
Abstract
Background
Comparative transcriptomics and functional studies of different Saccharomyces species have opened up the possibility of studying and understanding new yeast abilities. This is the case of yeast adaptation to stress, in particular the cold stress response, which is especially relevant for the food industry. Since the species Saccharomyces kudriavzevii is adapted to grow at low temperatures, it has been suggested that it contains physiological adaptations that allow it to rapidly and efficiently acclimatise after cold shock.
Results
In this work, we aimed to provide new insights into the molecular basis determining this better cold adaptation of S. kudriavzevii strains. To this end, we have compared S. cerevisiae and S. kudriavzevii transcriptome after yeast adapted to cold shock. The results showed that both yeast mainly activated the genes related to translation machinery by comparing 12°C with 28°C, but the S. kudriavzevii response was stronger, showing an increased expression of dozens of genes involved in protein synthesis. This suggested enhanced translation efficiency at low temperatures, which was confirmed when we observed increased resistance to translation inhibitor paromomycin. Finally, 35S-methionine incorporation assays confirmed the increased S. kudriavzevii translation rate after cold shock.
Conclusions
This work confirms that S. kudriavzevii is able to grow at low temperatures, an interesting ability for different industrial applications. We propose that this adaptation is based on its enhanced ability to initiate a quick, efficient translation of crucial genes in cold adaptation among others, a mechanism that has been suggested for other microorganisms.
Similar content being viewed by others
Background
Nowadays, there is a trend in winemaking that consists in decreasing fermentation temperatures to improve the aromatic profile of wines. However, lowering fermentation temperatures has its disadvantages, including prolonged process duration and a greater risk of halted or sluggish fermentation [1]. These problems can be avoided by providing better-adapted yeasts to ferment at low temperature. Although the wine industry already has yeasts that are sold as cryotolerant yeasts (QA23, Lallemand Inc. or Fermol Cryophile, Fermol Reims Champagne, AEB group), most do not offer desirable fermentation performance at low temperature (10-15°C Beltran et al. [2] performed a transcriptomic analysis using the commercial S. cerevisiae wine-making strain QA23 during industrial fermentations at low temperature. They showed how the expression profiles during wine fermentation at 13°C contrasted significantly with those at 25°C. In particular, the genes of the cell cycle, cell growth, cell fate and maintenance categories were less expressed in the exponential growth phase at 13°C than at 25°C, whereas those genes whose expression was activated in the exponential phase of growth at 13°C were essentially those involved in the environmental stress response [3].
Previous physiological and enological works from this laboratory have already indicated the tremendous advantage of S. kudriavzevii fermenting at low temperature, and have shown its well-established cryotolerant character [4, 5]. Its sugar consumption rate, similar to that of S. cerevisiae, makes this organism a serious candidate in the wine yeast industry to compete for a place at low fermentation temperatures. The lipid composition of this species presents some features that might enable it to adapt much better at low temperature [6]. Although it has not been found in natural wine fermentations, probably due to its low ethanol tolerance, S. cerevisiae – S. kudriavzevii natural hybrids, which combine optimal characteristics of both parents, are present in cold climate wineries [6, 7]. Although we have found several genome-wide expression analysis studies that used DNA microarray technology in S. cerevisiae, there is no equivalent information available on other species of the genus adapting to low temperature. Therefore, the use of cryotolerant yeasts to study adaptation to low temperature can help us to better understand this stress factor and to also discriminate if these adaptation strategies are species-specific or common to all the strains of the Saccharomyces genus.
In this study, we conducted a comparative genome-wide gene expression analysis between a well-known wine yeast strain belonging to the species S. cerevisiae (T73) and the type strain from S. kudriavzevii IFO1802, a cryotolerant yeast, in natural must fermentations. Significant differences were found in the expression of those genes related to translation machinery. Sensitivity of translation inhibitor paromomycin reflected the enhanced translation efficiency of S. kudriavzevii at low temperatures. Indeed, an increased translation rate of S. kudriavzevii was observed after cold shock, suggesting that the efficiency of protein synthesis is an important process for the adaptation of yeast cells to grow at low temperatures.
Results
Effect of low temperature stress
Previous studies in our laboratory have clearly shown growth differences among the species belonging to the genus Saccharomyces in colony development on GPY plates at low temperature [8]. To confirm this behaviour in the strains selected for this study, we performed a dropping assay at 12°C with IFO1802 (Saccharomyces kudriavzevii) and used the yeast T73 (Saccharomyces cerevisiae) (Additional file 1: Table S1) as a control to verify the enhanced growth of the S. kudriavzevii strain at the selected cold temperature. Colonies were grown on GPY plates incubated at 12°C and 28°C to test yeast tolerance against low temperature (Figure 1). Growth at 28°C was recorded after 36 h of incubation, while growth at 12°C was recorded after 6 days of incubation. At 28°C, both strains were able to grow until the last dilution. At 12°C, IFO1802 reached the last dilution, while T73 was clearly at a disadvantage since it displayed a strong low temperature effect.

S . kudriavzevii has increased grow abilities at low temperature. A drop test assay was performed in rich media GPY with S. cerevisiae strain T73 and S. kudriavzevii strain IFO1802 and plates were incubated at different temperatures (12 or 28°C).
Differential growth kinetics was also observed during wine micro-fermentations in vessels with 0.45 L of Tempranillo grape must. At 12°C, the S. kudriavzevii strain took around 50 hours to consume 15% of sugars, almost 3 times less than T73, which demonstrates the better adaptation of IFO1802 to low temperature. In addition, S. kudriavzevii strain maintained a good fermentation ratio until the end (11 days). T73 finished in 21 days (Table 1). Thus, these data not only prove that the selected temperature produces a cold stress in both yeasts, although it becomes much more critical in T73, and but also validate this temperature to produce differences in the adaptation of both strains to cold temperature.
Differential gene expression in S. cerevisiae and S. kudriavzeviiat low temperature
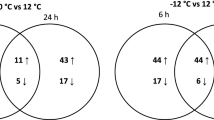
Changes in the global expression of genes during acclimation to low temperature in the wine fermentation of natural must were analysed with microarrays containing the annotated genome of the S. cerevisiae S288c strain. This comparative transcriptomic study of S. cerevisiae and S. kudriavzevii was carried out during wine micro-fermentations in 500 mL vessels of Tempranillo grape must at 12°C and 28°C with wine yeast strain T73 and IFO1802. The gene expression of these species was analysed at the beginning of the exponential phase by taking samples two generations after inoculation. After extracting RNA and verifying quality, the samples were retrotranscribed to cDNA, and were mixed in equal amounts and hybridised against the microarray based on the S288c genome. It should be borne in mind that an average of 86% of sequence similarity exists between S. cerevisiae and S. kudriavzevii. Thus, we used heterologous conditions in the hybridisation of the cDNA of both strains tested against the S. cerevisiae S288c microarrays. Genomic DNA hybridisations were done previously to assure the efficiency of the methodology; under our conditions, 95% of the total gene spots from the S288c array were hybridised by S. kudriavzevii DNA [9]. The use of non-restricted conditions increases the noise, but improves the hybridisation of the S. kudriavzevii samples. The gene expression of each strain was compared at both temperatures by performing biological triplicates. Significant genes with differential expressions were taken into account for further analysis using the SAM (Significance Analysis of Microarrays) test with an FDR below 5%. Table 2 shows the amount of the up- and down-regulated genes found in each species in relation to temperature and after comparing one species against the other. In S. cerevisiae strain T73, 177 genes were up-regulated, while 194 were down-regulated at 12°C, while for S. kudriavzevii, these numbers were 160 genes and 128, respectively. The transcriptome variation between temperatures (12-28°C) in S. cerevisiae was compared with the transcriptome variation between temperatures (12-28°C) in S. kudriavzevii, and an increased expression of 231 genes in IFO18802 and of 78 in T73 was observed (Additional file 2: Table S2).
The GO terms analysis (GO Term Finder) was performed to observe the functions of the significant up- or down-regulated genes (Table 2). At 12°C, T73 showed the GO-terms Cytoplasmic translation and localisation to be significantly up-regulated, whereas no functional group was found among the down-regulated genes. S. kudriavzevii also showed significantly up-regulated translation, together with the term Amino acid catabolic process via the Ehrlich pathway. Finally, the comparison made of both species between them ended up with only one significant functional group present in the IFO1802 up-regulated genes: Translation. The presence of this GO was confirmed with the FunSpec database that also detected other translation related functional groups (rRNA export from nucleus) and protein complexes (cytoplasmic ribosomal large and small subunit) (Addtional file 4: Table S4). This result suggests that, although both species increased the expression of those genes related to protein biosynthesis, S. kudriavzevii has an enhanced or faster response after cold shock.
In an attempt to find the putative signal transduction pathways involved in cold adaptation, we searched the transcription factors that regulate each up-regulated gene in the Yestract database [10]. In this search, we found that the main transcription factor was Sfp1p, which regulates the transcription of ribosomal proteins and biogenesis genes, and is related to 62.3% of the activated genes in IFO102 and to 49.7% of the activated genes in the T73 strain. Furthermore, Sfp1p was the principal regulator in the genes activated when comparing both species, with 50.4% of the genes. This result confirms not only the importance of the translation machinery in response to cold shock in both species, but also the enhanced response of S. kudriavzevii. We also found that 21.6% of the genes induced in IFO1802 and 24.5% in the T73 strain were regulated by Msn2p. This result is also interesting since the transcriptional network of the complex Msn2p/Msn4p has also been suggested to participate in adaptation to cold stress [11, 12].
Cold stress markers in adaptation to low temperature
Several genes that have been systematically found in different transcriptomic studies in response to low temperature stress are considered cold stress markers [11–13]. One example of a gene induced by low temperature and involved in translation initiation is NSR1, which encodes a nucleolar protein that binds nuclear localisation sequences and is required for pre-rRNA processing and ribosome biogenesis. Among the genes regulated by Msn2p/Msn4p, a paradigm group of genes systematically found in different transcriptomic studies in response to low temperature stress are different heat shock proteins related to oxidative stress. This is the case of HSP12, which encodes a chaperone involved in correct protein folding under many stress conditions, including cold shock. We observed that NSR1 and HSP12 were up-regulated in both yeasts after cold shock (Additional file 3: Table S4), thus confirming once more its implication in the cellular response against low temperature stress.
The comparison made between both species showed an increased expression of other cold shock markers in the IFO1802 strain if compared to T73: OLE1, TIP1, ERG1 and PAU4. OLE1 and ERG1 are implicated in lipid metabolism, whereas TIP1 and PAU4 belong to the DAN/TIR family of putative cell-wall mannoproteins and its sequence-related seripauperin (PAU) family, respectively. This observation supports the idea of an enhanced transcriptional response in S. kudriavzevii as compared to S. cerevisiae after cold shock.
NSR1expression profile during synthetic must fermentation
It was our intention to test the expression profile of one of the main cold markers genes defined for cold in other time points of fermentation and to know if NSR1 behaves similarly in both species. To do this, we did fermentations in synthetic must, a media that mimics natural must. The time points selected at both temperatures were: 2 h after inoculation, when the OD doubled the initial OD, in the middle of exponential phase, and finally at the start and at the end of the stationary phase (sugar exhaustion) (Figure 2). As previously described [14], cold marker NSR1 in the S. cerevisiae strain was induced at the beginning of growth with a much higher expression level at 12°C (Figure 2). This confirms the use of this gene as a cold marker. During the entrance in the stationary phase, it was repressed at 28°C and became less activated at 12°C in S. cerevisiae. In S. kudriavzevii, there a similar induction to S. cerevisiae occurred, but it was not repressed as it was in S. cerevisiae, and maintained greater activity during the rest of the process, especially at 12°C.

S. kudriavzevii has increased expression of the cold stress gene marker NSR1 at low temperature. Gene expression measured by qPCR technique after S. cerevisiae (T73) or S. kudriavzevii (IFO1802) cells were transferred to 12 (light grey) or 28°C (dark grey) synthetic grape must. Samples were taken 2 hours after the inoculation (A), when the OD600 reached the double of the initial OD600 (B), in the middle of exponential phase (C), and at the start (D) and at the end (E) of the stationary phase (sugar exhaustion). Average of biological triplicates was calculated and standard deviations were lower than 20%. Gene expression levels are shown as the changes in the concentration of the studied gene compare to the control sample and normalized with the concentration of the housekeeping ACT1 gene. Gene expression differences between 12 a 28°C were statistically significant, except in the case of time point A for S. kudriavzevii.
Translation efficiency at low temperature
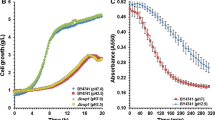
To demonstrate that S. kudriavzevii adaptation to low temperature is related to enhanced translation efficiency, we tested its sensitivity to paromomycin, a potent translation inhibitor [15]. We studied the paromomycin (0.4, 2 and 10 mg/ml) growth inhibition of yeast cells at either 28°C or 12°C for two S. cerevisiae strains (T73 and QA23) and two S. kudriavzevii strains (IFO1802 and CR85). A growth inhibition halo was observed under some conditions (Figure 3A). A summary of the results, presented in Table 3, indicates that S. cerevisiae strains T73 and QA23 were severely affected by paromomycin at 12°C whereas the S. kudriavzevii strains showed low (CR85) or no growth inhibition at all (IFO1802). This result confirms the better translation performance of S. kudriavzevii at 12°C. At 28°C, the S. cerevisiae strains showed no growth inhibition, whereas S. kudriavzevii strains presented a mild negative effect of paromomycin on cell growth. This result can be explained because 28°C is a high temperature for S. kudriavzevii[16] and this suboptimal situation, together with paromomycin inhibitory effects, can affect cell growth.

S. kudriavzevii presents increased translation efficiency at low temperatures. In panel A, the inhibitory effect of the translation inhibitor paromomycin was evaluated by measuring the halo diameter generated in S. cerevisiae (T73) or S. kudriavzevii (CR85) lawns growing in GPY plates at 28 or 12°C. In panel B, the translation efficiency was evaluated by measuring 35S-Methionine incorporation 16 h after transfer S. cerevisiae (Q23) or S. kudriavzevii (CR85) cells to cold (12°C) rich media. Cpm values were normalized with OD600, relativized to maximum value and represented against the time. Average of biological triplicates and standard deviations are shown.
Since the data obtained in this work suggested an enhanced response to cold shock in the S. kudriavzevii strain, especially in those genes related to translation machinery, we decided to study the translation kinetics of both species after cold shock by measuring the 35S-methionine incorporation rate before yeast cells started to grow (Figure 3B), which is an indication of the translation rate. Quantification of the 35S-methionine incorporation rate to nascent peptides after transfer yeast cells at either 28°C or 12°C culture media for two S. cerevisiae strains (T73 and QA23) and two S. kudriavzevii strains (IFO1802 and CR85) is presented in Table 4. The results show that the 35S-methionine incorporation rate of both S. kudriavzevii strains was significantly lower than any S. cerevisiae strain at 28°C (measured between 1–5 h after transferring cells). In contrast at 12°C (measured between 16–24 h after transferring cells), the situation was the opposite; the 35S-met incorporation rate of both S. kudriavzevii strains was significantly higher than any S. cerevisiae strain. These results support the notion that the cryotolerant species S. kudriavzevii has an enhanced translational response after cold shock, which allows cells to better adapt to the stressful condition and start to grow.
Discussion
Temperature fluctuations are an inevitable aspect of microbial life in exposed natural environments, although sub-optimal temperatures are also common in industrial processes. Low temperatures (10-15°C) are used in wine fermentations to enhance production and to retain flavour volatiles. Although S. cerevisiae is always predominant in wine fermentations, a drop in temperature affects its competitiveness. In contrast, S. kudriavzevii showed the lowest optimum growth temperature, which confirms that this species is more psychrophilic than S. cerevisiae, and that its competitiveness increased as the fermentation temperature dropped [17–19]. Thus, despite them being absent in fermentative environments, S. kudriavzevii strains have a great potential for being used in wine fermentations at low temperature. Moreover, if these strains are not sufficiently stress-tolerant to cope with the stress of wine fermentation conditions, they can be used to build artificial hybrids with S. cerevisiae strains. These hybrids might acquire some physiological properties of both parents. It should be noted that the natural hybrids of S. cerevisiae × S. kudriavzevii have already colonised central European wine fermentations [20]. The psychrophilic nature of the S. kudriavzevii strain has once again been supported by the drop test obtained at low temperature in this study.
In any case, S. kudriavzevii is a good model to study adaptation at low temperatures.. A transcriptome comparison with S. cerevisiae has shed light on the response of this cryotolerant yeast species. A common answer for both species is the presence of the up-regulated genes related to translational machinery (Table 2). There are a number of publications of genome-wide analysis at low temperature [12, 13, 21–23]. As Tai et al. [13] remarked, these studies present some inconsistencies; for instance, the different expression of ribosomal protein genes. Since we selected a condition where cells were already growing at both temperatures, we did not expect any differences in ribosome biogenesis or proteins synthesis because these categories are typical from the start of exponential growth. On the contrary, our results reveal that both species have up-regulated GO-terms related to translation, although S. kudriavzevii shows an enhanced response. We postulate that it could be the result of changes in the stability of a functional RNA conformation in relation to a competing structure [24]. It has been described that cold sensitive phenotypes, caused by hyper-stabilisation of RNA structures, can be found elsewhere in the spliceosome, where RNA structures must form and then disrupt for splicing to progress. If this plasticity of RNA structures is reduced by RNA stabilisation due to a cold environment, the maturation of ribosomes may be in danger [24–29].
Another result that relates low temperature adaptation and translation efficiency of the Saccharomyces species is the differential regulation of cold shock gene marker NSR1. This gene, together with other nucleolar proteins with small nucleolar RNAs (snoRNAs), is required for the normal processing of rRNA precursors. These snoRNAs associate with pre-rRNA as snoRNP complexes and participate in the assembly of ribosomal subunits [30]. The plasticity of these secondary RNA structures is essential to constitute snoRNP complexes, but secondary RNA structures are highly subjected to malfunction due to low temperatures. It is possible that some of these snoRNAs are more sensitive to cold than others, especially those required by NSR1p. Studies in prokaryotes have shown the induction of a set of cold shock proteins, which include RNA helicases [31]. It is possible that NSR1 can function as bacterial cold shock proteins (CSPs), which destabilise the RNA secondary structures that have been stabilised as an effect of low temperatures and acts as an RNA chaperone. CSPs also present greater protein resistance to unfoldment, and therefore play an important role in adaptation to low temperature [32, 33]. The interspecies differences observed in the NSR1 gene expression profile during a fermentative process between S. cerevisiae and S. kudriavzevii are an example of how different yeasts species adapt to low temperatures.
The different susceptibility to paromomycin suggests that other mechanisms might be implicated in S. kudriavzevii-enhanced adaptation to low temperatures. Paromomycin, a potent inhibitor of translation, is a member of the aminoglycoside family of antibiotics. This family is thought to reduce the dissociation rate of A-site tRNA from the ribosome [34]. Paromomycin increases the error rate of the ribosome, but it has been also described as a powerful inhibitor of ‘ribosome recycling’ [35]. Ribosome recycling represents the reaction to recycle the spent ribosome for the next round of translation of new mRNA. Kurata et al. [36] have recently reported that paromomycin negatively affects all different ribosome recycling steps to produce a dramatic effect on translation efficiency. Thus, increased resistance to paromomycin can be the result of enhanced translation efficiency due to an increased number of ribosomes available to a new round of mRNA translation. Since S. kudriavzevii is less affected by paromomycin at low temperatures than S. cerevisiae, our data suggest that this species has increased translation efficiency due to higher ribosome availability after adaptation to cold shock.
It is also worth mentioning the specific activation of the genes of the amino acid catabolic process via the Ehrlich pathway. Recently our group compared the metabolomic differences between S. cerevisiae and S. kudriavzevii[37]. The main differences between the metabolic profiling of both species were observed for amino acids. In fact, nitrogen metabolism is one of the most affected cellular processes at low temperature in S. cerevisiae. Pizarro et al. [38] reported that the physiological and transcriptional response of laboratories and wine yeast strains to stress at low temperature was similar to growth under nitrogen-limiting conditions. Thus, the stronger activity in S. kudriavzevii of the genes involved in one of the main amino acid pathways might represent this metabolic bottle-neck having been better surpassed in this species. As already reported Tronchoni et al. [6], a better adapted lipid membrane composition in S. kudriavzevii might enable better transport of nitrogen compounds and, therefore, more activity of amino acid metabolism.
Conclusions
Our results confirm that S. kudriavzevii is better adapted to grow at low temperatures and reveals an enhanced translation in S. kudriavzevii. Our data suggest that translation efficiency can be an important target of adaptative evolution when cells face changing environments. New studies of comparative genetics could shed light on the specific mechanism underlying enhanced translation efficiency at low temperatures.
Methods
Strains and media
The yeasts used in this study belong to the species S. cerevisiae and S. kudriavzevii. Additional file 1: Table S1 shows the references and origin of these yeasts. T73 (ATCC 90607) and QA23 are commercial strains, marketed by Lallemand S.A. (Canada), that have been used as a wine yeast S. cerevisiae model in many studies [16, 39]. IFO1802 (NBRC 1802) is the S. kudriavzevii type strain and CR85 was isolated in Spain [40]. Strains are available upon request. GPY medium (0.5% peptone, 2% glucose, 0.5% yeast extract) was used to propagate yeast strains.
Natural must fermentations
Yeast strains were cultivated in Erlenmeyer flasks containing 250 ml of GPY at 25°C in an agitated incubator (Selecta, Barcelona, Spain). At the end of the exponential phase, determined by absorbance at 600 nm, 2 × 106 cells ml−1 were inoculated in each grape must flask. Fermentations were carried out in triplicate using 450 ml of Tempranillo grape must at pH 3.5. Before fermentation, must was clarified by sedimentation for 24 h at 4°C in the presence of 60 mg l−1 of sulphur dioxide. After separation, chemically pure glucose and fructose were added to raise the sugar content to 250 g l−1. The must was then supplemented with 0.25 g l−1 of yeast nutrients (Lallemand, Montreal, QC, Canada). Yeast assimilable nitrogen was determined by the formol index method [41], and diammonium sulphate was added to reach a final concentration of 250 mg l−1. Finally, must was sterilised by adding dimethyl dicarbonate (Fluka, Buchs, St. Gallen, Switzerland) at a concentration of 1 ml l−1 must.
To follow wine fermentation kinetics, Tempranillo grape must was fermented at 12°C and 28°C. Fermentations were carried out in biological duplicate and monitored by sugar consumption. Glucose and fructose concentrations were determined enzymatically in duplicate using a commercial kit (AMS-SYSTEA) in an Echo-Enosys analyser (Tecnova S.A., Madrid, Spain). Fermentations were finished when the concentration of reducing sugars was lower than 2 g l−1.
Synthetic must fermentations
Experiments were carried out in a complex synthetic medium (MS300) to mimic a standard natural must previously described by Bely et al. [42]. Natural musts show a variable composition among different seasons that can influence yeast growth. For this reason, a defined synthetic must was chosen in this work as the most appropriate growth medium to overcome this variation. The sugar concentration (50% glucose, 50% fructose) was adjusted in distilled water according to the previously described natural must Tempranillo and was heated at 100°C for 15 min to prevent sugar caramelization. The stocks for the other components of the medium (mineral salts, vitamins, amino acids and anaerobic factors) were previously sterilised by filtration (0.2 mm) and were then added to the basal medium at the appropriate concentration [43]. Finally, pH was adjusted by aseptically adding tartaric acid (85%, wt/vol) according to the experimental design. We chose this organic acid because it is a compound that is normally found in grapes and wines, and it is very rarely metabolised by ascomycetous yeasts. Sterile glass bottles (500 ml of volume) were filled with 450 ml of synthetic must and were independently inoculated with 50 μl of the corresponding yeast saline suspension to reach an initial concentration of inoculum of about 2 × 106 cells ml−1 determined by absorbance at 600 nm. Bottles were incubated at 12°C and 28°C.
RNA extraction
Cells were collected by centrifugation (4000 rpm/min, 5 min) from three independent fermentations at 12°C and 28°C at the beginning of the exponential phase by taking samples two generations after inoculation. The RNA extraction method was based on consecutive treatments with phenol-tris, phenol-chloroform (5:1) and chloroform-isoamyl alcohol (24:1), and a final precipitation with ethanol and sodium acetate [44]. RNA concentrations and purity was determined using a Nanodrop spectrophotometer ND-1000 (Nanodrop Technologies™, Wilmington, DE). RNA integrity was determined by electrophoresis in 1% agarose gel.
Microarray hybridisation
Firstly, 2–4 μg of total RNA from each sample were linearly amplified using the Low RNA Input Fluorescent Linear Amplification kit (Agilent Technologies™, Ca, USA). 2–3 μg of amplified cRNA were used as a template for cDNA synthesis. cDNA was marked indirectly with the “SuperScript™ Indirect cDNA Labeling System” (Invitrogen™, San Diego, CA). The fluorophores used were Cy3 and Cy5 mono-reactive Dye (Amersham GE Healthcare™, Amersham, UK) and dye incorporation was monitored by a Nanodrop spectrophotometer. A mixture of 200–300 pmol of the two labelled samples was concentrated in a Concentrator Plus (Eppendorf™, Hamburg, Germany). Competitive hybridisation was performed on a Yeast 6.4 K Array, PCR-amplified ORFs of yeast S288c strain, (Microarray Centre, UHN, Toronto, Canada) in hybridisation chambers AHC (ArrayIt Corporation, CA, USA) at 42°C overnight.
The pre-hybridisation solution contained 3X SSC, 0.1% SDS and 0.1 mg/ml BSA; hybridisation solution contained 5X SSC, 0.1% SDS and 0.1 mg/ml of salmon DNA. Microarrays were washed manually with different solutions containing distinct SSC 20X and SDS 10% concentrations (Sol.1: 2X SSC-0.1% SDS; Sol.2: 0.1X SSC-0.1% SDS; Sol.3: 0.1 SSC; Sol4: 0.01X SSC). Signal intensities of Cy3 and Cy5 were acquired with an Axon GenePix 4100A scanner (Molecular Devices, CA, USA) using the GenePix Pro v.6.1 software at a resolution of 10 μm. Genomic DNA hybridisations were done previously to assure the efficiency of the methodology; under our conditions, 95% of the total gene spots from the S288c array were hybridised by S. kudriavzevii DNA [9]. The use of non-restricted conditions increases the noise, but improves the hybridisation of the S. kudriavzevii samples.
Microarray data were derived from three independent experiments for cDNA hybridisation. Raw data with a global background subtraction were generated from GenePix pro 6.0. The analyses were done using the Acuity 4.0 software (Molecular Devices, CA, USA). The individual data sets were normalised at a log2 ratio value of 1. After normalisation, data were filtered to remove the spots flagged as not found. Only those spots with at least two replicates were considered. The gene expression of each strain was compared at both temperatures by performing biological triplicates. Significant genes with differential expressions were taken into account for further analysis using the SAM test [45] with an FDR below 5% using MeV software [46]. GO term analysis was performed with the online tools of SGD [47] or FunSpec [48] Database selecting for significant functional groups (p < 0.01) with Bonferroni correction for false positives. Data was deposited in Gene Expression Omnibus (GEO) Database with the accession number: GSE52545.
Real-time qPCR
The PCR primers used in this study were TGGATTCCGGTGATGGTGTT - CGGCCAAATCGATTCTCAA for ACT1 and TTCAATGCTGACAGAGACGCTATT – GATACGGACGGAAACAACTTCAC for NSR1. All the amplicons were shorter than 100 bp, which ensured maximal PCR efficiency and, therefore, the most precise quantification. RNA extraction was done as previously described (see above). A relative quantification model with kinetic PCR efficiency correction was built [49]. Experiments were carried out in triplicate. The control sample was extracted from the inoculum of S. cerevisiae and S. kudriavzevii at 28°C in the stationary phase after overnight growth in GPY. The reference gene used was ACT1, which showed excellent uniformity in the expression levels in these fermentation conditions [50], and all the reactions were done in a LightCycler® 480 Real-Time PCR System. Average of biological triplicates was calculated and standard deviations were lower than 20%. The gene expression levels are shown as the changes in the concentration of the studied gene as compared to the control sample and were normalised with the concentration of the housekeeping ACT1 gene.
Paromomycin assays
For the halo assays, yeast cells were grown overnight in GPY and diluted the next morning. They were then grown until the mid-log phase (approximately 1 × 107 cells/ml) and then 175 μl were spread on each GPY plate. When the plate was absolutely dry, a filter (1 cm diameter) imbibed with different amounts of paromomycin (0.4, 2 or 10 μg of drug) was placed on the surface and plates were incubated at 30°C until the lawn was confluent. The measurement was taken from the point where colonies were grown. Inside the halo, there were only single cells and clumps. The assays were repeated twice.
Translation rate determination
An initial concentration of 2 × 106 cells ml−1, determined by absorbance at 600 nm, was used to inoculate 25 mL of synthetic must without methionine. Firstly 50 ml tubes were incubated at 12°C and 28°C, 10 μl of 35S-methionine (0.1 mCi, Hartmann Analytic GmbH, Germany) were added to the media and samples were taken at different time points. Next 2 ml of sample were mixed with 250 μl of 1 M NaOH in a glass test tube and were incubated at RT for 10 min. Two ml of cold TCA (25%) were added to the sample which was vortexed briefly. Samples were incubated on ice for 5 min. Vacuum filtration was used to collect the precipitated protein. 10% TCA pre-wet glass fibre filters were used to filter the sample with vacuum. Filters were rinsed 3X with cold 10% TCA and once with 95% ethanol to dry them and to prevent quenching. Dry filters were placed into scintillation vials with 2 ml scintillation fluid. Samples were measured in a scintillation counter after soaking the filters in scintillation fluid overnight. To determine the translation rate, cpm values were normalised with yeast growth (OD600). Translation rate was determined as the slope of linear regression calculation in normalised cpm versus the time graphs performed with the GraphPad Prism 5.03 software. Average and standard deviation was calculated from three independent biological replicates.
Statistical analysis
Data were analysed with the Excel software. Results are expressed as mean and standard deviation. To evaluate statistical significance, two tailed t-student test was applied with p-value < 0.01. Bonferroni correction was used for transcriptomic and GO analysis.
Availability of supporting data
The data set supporting the results of this article is available in the Gene Expression Omnibus (GEO) Database repository, GSE52545, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52545.
Also other data set supporting the results of this article are included within the article and Additional file 2: Table S2, Additional file 3: Table S4 and Additional file 4: Table S3.
References
Bisson LF: Stuck and sluggish fermentations. Am J Enol Vitic. 1999, 50: 107-119.
Beltran G, Novo M, Leberre V, Sokol S, Labourdette D, Guillamón JM, Mas A, François J, Rozès N: Integration of transcriptomic and metabolic analyses for understanding the global responses of low-temperature winemaking fermentations. FEMS Yeast Res. 2006, 6: 1167-1183. 10.1111/j.1567-1364.2006.00106.x.
Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO: Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000, 11: 4241-4257. 10.1091/mbc.11.12.4241.
Tronchoni J, Arroyo-Lopez FN, Gamero A, Querol A, Barrio E: Differences in the glucose and fructose consumption profiles in diverse Saccharomyces wine species and their hybrids during grape juice fermentation. Int J Food Microbiol. 2009, 134: 237-243. 10.1016/j.ijfoodmicro.2009.07.004.
Gamero A, Wesselink W, de Jong C: Comparison of the sensitivity of different aroma extraction techniques in combination with gas chromatography–mass spectrometry to detect minor aroma compounds in wine. J Chromatogr A. 2013, 1272: 1-7.
Tronchoni J, Rozès N, Querol A, Guillamón JM: Lipid composition of wine strains of Saccharomyces kudriavzevii and Saccharomyces cerevisiae grown at low temperature. Int J Food Microbiol. 2012, 155: 191-198. 10.1016/j.ijfoodmicro.2012.02.004.
Gonzalez SS, Gallo L, Climent MAD, Barrio E, Querol A: Enological characterization of natural hybrids from Saccharomyces cerevisiae and S. kudriavzevii. Int J Food Microbiol. 2007, 116: 11-18. 10.1016/j.ijfoodmicro.2006.10.047.
Belloch C, Orlic S, Barrio E, Querol A: Fermentative stress adaptation of hybrids within the Saccharomyces sensu stricto complex. Int J Food Microbiol. 2008, 122: 188-195. 10.1016/j.ijfoodmicro.2007.11.083.
Peris D, Lopes CA, Belloch C, Querol A, Barrio E: Comparative genomics among Saccharomyces cerevisiae × Saccharomyces kudriavzevii natural hybrid strains isolated from wine and beer reveals different origins. BMC Genomics. 2012, 13: 407-10.1186/1471-2164-13-407.
Abdulrehman D, Monteiro PT, Teixeira MC, Mira NP, Lourenço AB, SC S d, Cabrito TR, Francisco AP, Madeira SC, Aires RS, Oliveira AL, Sá-Correia I, Freitas AT: YEASTRACT: providing a programmatic access to curated transcriptional regulatory associations in Saccharomyces cerevisiae through a web services interface. Nucleic Acids Res. 2011, 39: D136-D140. 10.1093/nar/gkq964.
Kandror O, Bretschneider N, Kreydin E, Cavalieri D, Goldstein AL: Yeast adapt to near-freezing temperatures by STRE/Msn2,4-dependent induction of trehalose synthesis and certain molecular chaperones. Mol Cell. 2004, 13: 771-781. 10.1016/S1097-2765(04)00148-0.
Schade B, Jansen G, Whiteway M, Entian K, Thomas D: Cold adaptation in budding yeast. Mol Biol Cell. 2004, 15: 5492-5502. 10.1091/mbc.E04-03-0167.
Tai SL, Daran-Lapujade P, Walsh MC, Pronk JT, Daran J-M: Acclimation of Saccharomycescerevisiae to low temperature: a chemostat-based transcriptome analysis. Mol Biol Cell. 2007, 18: 5100-10.1091/mbc.E07-02-0131.
Kondo K, Kowalski LR, Inouye M: Cold shock induction of yeast NSR1 protein and its role in pre-rRNA processing. J Biol Chem. 1992, 267: 16259-16265.
Kurata S, Nielsen KH, Mitchell SF, Lorsch JR, Kaji A, Kaji H: Ribosome recycling step in yeast cytoplasmic protein synthesis is catalyzed by eEF3 and ATP. Proc Natl Acad Sci U S A. 2010, 106: 2571-2576.
Arroyo-Lopez FN, Perez-Torrado R, Querol A, Barrio E: Modulation of the glycerol and ethanol syntheses in the yeast Saccharomyces kudriavzevii differs from that exhibited by Saccharomyces cerevisiae and their hybrid. Food Microbiol. 2010, 27: 628-637. 10.1016/j.fm.2010.02.001.
Arroyo-Lopez FN, Pérez Través L, Querol A, Barrio E: Exclusion of Saccharomyces kudriavzevii from a wine model system mediated by Saccharomyces cerevisiae. Yeast. 2011, 28: 423-435. 10.1002/yea.1848.
Salvado Z, Arroyo-Lopez FN, Guillamón JM, Salazar G, Querol A, Barrio E: Temperature adaptation markedly determines evolution within the genus Saccharomyces. Appl Environ Microbiol. 2011, 77: 2292-2302. 10.1128/AEM.01861-10.
Sampaio JP, Goncalves P: Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl Environ Microbiol. 2008, 74: 2144-2152. 10.1128/AEM.02396-07.
Gangl H, Batusic M, Tscheik G, Tiefenbrunner W, Hack C, Lopandic K: Exceptional fermentation characteristics of natural hybrids from Saccharomyces cerevisiae and S. kudriavzevii. N Biotechnol. 2009, 25: 244-251. 10.1016/j.nbt.2008.10.001.
Sahara T: Comprehensive expression analysis of time-dependent genetic responses in yeast cells to low temperature. J Biol Chem. 2002, 277: 50015-50021. 10.1074/jbc.M209258200.
Homma T, Iwahashi H, Komatsu Y: Yeast gene expression during growth at low temperature. Cryobiology. 2003, 46: 230-237. 10.1016/S0011-2240(03)00028-2.
Murata Y, Homma T, Kitagawa E, Momose Y, Sato M, Odani M, Shimizu H, Hasegawa-Mizusawa M, Matsumoto R, Mizukami S: Genome-wide expression analysis of yeast response during exposure to 4 C. Extremophiles. 2006, 10: 117-128. 10.1007/s00792-005-0480-1.
Zavanelli M, Britton J, Igel A, Ares M: Mutations in an essential U2 small nuclear RNA structure cause cold-sensitive U2 small nuclear ribonucleoprotein function by favoring competing alternative U2 RNA structures. Mol Cell Biol. 1994, 14: 1689-
Fortner DM, Troy RG, Brow DA: A stem/loop in U6 RNA defines a conformational switch required for pre-mRNA splicing. Genes Dev. 1994, 8: 221-233. 10.1101/gad.8.2.221.
Li Z, Brow DA: A spontaneous duplication in U6 spliceosomal RNA uncouples the early and late functions of the ACAGA element in vivo. RNA. 1996, 2: 879-894.
Staley JP, Guthrie C: An RNA switch at the 5’ splice site requires ATP and the DEAD box protein Prp28p. Mol Cell. 1999, 3: 55-64. 10.1016/S1097-2765(00)80174-4.
Hilliker AK, Mefford MA, Staley JP: U2 toggles iteratively between the stem IIa and stem IIc conformations to promote pre-mRNA splicing. Genes Dev. 2007, 21: 821-834. 10.1101/gad.1536107.
Perriman RJ, Ares M: Rearrangement of competing U2 RNA helices within the spliceosome promotes multiple steps in splicing. Genes Dev. 2007, 21: 811-820. 10.1101/gad.1524307.
Fournier MJ, Stuart Maxwell E: The nucleolar snRNAs: catching up with the spliceosomalsnRNAs. Trends Biochem Sci. 1993, 18: 131-135. 10.1016/0968-0004(93)90020-N.
Jones P, Inouyej M: RbfA, a 30S ribosomal binding factor, is a cold-shock protein whose absence triggers the cold-shock response. Mol Microbiol. 1996, 21: 1207-1218. 10.1111/j.1365-2958.1996.tb02582.x.
Ermolenko DN, Makhatadze GI: Bacterial cold-shock proteins. Cell Mol Life Sci. 2002, 59: 1902-1913. 10.1007/PL00012513.
Horn G, Hofweber R, Kremer W, Kalbitzer HR: Structure and function of bacterial cold shock proteins. Cell Mol Life Sci. 2007, 64: 1457-1470. 10.1007/s00018-007-6388-4.
Karimi R, Ehrenberg M: Dissociation rate of cognate peptidyl-tRNA from the A-site of hyper-accurate and error-prone ribosomes. Eur J Biochem. 1994, 226: 355-360. 10.1111/j.1432-1033.1994.tb20059.x.
Hirokawa G, Kiel MC, Muto A, Selmer M, Raj VS, Liljas A, Igarashi K, Kaji H, Kaji A: Post-termination complex disassembly by ribosome recycling factor, a functional tRNA mimic. EMBO J. 2002, 21: 2272-2281. 10.1093/emboj/21.9.2272.
Kurata S, Shen B, Liu JO, Takeuchi N, Kaji A, Kaji H: Possible steps of complete disassembly of post-termination complex by yeast eEF3 deduced from inhibition by translocation inhibitors. Nucleic Acids Res. 2013, 41: 264-276. 10.1093/nar/gks958.
López-Malo M, Querol A, Guillamón JM: Metabolomic comparison of Saccharomyces cerevisiae and the cryotolerant species S. bayanus var. uvarum and S. kudriavzevii during wine fermentation at low temperature. PLoS One. 2013, 8: e60135-10.1371/journal.pone.0060135.
Pizarro FJ, Jewett MC, Nielsen J, Agosin E: Growth temperature exerts differential physiological and transcriptional responses in laboratory and wine strains of Saccharomyces cerevisiae. Appl Environ Microbiol. 2008, 74: 6358-6368. 10.1128/AEM.00602-08.
Gómez-Pastor R, Perez-Torrado R, Cabiscol E, Ros J, Matallana E: Reduction of oxidative cellular damage by overexpression of the thioredoxin TRX2 gene improves yield and quality of wine yeast dry active biomass. Microb Cell Fact. 2010, 9: 9-10.1186/1475-2859-9-9.
Lopes CA, Barrio E, Querol A: Natural hybrids of S. cerevisiae × S. kudriavzevii share alleles with European wild populations of Saccharomyces kudriavzevii. FEMS Yeast Res. 2010, 10: 412-421. 10.1111/j.1567-1364.2010.00614.x.
Aerny J: Composés azotes des moûtset des vins. Revue Suisse de Viticulture, arboriculture et Horticulture. 1997, 28: 161-165.
Bely M, Sablayrolles J-M, Barre P: Automatic detection of assimilable nitrogen deficiencies during alcoholic fermentation in oenological conditions. J Ferm Bioeng. 1990, 70: 246-252. 10.1016/0922-338X(90)90057-4.
Rossignol T, Dulau L, Julien A, Blondin B: Genome-wide monitoring of wine yeast gene expression during alcoholic fermentation. Yeast. 2003, 20: 1369-1385. 10.1002/yea.1046.
Garcia-Martinez J, González-Candelas F, Perez-Ortin JE: Common gene expression strategies revealed by genome-wide analysis in yeast. Genome Biol. 2007, 8: R222-10.1186/gb-2007-8-10-r222.
Tusher VG, Tibshirani R, Chu G: Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001, 98: 5116-5121. 10.1073/pnas.091062498.
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J: TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003, 34: 374-378.
Skrzypek MS, Hirschman J: Using the Saccharomyces Genome Database (SGD) for analysis of genomic information. Curr Protoc Bioinformatics. 2011, Chapter 1:Unit 1.20.1-23. doi:10.1002/0471250953.bi0120s35
Robinson MD, Grigull J, Mohammad N, Hughes TR: FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics. 2002, 3: 35-10.1186/1471-2105-3-35.
Pfaffl MW, Horgan GW, Dempfle L: Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30: e36-10.1093/nar/30.9.e36.
Chiva R, López-Malo M, Salvadó Z, Mas A, Guillamón JM: Analysis of low temperature-induced genes (LTIG) in wine yeast during alcoholic fermentation. FEMS Yeast Res. 2012, 12: 831-843. 10.1111/j.1567-1364.2012.00834.x.
Acknowledgements
J. Tronchoni was a recipient of a FPI fellowship from the Spanish Ministry of Education and Science. R. Pérez-Torrado was supported from JAE-DOC postdoctoral programme (IATA-CSIC) co-funded by FSE. This work was supported by CICYT grants (ref. AGL2012-39937-CO2-01, AGL2012-39937-CO2-02 and AGL2010-22001-C02-01) from the Spanish Ministry of Education and Science and by grant PROMETEO (project PROMETEO/2009/019) from -Generalitat Valenciana.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JT performed the transcriptomic studies, must fermentations and cold stress assays, analysed the data and wrote the manuscript. VM performed the translation assays. JMG and AQ analysed the data, participated in the design of the study and wrote the manuscript. RPT conceived the study, analysed the data, participated in the design of the study and wrote the manuscript. All the authors have read and approved the final manuscript.
Electronic supplementary material
12864_2013_6110_MOESM2_ESM.xlsx
Additional file 2: Table S2: Differential gene expression comparing S. kudriavzevii (12-28°C) with S. cerevisiae (12-28°C) response. (XLSX 27 KB)
12864_2013_6110_MOESM3_ESM.xlsx
Additional file 3: Table S4: Results from FunSpec database comparing S. kudriavzevii (12-28°C) with S. cerevisiae (12-28°C) response. (XLSX 15 KB)
12864_2013_6110_MOESM4_ESM.xlsx
Additional file 4: Table S3: Differential gene expression comparing 12 with 28°C in S. kudriavzevii and S. cerevisiae.(XLSX 305 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Tronchoni, J., Medina, V., Guillamón, J.M. et al. Transcriptomics of cryophilic Saccharomyces kudriavzevii reveals the key role of gene translation efficiency in cold stress adaptations. BMC Genomics 15, 432 (2014). https://doi.org/10.1186/1471-2164-15-432
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-432