17 days (398 hours). The control strain was cultured at the same temperature in an incubator on earth. After spaceflight, the antibiotic resistance and pathogenicity of the strains were examined. Based on these analyses, the LCT-KP289 strain obtained after spaceflight was selected and compared to the control strain LCT-KP214. The genomes of LCT-KP289 and LCT-KP214 were sequenced to compare their genomic variations. These analyses revealed genes potentially related to drug resistance, and analysis of the putative drug-resistance genes revealed variations in the homologous genes in the two strains. Studies on these candidate resistance genes will be important to improve understanding of the drug resistance of K. pneumoniae.
Results
Genomic features of the strains LCT-KP214 and LCT-KP289
The genome sequences of LCT-KP214 and LCT-KP289 were deposited in NCBI under the GenBank accession numbers AJHE00000000 and ATRO00000000, respectively. The genomes are described in Table 1. Genome alignments revealed that most regions were present in both strains, including 339 alignment blocks that covered 95.90% of the LCT-KP214 genome and 91.45% of the LCT-KP289 genome. The strain LCT-KP289 exhibited a larger genome size, and the extra sequences mainly comprised repetitive sequences (including tandem repeat fragments and interspersed repeated sequences).
Based on the assembly results, the genes in strains LCT-KP214 and LCT-KP289 were predicted and annotated using COGs (Cluster of Orthologous Groups). Using this analysis, 3,791 and 3,935 genes were assigned to specific gene functions in strains LCT-KP214 and LCT-KP289, respectively (Figure 1). Genome alignment revealed that most genes in LCT-KP289 were present in LCT-KP214 and that the additional genes in LCT-KP289 were most likely repeated sequences. The unique genes in these strains are discussed below. The predicted genes were annotated using the ARDB (Antibiotic Resistance Genes Database), and 26 and 30 genes were assigned antibiotic-resistance functions in LCT-KP214 and LCT-KP289, respectively. The assembly results were then annotated using plasmid databases, and some scaffolds in LCT-KP214 and LCT-KP289 were homologous to plasmid genomes. The plasmid annotation analysis revealed that some plasmids did not belong to K. pneumoniae (Table 2). However, the presence and origin of these plasmids require further analysis.

Genomic structural variation and distribution of paired genes. The structural variations in the genomes and paired genes are shown. The circles represent (inner to outer) the LCT-KP214 GC-skew distribution, LCT-KP214 COG distribution, and the structural variations between LCT-KP214 and LCT-KP289, respectively. LCT-KP214 GC-skew distribution: a 1,000-bp sliding window was used on the genome of LCT-KP214, and the GC-skew was recorded in each window. GC-skew = (G-C)/(G + C); purple and green indicate positive and negative values, respectively. The junction between purple and green represents the potential transcription initiation site. LCT-KP214 COG distribution: the inner, second and third circles represent the COG functional distribution in LCT-KP214. Different functions are indicated by different colors, and the captions are listed on the right. Structural variations between LCT-KP214 and LCT-KP289: the circles indicate the genomic structural variation, syntenic regions and gene pairs in LCT-KP214 and LCT-KP289 (the genes located on the forward strand are indicated in sky blue, and the genes located on the reverse strand are indicated in orange). The outer circles represent LCT-KP214, and the inner circles represent LCT-KP289. The lines connecting the circles indicate paired genes in LCT-KP214 and LCT-KP289, and the different colors indicate structural variations, transfer elements and duplicated sequences.
Detection of genomic structural variations and functional enrichment of variant genes
The genomic variations in LCT-KP214 and LCT-KP289 were analyzed and the genomic differences, including sequence variants, were identified (Figure 1). We defined variant genes as those that contained SNPs or InDels, were partly located in syntenic or repeat regions, or were unpaired (genes present at similar loci in both strains were considered paired). Subsequently, the variant genes in strains LCT-KP214 and LCT-KP289 were annotated using the COG/GO/KEGG databases and classified by their functions (Figure 2A).Analysis of genome variation revealed 1 inversion, 25 deletions, 59 insertions, 1 translocation and 6 translocations with inversions (Figure 1). Genes outside syntenic regions (mainly separated by large insertions or deletions) were identified, including 389 and 629 genes, respectively, in LCT-KP214 and LCT-KP289. In addition, the two strains contained some homologous genes. After filtering the homologous genes, we observed 155 and 400 unique genes in LCT-KP214 and LCT-KP289, respectively. Based on COG annotations, the functional categories “Lipid transport and metabolism”, “Inorganic ion transport and metabolism” and “Secondary metabolites biosynthesis, transport and catabolism” were significantly enriched in the unique genes in LCT-KP289; the functional category “Inorganic ion transport and metabolism” was significantly enriched in the unique genes in LCT-KP214 (Figure 2B). Furthermore, we detected 11 interspersed repeated sequences with duplication units longer than 400 bp, and the copy numbers of the repeated sequences were different. The genes located in the repeat regions were not significantly enriched in COG or KEGG functional categories; however, 9 gene functional categories were enriched based on GO annotations, including “fatty acid metabolic process”, “peptidoglycan biosynthetic process”, and “peptidoglycan-based cell wall” (Figure 2D).

Functional enrichment of variant genes after KEGG/COG/GO annotation. Picture A: Analysis of functional enrichment. The red and green bars represent LCT-KP214 and LCT-KP289, respectively. The genes containing SNPs or InDels, those located in repeat regions or those unique to each strain were analyzed separately. Each classification square contained three paired bars, indicating the number of enriched functional items, the number of annotated variant genes, and the number of all annotated genes in the strain from left to right. Pictures B, C and D indicate the detailed enrichment results of variant genes after COG, KEGG and GO annotation. In pictures B, C, and D, the annotation axis is classified by functional items. The hollow columns represent the number of all annotated genes in the strain. The solid columns represent the number of annotated variant genes. The arrows in pictures B and C and the asterisk in picture D indicate significantly enriched functional items.
In addition to the large-scale genome variations, 770 SNPs were identified (3,133 raw SNPs; 2,363 SNPs containing N bases, nearly N bases or with low sequencing depth were filtered). To analyze the genes affected by SNPs, we selected SNPs located in CDS or intergenic regions no further than 300 bp from the nearest gene. We observed that 334 and 330 genes were affected by SNPs in LCT-KP289 and LCT-KP214, respectively. Furthermore, 99 InDels were also identified (133 raw InDels; 34 InDels containing Ns or with low sequencing depth were filtered). InDel-affected genes were identified using the method described for SNPs; 299 and 304 genes were affected by InDels in LCT-KP289 and LCT-KP214, respectively (not all InDels were located in CDS regions in both strains). Based on the GO annotations, 48 and 49 functional categories were enriched in the genes containing SNPs and InDels, respectively, in the strain LCT-KP289. Most of the affected genes encoded molecular functions related to transmembrane transporters (Additional file 1: Table S1). Based on KEGG annotations, the unique gene KP214_00701 in LCT-KP214 was assigned the function “Glycerolipid metabolism (ko00561)” [Figure 2C]; this gene contained 12 SNPs and 5 InDels. KP214_00701 encodes dihydroxyacetone kinase (dha), which is required to generate ATP and NADH for microbial growth [12]. However, this gene was not functional in LCT-KP289 because of a nonsense mutation (GAA- > TAA). This mutation in LCT-KP289 might have been caused by the severe environmental changes during spaceflight.
Analysis of phylogenetic relationships
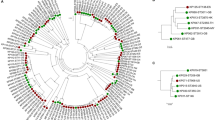
To understand the effect of the environment on strain evolution, the phylogenetic relationship between LCT-KP214 and LCT-KP289 was analyzed, and the base substitution rates were compared. We downloaded five other K. pneumoniae genome sequences from NCBI (ftp://ftp.ncbi.nih.gov/genbank/genomes/Bacteria) and used Bayesian analysis to generate phylogenetic trees at the whole-genome (Figure 3A) and gene levels (Figure 3B). The base substitution rates in LCT-KP214 and LCT-KP289 were 3.4e-05 and 4.6e-05, respectively, at the genome level, and 5e-06 and 5.7e-04, respectively, at the gene level. Therefore, LCT-KP289 had higher base substitution rates at the genome and gene levels. We used the phylogenetic trees and 3,678 core genes of the seven K. pneumoniae strains (Figure 3C) to calculate the dN/dS ratios in LCT-KP214 and LCT-KP289 using the CODONML software (in PAML version 4.4, January 2010) with the GY-HKY model. Comparison of LCT-KP214 and LCT-KP289 revealed that most genes in these strains shared the same selection pressure, and the dN/dS value of most of the genes was greater than 1 (Figure 3D), which indicated that the strains tended to have greater diversity under special circumstances, such as spaceflight.

Phylogenetic analysis. Picture A: A phylogenetic tree was constructed at the genome level, and the phylogenetic relationships among these 7 strains are shown. Picture B: A phylogenetic tree was constructed at the gene level. The base substitution rates of the genes in LCT-KP289 were higher compared to those in LCT-KP214. Picture C: The distribution of the number of genes in each K. pneumoniae strain and the number of their core genes. Picture D: dN/dS comparison between LCT-KP214 and LCT-KP289. For the paired samples, the p value was 0.001989 by the Wilcoxon test and 2.66e-06 by Student's t-test.
Furthermore, heterozygotic SNPs, which might reflect differential rates of evolution, were observed in these two strains. We mapped the reads to the assembly results and selected bases with a query value greater than 20 to analyze SNP heterozygosis. We selected heterozygotic SNPs that satisfied the following criteria: frequency of the second-most abundant nucleotide greater than 0.05 and total depth greater than 50X. The comparison of the base frequencies in the core genomes of strains LCT-KP214 and LCT-KP289 is shown in Figure 4. We observed that most base types were stable in LCT-KP214, but strain LCT-KP289 had increased base-type diversity. The statistics of the annotation results in Table 3 indicated that most heterozygotic SNPs were nonsynonymous. The analysis also revealed that the K. pneumoniae bacterial colonies had greater genome variation after spaceflight.

Base frequencies of the core genomes of LCT-KP214 and LCT-KP289. Homo indicates that the base pair was homozygous at the locus, and hete indicates that the base pair was heterozygous at the locus. Homo:Hete indicates that the base-types were stable in LCT-KP214, but not in LCT-KP289; Hete:Homo indicates that the base-types were stable in LCT-KP289, but not in LCT-KP214; Hete:Hete indicates that the base-types were not stable in LCT-KP214 or LCT-KP289.
Characterization of drug-resistance and related genes
Spaceflight can cause genome variation that leads to alterations in bacterial drug resistance [13, 14]. We measured the growth rates and characterized the drug resistance phenotypes of strain LCT-KP289 after spaceflight and of the ground-control strain LCT-KP214. Both strains had similar growth curves (Figure 5A). However, the two strains differed in their resistance to sulfamethoxazole; LCT-KP289 was resistant to this compound, but LCT-KP214 was not. Both strains exhibited similar resistance to benzylpenicillin, ampicillin, lincomycin, vancomycin, chloramphenicol, and streptomycin (Figure 5B).

Phenotypic analyses. Picture A: The growth curves of LCT-KP214 and LCT-KP289 were determined by measuring OD600 values, which represent the concentration of the bacterial population. The growth curves of LCT-KP214 and LCT-KP289 were nearly identical. Picture B: The antibiotic resistance phenotypes were characterized by diffusion tests. The diameter of the zone of inhibition was measured for each antibiotic, and the drug was applied in a 0.6-cm diameter circle. If the diameter of the zone of inhibition of an antibiotic was 0.6 cm after incubation of the bacterial plate, the strain was considered resistant to the antibiotic. The data showed that LCT-KP289 was resistant to sulfamethoxazole, but LCT-KP214 was not.
The sulfonamide sulfamethoxazole might inhibit dihydrofolate synthetase and repress DNA synthesis in K. pneumoniae. To identify the sulfamethoxazole-resistance mechanism in LCT-KP289, we analyzed the ARDB annotation results and observed that the sul1 gene, which is important for sulfonamide resistance, was duplicated. LCT-KP214 had only one copy of sul1 (in Scaffold4), but LCT-KP289 had two copies of this gene (in Scaffold4 and Scaffold7). sul1 encodes an alternative dihydrofolate synthetase that is not inhibited by sulfonamide; therefore, sulfonamides cannot block the synthesis of dihydropteroate, leading to sulfonamide resistance [15]. Plasmid annotation analysis revealed that the scaffolds containing the sul1 gene were homologous to plasmid genomes (Table 2). As the sul1 gene is mostly associated with class 1 integrons, we decided to explore the genetic context of sul1. In both strains, the sul1 gene was part of a class 1 integron (Figure 6). Integrons can capture gene cassettes by site-specific recombination and drive the expression of these genes. Integrons are present in plasmids or transposons [16], which leads to the spread of drug-resistance genes. In addition to the integrase (intI) gene, Class 1 integrons usually contain genes encoding resistance to quaternary ammonium compounds, ethidium bromide (qacED) and sulfonamides (sul1) [17–19]. In this study, LCT-KP214 and LCT-KP289 contained resistance integrons (RI), which included the gene emrE (encoding the ethidium bromide-methyl viologen resistance protein) and other resistance genes such as ant (ant2ia and ant3ia encode streptomycin 3’-adenylyltransferase and aminoglycoside o-nucleotidyl transferase, respectively, which can modify aminoglycosides such as spectinomycin, streptomycin, tobramycin, kanamycin, sisomicin, dibekacin and gentamicin by adenylation) (Figure 6). In addition to integrons, transposon resolvase (Tn21) and the gene ampC were observed near the sul1 gene. ampC encodes a Class A beta-lactamase, which confers resistance to penicillin, cephalosporin-ii and cephalosporin-i. In addition, 30 genes in LCT-KP289 and 26 genes in LCT-KP214 were assigned to ARDB, and these genes are important for K. pneumoniae antibiotic resistance (Additional file 2: Table S2).

Gene elements near the sul1 g ene. Two copies of the sul1 gene were observed in LCT-KP289, but only one copy was observed in LCT-KP214. We observed two integrases in the vicinity of the sul1 gene in LCT-KP214 and LCT-KP289. Between these genes, other resistance genes, such as ant, which confers resistance to spectinomycin, streptomycin, tobramycin, kanamycin, sisomicin, dibekacin and gentamicin, were observed. The gene ampC, which confers resistance to penicillin, cephalosporin-ii and cephalosporin-I, was also observed near the sul1 gene. The different-colored arrows indicate the direction and length of the genes.
Discussion
Based on the assembly results, the genome of LCT-KP289 was larger than that of LCT-KP214; genome alignment and prediction of genome elements revealed that the extra sequences in LCT-KP289 were from tandem repeat fragments and interspersed repeated sequences. Analysis of genome variation revealed that LCT-KP289 contained more unique genes than LCT-KP214. GO functional annotation revealed that the unique genes of LCT-KP289 were enriched in 9 functional items, including oxidation-reduction, biofilm formation, and arginine catabolism. Therefore, the genomic changes in LCT-KP289 were related to environmental adaptation. We analyzed the evolutionary rate and phylogenetic relationships of LCT-KP289. The base frequencies of the core genomes revealed a greater number of heterozygotic SNPs in LCT-KP289 compared to LCT-KP214. The phylogenetic trees at the genome and gene levels indicated that LCT-KP289 had a greater base-substitution rate. The dN/dS analysis revealed that most genes in LCT-KP289 had dN/dS values greater than 1. Together, these data indicated that special environmental factors can stimulate base substitutions in bacteria and that environmental selection pressure might influence the direction of bacterial evolution [20].
To identify the genes that were most affected by the environment, the variant genes in LCT-KP214 and LCT-KP289 were annotated using the COG, KEGG and GO databases. These annotations revealed that some functional categories were enriched in LCT-KP289 compared to LCT-KP214; these categories were related to bacterial metabolism such as “Lipid transport and metabolism”, “Inorganic ion transport and metabolism” and “Secondary metabolites biosynthesis, transport and catabolism”. However, using KEGG annotation, LCT-KP289 lacked the dihydroxyacetone kinase gene, which is important for the generation of ATP and NADH, because of a premature stop codon [21]. Therefore, we propose that, in LCT-KP289, mutations occurred more frequently in genes related to environmental adaptation.
K. pneumoniae is highly pathogenic and encodes multiple types of drug resistance genes. In this study, we analyzed the drug-resistance phenotypes of LCT-KP214 and LCT-KP289. Both strains were resistant to several drugs, except sulfamethoxazole, and LCT-KP289 was more resistant to this drug than was LCT-KP214. Based on genome alignment and ARDB annotation, we observed that LCT-KP289 contained an extra copy of the sul1 gene in scaffold7. sul1 encodes an alternative form of dihydrofolate synthetase, which cannot be inhibited by the drug [22]. Therefore, environmental selection pressure might cause genome variations, leading to an additional copy of the sul1 gene in LCT-KP289 and subsequent enhanced drug resistance.
Furthermore, we observed gene elements related to antibiotic resistance in K. pneumoniae. Integron determinants, including gene cassettes and several antibiotic-resistance genes (e.g., emrE, ant and sul1), were observed in both strains. Antibiotics can be divided into four classes based on their mechanism of action: i) repression of bacterial cell-wall synthesis, ii) destruction of the cell membrane structure, iii) repression of protein synthesis, and iv) repression of DNA synthesis. emrE encodes an efflux pump that confers multidrug resistance [23]. The protein encoded by the gene ant potentially modifies aminoglycosides by adenylation, conferring resistance to protein synthesis inhibitors such as spectinomycin, streptomycin, tobramycin, kanamycin, and gentamicin [24]. Because integrons are mobile genetic elements, the antibiotic genes contained in integrons can be transferred among different strains. Based on plasmid annotation, we observed that the integrons in LCT-KP289 and LCT-KP214 might originate from Escherichia coli plasmids. We observed the gene ampC, which encodes beta-lactamase, flanking the integrons. This enzyme can inactivate cell-wall synthesis inhibitors such aspenicillin and cephalosporin [25]. Furthermore, the Tn21 resolvase was observed near the integrons, which might enhance the mobility of the antibiotic-resistance genes. Other antibiotic-resistance genes, including those related to polymyxin and bacitracin, were dispersed in the genome of K. pneumoniae.
Conclusion
In this study, comparative genomics was used to analyze the strain LCT-KP289, which was selected after spaceflight, and the ground-control strain LCT-KP214 to identify the relationship between the unique space environment and genome variation; furthermore, the potential rate of evolution was studied using phylogenetic analysis. We observed that the space environment affected the evolutionary rates of environment-related genes. Phenotypic analysis revealed that LCT-KP289 had increased resistance to sulfonamides, potentially due to an increased copy number of the gene sul1. Furthermore, the observation of integrons in K. pneumoniae provides insight into the mechanism of multidrug resistance. The HMP data have shown that the bacteria were multidrug resistant and highly pathogenic. Therefore, this study paves the way for future studies on the effect of spaceflight on drug resistance and pathogenicity.
Methods
Bacterial strains and growth conditions
The K. pneumoniae strain (CGMCC 1.1736) used in this study was obtained from the Chinese General Microbiological Culture Collection Center and was carried in the Shenzhou VIII unmanned spacecraft for more than 17 days (398 hours). This strain is NCTC 5056, which was clinically isolated from patients diagnosed with pneumonia. The strains were cultured in LB, which contained tryptone (10 g/liter), yeast extract (5 g/liter), NaCl (10 g/liter), and agar powder (15 g/liter). The pH of the culture medium was adjusted to 7.0-7.2. The culture broth was identical to the culture medium but lacked Bacto Agar. The Shenzhou VIII unmanned spacecraft was launched by a Long March 2 F rocket at 5:58 on November 1, 2011 (GMT + 8). It arrived at the Tiangong-1 space station (approximately 340 km apogee distance) and successfully docked with the space station at 17:28(GMT + 8). After docking, the Shenzhou VIII spacecraft and Tiangong-1 remained connected for 12 days at an approximate apogee distance of 350 km. The Shenzhou VIII spacecraft successfully separated from the Tiangong-1 space station and docked again on November 14. Subsequently, the Shenzhou VIII spacecraft remained with the Tiangong-1 space station for three more days. The return capsule of the Shenzhou VIII spacecraft completed the experimental task and left the Tiangong-1 space station on November 17. It landed in the Inner Mongolia region at 20:38 on November 17 (GMT + 8). Microbiological samples were quickly removed and transported to Beijing by military helicopters. The samples arrived at the laboratory of the Chinese PLA General Hospital at 7:17 on November 18 (GMT + 8). As a control, the same K. pneumoniae strain was maintained in a laboratory incubator on earth under the same temperature conditions as the cabin of the Shenzhou VIII spacecraft. The temperature conditions were adjusted according to the spacecraft conditions. During spaceflight, the temperature fluctuated between 16-21°C. Because the warehouse was not equipped with a radiation-measuring device, we were unable to obtain relevant data. After the Shenzhou VIII spacecraft landed, the bacterial colonies were randomly selected from plates coated with K. pneumoniae. Phenotypic analyses, including disk diffusion tests and growth curves, were performed to compare the spaceflight clones and the ground control strain (Additional file 3: Figure S1). The ground control strain was named LCT-KP214. One clone that was obtained after spaceflight and was significantly different from LCT-KP214 was named LCT-KP289.
Growth curve and antibiotic-resistance analyses
The strains were grown on LB liquid medium for 18 h at 37°C. Approximately 20-μl suspensions were inoculated into microtiter plates (honeycomb plates) containing 350 μl LB broth and detected by Bioscreen C (Lab Systems, Helsinki, Finland) at 37°C with continuous shaking. The growth of each sample was monitored by measuring the optical density at 600 nm (OD600) at three time points. A well containing 370 μl LB was included as a negative control. The growth curve of each strain was generated based on the OD600 measurements. The K. pneumoniae strains were transferred from the plate culture system to 1.5-ml centrifuge tubes containing 1 ml of physiological saline, and the concentrations of the bacterial suspension were diluted to 107 ~ 108 bacteria per ml. Culture plates were then coated with 100 μl of bacterial suspension of each strain. Tablets of filter paper were moistened with 17 different antibiotics, including benzylpenicillin, ampicillin, cefazolin, ceftazidime, ceftriaxone sodium, azithromycin, ciprofloxacin, lincomycin, vancomycin, the pediatric compound sulfamethoxazole, chloramphenicol, cefoperazone sodium, amikacin, streptomycin, minocycline, meropenem, and piperacillin. The tablets were then placed on the surface of the culture plates. Each plate contained three types of antibiotics, and two tablets were used for each antibiotic. The plates were then incubated at 37°C for 18 ~ 24 h, and the diameters of the inhibition zones were measured and recorded. The diameter of the tablets was 6 mm.
DNA sample preparation and sequencing
K. pneumoniae genomes were sequenced using an Illumina Hiseq2000 with a multiplexed protocol. Paired-end reads of 90 bp each were generated from 500-bp and 6-kb random sequencing libraries for the control strain LCT-PK214 and the strain obtained after spaceflight, LCT-PK289. We filtered the raw data in four steps: removing reads with 5 bp of Ns, removing reads with 20 bp of low-quality (≤Q20) bases, removing adapter contamination, and removing duplicate reads. Finally, 100X and 50X filtered paired-end reads were obtained for the 500-bp and 6-kb libraries, respectively. The assembly was performed using the SOAPde novo algorithm [26, 27] (http://soap.genomics.org.cn/soapdenovo.html, version: 2.04). Local assembly and gap closure were performed on paired-end reads located in gaps. For highly complex regions, PCR gap closure was performed to obtain sequences without outer gaps. Finally, the SOAPaligner/soap2 software was used for error correction [28, 29] (http://soap.genomics.org.cn/soapaligner.html, version 2.21). The reads were mapped to the sequence, mapping information was recoded, and single-base and local proofreading were performed to analyze the assembly results.
Analysis of genomic components and identification of variations
The sequence of the query strain LCT-PK289 was compared to the reference sequence LCT-PK214 using Mummer [30] (http://mummer.sourceforge.net, version 3.22) and LASTZ [31, 32] (http://www.bx.psu.edu/miller_lab/dist/README.lastz-1.02.00, Version: 1.01.50). We used Mummer for chain stander and start site selection, and LASTZ was used for detailed alignment. Next, the syntenic regions and structural variations, including deletions, insertions, inversions and translocations, were identified in the alignment blocks [33]. SNPs were identified by measuring the distances between mismatched sites in syntenic regions. SNPs located in sequence gaps, repeat regions, or scaffold ends were discarded. To validate the results of the non-redundant candidate SNPs in the genomes, the high-quality, paired-end reads were first mapped to the corresponding genomes using SOAPaligner [28, 29] (http://soap.genomics.org.cn, version 2.21). Next, the numbers of the most abundant (n1) and second-most abundant (n2) nucleotides at each SNP position in each strain (counted according to the number of reads in each strain supporting this nucleotide) were examined. High quality SNPs were defined as SNPs that satisfied the criteria n1 + n2 ≥ 10 and n1/n2 ≥ 5 and for which the quality score of each mapped base was >20. Raw small InDels shorter than 50 bp were predicted from the regions recognized as gaps in the alignment of syntenic regions. InDels containing more than one mismatch 5 bp upstream or downstream were eliminated. Read validation was then performed on the remaining InDels, and the InDels for which ≥3 query reads mapped to the InDel-removed sequence of the subject were retained.
Function enrichment of variant genes
We analyzed the relationships between all gene function variations (genes at SV regions or those containing SNPs or InDels) using different gene/protein databases (COG/GO/KEGG). This allowed us to compute the numbers of proteins for each corresponding COG/GO/KEGG term. We determined the GOG/GO/KEGG enrichment terms for the variant genes using a hypergeometric test [34, 35] and calculated the P-value. P ≤ 0.05 was considered a significant enrichment of the GOG/GO/KEGG term for the variant gene/protein. We determined the main biological function of differential proteins using function enrichment analysis.
Phylogenetic analysis
We converted the protein sequence alignment results into multiple amino acid sequences in the CDS regions and aligned multiple sequences in the clustered gene family using the Muscle software (http://www.drive5.com/muscle, v3.8.31). Finally, we generated the gene family tree by analyzing the multiple-sequence alignment results based on Muscle using the Bayes method with the MrBayes software (MrBayes v3.1.2). The dN/dS ratios were calculated using the CODONML software (in paml version 4.4, January 2010) with the GY-HKY model [36].
Accession numbers
K. pneumoniae strains LCT-KP214 and LCT-KP289 genome sequences have been deposited in GenBank under accession numbers AJHE00000000 and ATRO00000000, respectively.
Abbreviations
- SNP:
-
Single nucleotide polymorphism
- InDel:
-
Insertion-deletion
- NR:
-
Non-redundant protein database
- COG:
-
Cluster of orthologous groups of proteins
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- GO:
-
Gene ontology
- ARDB:
-
Antibiotic resistance genes database
- RI:
-
Resistance integrin.
References
Paul AL, Wheeler RM, Levine HG, Ferl RJ: Fundamental plant biology enabled by the space shuttle. Am J Bot. 2013, 100 (1): 226-234.
Girdhani S, Sachs R, Hlatky L: Biological effects of proton radiation: what we know and don't know. Radiat Res. 2013, 179 (3): 257-272.
Girdhani S, Lamont C, Hahnfeldt P, Abdollahi A, Hlatky L: Proton irradiation suppresses angiogenic genes and impairs cell invasion and tumor growth. Radiat Res. 2012, 178 (1): 33-45.
Gao H, Liu Z, Zhang L: Secondary metabolism in simulated microgravity and space flight. Protein Cell. 2011, 2 (11): 858-861.
Moeller R, Reitz G, Horneck G, Nicholson The Protect Team WL: Mutagenesis in bacterial spores exposed to space and simulated martian conditions: data from the EXPOSE-E spaceflight experiment PROTECT. Astrobiology. 2012, 12 (5): 457-468.
Tixador R, Gasset G, Eche B, Moatti N, Lapchine L, Woldringh C, Toorop P, Moatti JP, Delmotte F, Tap G: Behavior of bacteria and antibiotics under space conditions. Aviat Space Environ Med. 1994, 65 (6): 551-556.
Podschun R, Ullmann U: Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev. 1998, 11 (4): 589-603.
Schwarz E, Oesterhelt D: Cloning and expression of Klebsiella pneumoniae genes coding for citrate transport and fermentation. EMBO J. 1985, 4 (6): 1599-1603.
Bott M: Anaerobic citrate metabolism and its regulation in enterobacteria. Arch Microbiol. 1997, 167 (2/3): 78-88.
Kaspar S, Perozzo R, Reinelt S, Meyer M, Pfister K, Scapozza L, Bott M: The periplasmic domain of the histidine autokinase CitA functions as a highly specific citrate receptor. Mol Microbiol. 1999, 33 (4): 858-872.
Meyer M, Dimroth P, Bott M: Catabolite repression of the citrate fermentation genes in Klebsiella pneumoniae: evidence for involvement of the cyclic AMP receptor protein. J Bacteriol. 2001, 183 (18): 5248-5256.
Huang Y, Li Z, Shimizu K, Ye Q: Simultaneous production of 3-hydroxypropionic acid and 1,3-propanediol from glycerol by a recombinant strain of Klebsiella pneumoniae. Bioresour Technol. 2012, 103 (1): 351-359.
Juergensmeyer MA, Juergensmeyer EA, Guikema JA: Long-term exposure to spaceflight conditions affects bacterial response to antibiotics. Microgravity Sci Technol. 1999, 12 (1): 41-47.
Taylor PW, Sommer AP: Towards rational treatment of bacterial infections during extended space travel. Int J Antimicrob Agents. 2005, 26 (3): 183-187.
Grape M, Sundstrom L, Kronvall G: Sulphonamide resistance gene sul3 found in Escherichia coli isolates from human sources. J Antimicrob Chemother. 2003, 52 (6): 1022-1024.
Cameron FH, Groot Obbink DJ, Ackerman VP, Hall RM: Nucleotide sequence of the AAD(2'') aminoglycoside adenylyltransferase determinant aadB. Evolutionary relationship of this region with those surrounding aadA in R538-1 and dhfrII in R388. Nucleic Acids Res. 1986, 14 (21): 8625-8635.
Fluit AC, Schmitz FJ: Class 1 integrons, gene cassettes, mobility, and epidemiology. Eur J Clin Microbiol Infect Dis. 1999, 18 (11): 761-770.
Levesque C, Piche L, Larose C, Roy PH: PCR mapping of integrons reveals several novel combinations of resistance genes. Antimicrob Agents Chemother. 1995, 39 (1): 185-191.
Guerra B, Soto SM, Arguelles JM, Mendoza MC: Multidrug resistance is mediated by large plasmids carrying a class 1 integron in the emergent Salmonella enterica serotype [4,5,12:i:-]. Antimicrob Agents Chemother. 2001, 45 (4): 1305-1308.
Le Gac M, Cooper TF, Cruveiller S, Medigue C, Schneider D: Evolutionary history and genetic parallelism affect correlated responses to evolution. Mol Ecol. 2013, 22 (12): 3292-3303.
Wei D, Wang M, Jiang B, Shi J, Hao J: Role of dihydroxyacetone kinases I and II in the dha regulon of Klebsiella pneumoniae. J Biotechnol. 2014, 177C: 13-19.
Skold O: Resistance to trimethoprim and sulfonamides. Vet Res. 2001, 32 (3–4): 261-273.
Beketskaia MS, Bay DC, Turner RJ: Outer membrane protein OmpW participates with small multidrug resistance protein member EmrE in quaternary cationic compound efflux. J Bacteriol. 2014, 196 (10): 1908-1914.
Kotra LP, Haddad J, Mobashery S: Aminoglycosides: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob Agents Chemother. 2000, 44 (12): 3249-3256.
Gupta G, Tak V, Mathur P: Detection of AmpC beta Lactamases in Gram-negative Bacteria. J Lab Physicians. 2014, 6 (1): 1-6.
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, Li S, Yang H, Wang J, Wang J: De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20 (2): 265-272.
Li R, Li Y, Kristiansen K, Wang J: SOAP: short oligonucleotide alignment program. Bioinformatics. 2008, 24 (5): 713-714.
Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K: SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009, 19 (6): 1124-1132.
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J: SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009, 25 (15): 1966-1967.
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL: Versatile and open software for comparing large genomes. Genome Biol. 2004, 5 (2): R12-
Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W: Human-mouse alignments with BLASTZ. Genome Res. 2003, 13 (1): 103-107.
Dewey CN, Pachter L: Evolution at the nucleotide level: the problem of multiple whole-genome alignment. Hum Mol Genet. 2006, 15 Spec No 1: R51-R56.
Li Y, Zheng H, Luo R, Wu H, Zhu H, Li R, Cao H, Wu B, Huang S, Shao H, Ma H, Zhang F, Feng S, Zhang W, Du H, Tian G, Li J, Zhang X, Li S, Bolund L, Kristiansen K, De Smith AJ, Blakemore AI, Coin LJ, Yang H, Wang J, Wang J: Structural variation in two human genomes mapped at single-nucleotide resolution by whole genome de novo assembly. Nat Biotechnol. 2011, 29 (8): 723-730.
Rivals I, Personnaz L, Taing L, Potier MC: Enrichment or depletion of a GO category within a class of genes: which test?. Bioinformatics. 2007, 23 (4): 401-407.
Zhou X, Su Z: EasyGO: gene ontology-based annotation and functional enrichment analysis tool for agronomical species. BMC Genomics. 2007, 8: 246-
Yang Z, Kumar S, Nei M: A new method of inference of ancestral nucleotide and amino acid sequences. Genetics. 1995, 141 (4): 1641-1650.
Acknowledgements
This work was supported by the National Basic Research Program of China (973 program) No. 2014CB744400, the Program of Manned Spaceflight No. 040203, Key Program of Medical Research in the Military ‘12th 5-year Plan’, No. BWS12J046, Key Pre-Research Foundation of Military Equipment of China No. 9140A26040312JB10078, and Beijing Novel Program, No.Z121107002512128.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the project: CL, YG, LS, WL and WD. Experiments: DC, LS, YG, XF, JW, TL and CF. Bioinformation analysis: WL, YL, YY, TW and LS. Wrote the paper: YG, LS, YL and WL. All authors read and approved the final manuscript.
Yinghua Guo, Yinhu Li, Longxiang Su contributed equally to this work.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Guo, Y., Li, Y., Su, L. et al. Comparative genomic analysis of Klebsiella pneumonia (LCT-KP214) and a mutant strain (LCT-KP289) obtained after spaceflight. BMC Genomics 15, 589 (2014). https://doi.org/10.1186/1471-2164-15-589
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-589