
Abstract
Background
The two main genetic types in Iberian pig production show important phenotypic differences in growth, fattening and tissue composition since early developmental stages. The objective of this work was the evaluation of muscle transcriptome profile in piglets of both genetic types, in order to identify genes, pathways and regulatory factors responsible for their phenotypic differences. Contemporary families coming from pure Iberian pigs (IB) or from crossing with Duroc boars (DU×IB) were generated. Piglets (14 from each genetic type) were slaughtered at weaning (28 days) and longissimus dorsi was sampled for composition and gene expression studies. RNA was obtained and hybridized to Affymetrix Porcine Genechip expression arrays.
Results
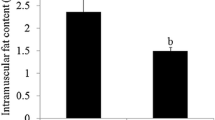
Loin muscle chemical composition showed significant differences between genetic types in intramuscular fat content (6.1% vs. 4.3% in IB and DUxIB animals, respectively, P = 0.009) and in saturated (P = 0.019) and monounsaturated fatty acid proportions (P = 0.044). The statistical analysis of gene expression data allowed the identification of 256 differentially expressed (DE) genes between genetic types (FDR < 0.10), 102 upregulated in IB and 154 upregulated in DU×IB. Transcript differences were validated for a subset of DE genes by qPCR. We observed alteration in biological functions related to extracellular matrix function and organization, cellular adhesion, muscle growth, lipid metabolism and proteolysis. Candidate genes with known effects on muscle growth were found among the DE genes upregulated in DU×IB. Genes related to lipid metabolism and proteolysis were found among those upregulated in IB. Regulatory factors (RF) potentially involved in the expression differences were identified by calculating the regulatory impact factors. Twenty-nine RF were found, some of them with known relationship with tissue development (MSTN, SIX4, IRX3), adipogenesis (CEBPD, PPARGC1B), or extracellular matrix processes (MAX, MXI1). Correlation among the expression of these RF and DE genes show relevant differences between genetic types.
Conclusion
These results provide valuable information about genetic mechanisms determining the phenotypic differences on growth and meat quality between the genetic types studied, mainly related to the development and function of the extracellular matrix and also to some metabolic processes as proteolysis and lipid metabolism. Transcription factors and regulatory mechanisms are proposed for these altered biological functions.
Similar content being viewed by others

Background
Intramuscular fat (IMF) content and fatty acid (FA) composition are critical aspects of pig meat because of their influence on the sensorial and technological aspects of meat quality such as juiciness, flavor, tenderness and overall desirability of meat [1, 2]. Consequently, research on muscle lipid deposition is currently one of the most important fields of study in meat science [3]. Recently, the research interest on this topic includes the complex physiological and genetic mechanisms of IMF deposition and gene expression patterns and interactions along development [4].
Within skeletal muscle, lipids are stored as droplets both in myofiber cytoplasm and in adipocytes (interspersed between fiber fasciculi) with IMF deposition being highly dependent on the number of intramuscular adipocytes [5]. Although adipocyte differentiation starts early in fetal stages, the increase in adipose cell number and size is maintained along early postnatal growth and later development [6]. IMF is considered a late-developing depot because hyperplasia and not only hypertrophy occurs postnatally, as demonstrated in both pigs and cattle [6, 7]; and hypertrophy by lipid filling of adipocytes persists in late stages [8]. Nevertheless, regulation of intramuscular adipocyte differentiation and growth and triglyceride storage is not completely understood and previous studies suggest that preadipocyte differentiation and lipogenesis exhibit breed-related scheduling [9]. Among different breeds, the adipocyte depots can be regulated differently, and may have a different propensity to metabolize lipids.
Comparison of transcriptome of skeletal muscles and other tissues between phenotypically different pig breeds has been proposed to improve the understanding of the genetic mechanisms underlying differences in growth and meat quality [10]. Several transcriptome studies have compared lean (Landrace, Large-White) vs. fatty breeds (mainly Chinese and other local obese breeds) [11–16]. Also the Duroc breed has been compared to the Pietrain lean breed in prenatal stages [17, 18] and to the Chinese obese Taoyuan breed [19]. Some of these previous works have been focused on the study of myogenesis and thus different stages of fetal development were studied, since myogenesis is assumed to be a predominantly prenatal process [20]. Nevertheless, recent work has shown that hyperplastic muscle growth also occurs from birth to weaning in pigs [21].
The Iberian pig breed is a fatty breed with clear differences in growth rate, adipogenic potential and meat quality with respect to many other porcine breeds [22]. Meat and dry-cured Iberian pig products, characterized by its high prices, come from two main genetic types: purebred Iberian and crossbred Iberian with Duroc boars. In the crossbred animals, the growth performance and primal cuts yield are improved, but conversely, meat quality decreases because their muscles contain lower proportions of IMF and monounsaturated fatty acids (MUFA), as oleic acid [23]. Phenotypic differences between both types in lean and adipose tissue growth are visible from early developmental stages [24]. Regarding the analysis of the Iberian pig transcriptome, a comparison with those of Large-White and other breeds for several tissues at 3 m of age has been reported [25, 26], but no comparison has been performed on the muscle transcriptome between the Iberian and Duroc breeds or their crosses.
Our objective was to evaluate the muscle genome expression profiles of Iberian and Duroc × Iberian genetic types in order to identify the genes and molecular pathways involved in their phenotypic differences. In contrast to previous studies with extreme breeds, the genetic types here compared share the same production system, being genetically closer and phenotypically more similar. We selected the longissimus dorsi muscle because it is a prime cut of high economic relevance for fresh and cured pork production. Muscle transcriptome was studied at weaning (28d), as this developmental stage is highly proliferative and relevant for the differentiation of muscular and adipose cells. Additionally, transcriptome information was employed for the identification of transcriptional regulators potentially involved in the different gene expression profiles observed in both genetic types.
Results and discussion
Phenotypic differences between genetic types
At weaning, 28 male piglets (14 of each genetic type) were slaughtered and loin muscle was sampled for composition and gene expression studies. Mean live weight at slaughter was 8.03 kg (SD = 1.59 kg). There was no significant difference in live weight between both genetic types. The percentage of loin IMF was higher in purebred Iberian than in crossbred animals (P = 0.009, Table 1). Differences in muscle fatty acid composition were also observed, with a higher MUFA content in IB and a higher saturated FA content in DU×IB (Table 1). These results confirm the differential trend in fattening and meat quality traits which is evident since this very early growth stage.
Transcriptome study: identification and functional characterization of differentially expressed genes associated with pure Iberian or crossbred genetic types
Among the platforms currently available, the Affymetrix Porcine array is the most sensitive and reproducible microarray for swine genomic studies [27]. Employing this platform, we detected 271 differentially expressed (DE) probes according to genetic type, exceeding the threshold PP-value corresponding to a FDR < 0.10 (Posterior Probability =0.006) (Additional file 1). These correspond to 256 known genes. Ten DE genes were represented by more than one DE probe (CASQ2, ERO1L, IGF2, MTUS2, LOX, MAP1B, ME1, PTPRD, SORT1, SVIP). Out of the 256 DE genes, 154 were overexpressed in DU×IB and 102 were overexpressed in IB. Regarding the size of the effects, the DE genes upregulated in DU×IB ranged between 1.23 and 5.98 fold-changes, and the ones upregulated in IB ranged between 1.21 and 7.87.
Real-time quantitative PCR (qPCR) was employed to assess the expression of 18 genes (eight upregulated in DU×IB, eight upregulated in IB and two unchanged ones), selected to represent different magnitudes of the differential expression detected in the microarray study. Twelve out of the 16 DE genes and one coming from the non-DE group were successfully validated as DE; and two out of the four non-validated genes (PLA1A and CASP4) were close to statistical significance (Table 2). The correlations among gene expression values obtained with microarray and qPCR were significant in 15 out of the 18 selected genes (Table 2). Overall, qPCR results were in the same direction and similar magnitude compared to the microarray. The ELOVL6 gene showed the lowest agreement between methods, which could be due to the detection of different splice variants, as up to 13 different transcripts have been described for this gene in humans. Interestingly, the SCD gene, which was selected as a control non-DE (1.5× higher expression in IB, but without statistical significance), was observed to be significantly DE in the qPCR validation step (2× upregulation in IB, P = 0.03). The substantial value of Concordance Correlation Coefficient calculated between microarray and qPCR fold-change values for these 18 genes (CCC = 0.863) indicates a remarkable agreement between both measures, validating the global reproducibility of microarray results [28].
Functions and pathways altered by genetic type were explored by studying overrepresentation of gene ontology (GO) terms on the three main family categories with DAVID tool (Database for Annotation, Visualization and Integrated Discovery) [29] (Additional file 2). Most enriched terms were related to main biological processes including: extracellular structure organization, developmental process, lipid metabolic process and muscle organ development. Regarding the cellular compartment GO category, the extracellular matrix (ECM) part was highly enriched in the list of DE genes.
The lists of genes upregulated in IB and DU×IB were also separately explored (Tables 3 and 4). In order to reduce the redundancy, a functional annotation clustering was performed with DAVID tool. Genes upregulated in IB affected GO terms grouped in three main annotation clusters, which are related to lipid metabolic process, transcriptional regulation and proteolysis (Table 3). Genes upregulated in DU×IB affected biological process terms clustered in annotation groups related to development, ECM organization, response to stimulus and cell migration and proliferation (Table 4). DAVID tool also allowed for the identification of KEGG pathways significantly enriched in the list of genes upregulated in each genetic type (Additional file 2). Ubiquitin mediated proteolysis was significantly enriched in IB, while several KEGG pathways were overrepresented in the DU×IB type. Among them the most significant ones were ECM-receptor interaction and Focal adhesion.
The main biological functions identified by Ingenuity Pathway Analysis (IPA) in the comparative dataset of the IB and DU×IB groups included categories related to Cell-To-Cell Signaling and Interaction (P = 0.00004, n = 35), Lipid Metabolism (P = 0.0001, n = 43) and Small Molecule Biochemistry (P = 0.0001, n = 48). Furthermore, transcripts related to lipid metabolism were mainly upregulated in IB group, while transcripts related to Cell-To-Cell Signaling and Interaction were mainly upregulated in DU×IB group. The specific functions of cell movement/migration and attachment/adhesion of cells were significantly predicted to be decreased in IB samples (P = 0.00004 and P = 0.0003, respectively). The canonical pathways significantly over-represented in the DE genes are reflected in Figure 1 and the most significant interaction networks (NW) are displayed in Figures 2, 3 and 4. Results concerning the separate analysis of the genes upregulated in each one of the genetic types provide potential specific mechanisms to underlie the biological functions and phenotypic traits changing between them.

Functional categorization analysis with IPA. Canonical pathways significantly enriched in the three sets of genes are shown (P < 0.05): A) Genes DE between both genetic types; B) Genes upregulated in IB; C) Genes upregulated in DU×IB. Signaling pathways are indicated with dark bars and metabolic pathways with light bars.

Gene network 1: Cellular Development, Cellular Growth and Proliferation, Embryonic Development (score 51). Molecules are represented as nodes and the biological relationships between nodes are represented as edges. Genes upregulated in IB are indicated in red and the ones upregulated in DU×IB are shown in green.

Gene network 2: Connective Tissue Disorders, Dermatological Diseases and Conditions, Cellular Assembly and Organization (score 47). Molecules are represented as nodes and the biological relationships between nodes are represented as edges. Genes upregulated in IB are indicated in red and the ones upregulated in DU×IB are shown in green.

Gene network 3: Protein Degradation, Protein Synthesis, Cell Morphology (score 41). Molecules are represented as nodes and the biological relationships between nodes are represented as edges. Genes upregulated in IB are indicated in red and the ones upregulated in DU×IB are shown in green.
The main altered functional patterns observed with the different tools are discussed individually below:
Tissue development and ECM organization
Among the DE genes affected by genetic type we found several widely known ones related to muscle development, including IGF2 (with 5× upregulation in DU×IB), which is the most significant DE gene with seven probes showing differential expression. The growth factor coded by this gene has a major function in muscle promoting fiber differentiation. This locus is paternally imprinted, and a nucleotide substitution in its intron 3 has been described, which abrogates in vitro interaction with a nuclear repressor factor. This substitution affects transcriptional regulation in a way that pigs inheriting the mutation from their sire have a threefold increase in IGF2 messenger RNA expression in postnatal muscle [30]. This mutation is absent in Iberian pig populations and at very high frequency in the Duroc sire lines employed for crossing with Iberian pigs. In fact, our animals were genotyped for this polymorphism and all DU×IB piglets showed the inheritance of the mutant allele from their Duroc sire, in agreement with the differences observed in gene expression.
We also found other DE genes with roles on myogenesis or muscle development as amyloid beta precursor protein (APP) and Fibrillin-2 (FBN2). The APP gene has a central role in the most significant gene network detected in this work (Figure 2), related to tissue development. The APP appears to promote cell adhesion, acting in an integrin-like manner [31]. Evidence of interaction with laminin and collagen provides further evidence of adhesion-promoting properties. Also studies suggest that peptides derived from the amyloid precursor protein can promote transcriptional activation and can have growth-promoting properties both before and after birth [32]. In fact, APP-deficient mice obtained by gene targeting are lighter in body mass. The FBN2 gene may influence the formation and maintenance of extracellular microfibrils [33], and it has been proposed to play an important role in muscle development being considered a candidate for muscling traits [14, 34]. Another interesting result is the upregulation in the DU×IB muscles of AE binding protein 1 (AEBP1) gene, which encodes a member of the carboxypeptidase A protein family. This protein may function as a transcriptional repressor in adipogenesis and muscle cell differentiation, playing a key role in modulation of in vivo adiposity and regulation of energy balance [35]. This protein downregulates PTEN, PPARγ1 and LXRα expression and transcriptional activity [36], and influence intracellular lipid accumulation. It promotes proliferation of preadipocytes and inhibits their differentiation into mature, fat-filled adipocytes [37].
Regarding the functions and pathways affected by genetic type, the most significantly affected biological function, according to all the employed tools, is ECM function and organization, which is narrowly related to tissue development (Additional file 2, Table 4, Figures 1 and 3). Genes involved in cell adhesion and extracellular structures were found upregulated in DU×IB, mainly several integrin and collagen genes and other interacting molecules such as POSTN, LOX, MATN2 or THBS4 (Figure 3). The extracellular matrix consists of a dynamic mixture of structural and functional macromolecules and serves an important role in tissue and organ morphogenesis and in the maintenance of cell and tissue structure and function [38]. It has been shown to be very relevant in cellular signalling because specific interactions between cells and the ECM have pivotal roles in the regulation of muscle cell proliferation and differentiation [39]. These interactions are mediated by transmembrane molecules, mainly integrins and also other cell-surface-associated components, and lead to a direct or indirect control of cellular activities such as adhesion, migration, differentiation, proliferation, and apoptosis [40]. Proliferation determines the pool of muscle cells available for differentiation and thus it influences the potential for muscle growth. Moreover, the extracellular environment regulates the proliferation and differentiation of mesenchymal stem cells and satellite cells [41], which can follow adipogenic differentiation thus increasing the intramuscular adipocytes, fibrogenic differentiation increasing connective tissue content, and of course myogenic differentiation which increases muscle mass. Thus, function of ECM has relevant influence not only on muscle development but also in future meat quality by affecting intramuscular fat deposition and connective tissue abundance [42].
Integrins function as mechanoreceptors and provide a force-transmitting physical link between the ECM and the cytoskeleton and it is proposed that these proteins also regulate angiogenesis [43]. There is considerable evidence of the close relationship between developing adipocytes and vasculature, and hence the influence of integrins on angiogenesis may have an impact on adipogenesis as well. In fact, adipogenesis and angiogenesis are reciprocally regulated [44]. Three different Integrin genes were upregulated in DU×IB (ITGAV, ITGA2 and ITGA6), which show central positions in NW2 related to Cellular Assembly and Organization, especially the ITGA2 gene (Figure 3). ITGA is considered a myogenesis inhibitor which has been suggested to delay muscle differentiation, contributing to a later but higher differentiation of muscle fibers in lean breeds [14], in agreement with our findings. ITGAV is also related to negative regulation of lipid transport and storage. Integrins are narrowly related to collagens and both types of molecules interact in the maintenance of the extracellular matrix. In fact, the DE gene ITGA2 encodes the alpha subunit of a transmembrane receptor for collagens and related proteins. Six different collagen genes were upregulated in DU×IB piglets (COL1A1, COL5A1, COL12A1, COL14A1, COL16A1, COL24A1). Collagen deposition in intramuscular locations starts in fetal stages, and this deposition is predictive of collagen deposition in intramuscular locations in growing pigs [45, 46]. Several works indicate that IGF system, PPARγ and myostatin may regulate the deposition of collagen in pig intramuscular locations [47, 48]. Muscle collagen content contributes to toughness of meat, influences texture and is associated with growth rate [49]. Moreover, collagen development is negatively related to adipocyte development in the ECM: extensive collagen deposition restricts local fat cell clusters growth, and conversely, removal of collagen stimulates the metabolism and survival of adipocytes [50]. Therefore, the higher collagen gene expression in DU×IB muscle is in agreement with the reported higher lean growth but lower meat tenderness and intramuscular fat content of fattened pigs [24].Canonical pathways upregulated in DU×IB (Figure 1) are narrowly related to the effects on ECM. Main signaling pathways are involved in assembly of actin cytoskeleton, cell adhesion to the extracellular matrix, cell-cell adhesion and cell motility and growth (Rho GTPase signaling, PAK signaling, Paxillin signaling, actin cytoskeleton signaling, Gα12 signaling).
Some of our individual DE genes have been previously reported to be DE between phenotypically different breeds or experimental animals, as IGF2[16]. Also, ECM components (mainly collagen genes) and functions have been detected as differential in comparison studies among populations differing in intramuscular fat composition, due to breed or dietary factors [15, 17, 51, 52]. These previous results together with the present ones reinforce the ECM biological importance in determining composition and organization of muscle tissue, as well as its significance in the regulation of IMF deposition.
Lipid metabolism and protein catabolism
The higher lipid deposition observed in muscle of IB pigs (Table 1), was coincident with the upregulation of genes related to lipid metabolism in this genetic type (Additional file 2 and Table 3). Most genes were involved in lipid and phospholipid biosynthetic processes (for example ELOVL6, ME1, PTGES3, AGPAT5, GNPAT) but also some of them had a role in lipid and fatty acid hydrolysis (PON3, PLA1A). This is consistent with previous results showing that fatter animals have higher mRNA levels for both lipogenic and lipolytic enzymes, with net lipid deposition being regulated by their ratio rather than enhancing only one of these pathways [10]. The SCD (Stearoyl-CoA desaturase) gene was also identified to be upregulated in IB muscle in the qPCR validation step (Table 2). This gene is highly relevant regarding meat quality because its product catalyzes the biosynthesis of monounsaturated fatty acids from saturated fatty acids. The higher expression of this gene in pure Iberian pig muscle is in agreement with the differences in fatty acid contents observed (Table 1), which constitute the main differential trait regarding tissue composition between the analysed genetic types also in adulthood. IPA analysis also indicated that Lipid Metabolism was one of the main biological functions in the comparative dataset. Moreover, the most significant canonical signaling pathway represented by the genes upregulated in IB (Figure 1) was involved in the regulation of lipid metabolism, inflammation, and cholesterol to bile acid catabolism (LXR/RXR activation) and the three metabolic pathways detected are related to the biosynthesis of lipids and fatty acids: Triacylglycerol biosynthesis, Prostanoid biosynthesis and Stearate biosynthesis.
When studying genome-wide transcriptional profile on heterogeneous samples as animal tissues it is necessary to take into account the cellular heterogeneity. Any observed differences may be strongly confounded by differences in cell type compositions between samples [53]. In this sense, results regarding lipid metabolism genes are consistent with an influence of differential cellularity between genetic types. Some of the previously mentioned expression differences are in agreement with an earlier adipocyte development in IB muscles, for instance, the DE observed for AEBP1 gene suggests a higher content of preadipocytes in DU×IB piglets vs. a higher content of mature adipocytes in IB. Also, the higher expression of LOX, collagens and other ECM proteins in crossbred muscles may be related to a higher content of preadipocytes [54]. In order to study the potential contribution of this differential cellularity to the expression differences observed, we performed the qPCR quantification of the expression of the preadipocyte marker DLK1 gene. This gene is highly expressed in preadipocytes and absent after adipocyte differentiation [55], and it is not present in the Affymetrix array. In spite of its low expression level in muscle, we detected significant DE in the gene according to genetic type. Muscles from crossbred piglets showed 1.5- fold upregulation (P < 0.023). This interesting result joint with the other DE genes detected supports the hypothesis of higher content of preadipocytes in crossbred animals’ vs. higher content of mature adipocytes in Iberian piglets, and thus earlier adipogenesis in the last ones. On the other hand, a higher lipogenic capacity in muscle of pure IB animals being reflected at the genetic level could be suggested.
Gene ontology enrichment and clustering analyses show that protein catabolic processes are significantly enriched in the dataset of genes upregulated in IB (Table 3 and Additional file 2). Ubiquitin mediated proteolysis KEGG pathway is predicted to be upregulated in IB. Also, according to IPA, the second main canonical pathway affected by the genes upregulated in IB is Protein ubiquitination pathway (Figure 1). The protein ubiquitination pathway plays a major role in the degradation of short-lived or regulatory proteins involved in a variety of cellular processes [56]. DE genes affecting protein ubiquitination include UBE2D4, NEDD4, PSMD11, CUL9, KLHL15, ASB11, RNF19B, UBE2QL1, MYLIP, MARCH6. Apart from this specific pathway which is modification-dependent, gene ontology analysis allows the detection of other proteolytic enzymes as C4BPA, CASP4, CASP7 or CTSL2. IPA also provides a significant gene NW related to proteolysis (Figure 4), composed exclusively of genes upregulated in IB.
Protein degradation may be the main cause of the poor muscle development of purebred Iberian pigs. According to previously reported results of a comparison with Landrace pigs, Iberian pigs show about 20-30% greater rates of muscle protein synthesis, but lower relative and absolute weight of biceps femoris, longissimus and semimembranosus muscles than Landrace [57]. Under similar nutritional and physiological conditions, protein turnover as well as the protein synthesis to protein deposition ratio may differ between Iberian and leaner breeds, resulting in dissimilar protein deposition rates [58]. Our results of differential gene expression support this hypothesis and further specify the potential pathways and genes responsible of the metabolic differences observed.
In summary, the performed comparison of the muscle transcriptome of crossbred and purebred IB piglets allows to highlight several important biological functions narrowly related to their respective muscle characteristics. Muscle tissue development and ECM organization are strongly upregulated in DU×IB, in concordance with greater muscle and connective tissue development, characteristic of this genetic type. Genes and functions upregulated in IB are related to lipid and protein metabolism and are also in agreement with phenotypic traits. Higher lipid metabolism and protein catabolism are coherent with the higher fattening and lower protein deposition in IB muscles. These results provide potential mechanisms to explain the singular growth and fattening phenotype of Iberian pigs, consistent in an earlier adypocite differentiation and hypertrophy and lower protein deposition to synthesis ratio of their muscles. Time-course studies of the differential expression along growth would help to improve the understanding of the metabolic and development differences between genetic types here observed.
Identification of transcriptional regulators potentially involved in the expression changes between genetic types
The magnitude of differential expression does not necessarily indicate biological significance [59]. A very small change in expression of a particular gene may have important physiological consequences if the protein encoded by this gene plays a regulatory role. Downstream genes usually amplify the signal produced by this regulator, thereby increasing their chance to be detected as DE by standard statistical methods. But the chance of a regulatory gene for being selected is small when focussing on the magnitude of differential expression. Regulatory Impact Factor (RIF) metrics have been developed to infer transcriptional regulation from gene expression data by identifying critical regulatory factors (RF) to explain the expression differences observed. RIF metrics are not dependent on the differential expression of the RF, increasing the biological knowledge that can be derived from gene expression experiments [60, 61]. RIF assigns extreme scores to those RF that are consistently most differentially co-expressed with highly abundant and highly DE genes (RIF1), or to those RF with the most altered ability to predict the abundance of DE genes (RIF2). We performed the prediction of RF with extreme RIF z-scores for the whole dataset of DE genes, and also for the genes included in each of the main gene networks detected with IPA.
a) Prediction of regulatory factors for the whole dataset of DE genes (256)
Regulatory factors potentially involved in the regulation of the metabolic processes differing in both genetic types were identified by their RIF1 and RIF2 z-scores. We performed the RIF study for 310 RF (309 TF and MSTN gene, see methods for description) included in the filtered array. We identified 29 RF with extreme z-scores for RIF1 and/or RIF2 parameters (Table 5). Sixteen RF showed extreme values for RIF1 and sixteen for RIF2. KLF11 showed the most extreme score according to RIF1 (-3.79 SD units), and ZHX2 had the most extreme score according to RIF2 (3.45 SD units). The genes SIX4, EYA2 and KLF11 were found to have extreme scores for both RIF1 and RIF2.
RIF metrics allow the identification of relevant RF even not being DE. The critical RF identified in this study were not DE except MAX gene, which showed 1.4× upregulation in the IB group. As expected, RIF metrics identified RF which have been previously shown to have an important role in regulating myogenesis and adipogenesis, but also other ones with less known function. Myostatin (MSTN) is a member of the transforming growth factor beta (TGFβ) superfamily that inhibits muscle differentiation and growth during myogenesis [62]. Its expression is negatively related with muscle mass. In fact, although Myostatin is not a transcription factor, it transmits the signal of TGF-beta cytokines activating multiple intracellular pathways resulting in phosphorylation and activation of downstream Smad proteins and other signalling molecules (e.g. Akt, MAPK, mTOR and Src). These molecules translocate to the nucleus, bind to DNA and regulate transcription of many genes by direct binding to the target gene promoter or through the interaction with transcriptional cofactors in a cell-type-specific manner [63, 64]. Although we do not detect its differential expression between genetic types, MSTN gene is predicted to be a main regulator of the transcriptome differences observed between them. This is in agreement with findings obtained in cattle by Hudson et al. [61, 65], which showed that MSTN is the RF with the highest evidence of contributing to differential expression in muscle in the absence of any demonstrable differential expression of the regulator itself. GATA3 transcription factor is crucial in a variety of developmental processes including adipogenesis [66], and a negative regulator of TGFβ- and Smad4 signaling [67]. Five RF showing extreme RIF1 or RIF2 z-scores have been reported as key regulators of myogenesis, muscle cell differentiation and growth: SIX4 and EYA2 which show a joint and interactive role on activating key muscle determination genes [68, 69], KLF11[70], SOX4[71], and HDAC1[72]. Interestingly, three TFs known to be functionally related and involved in transcriptional regulation of cell proliferation are detected as potential regulators. These are the MAX, MXI1 and MYCN genes. The MXI1 and MYCN proteins compete for interacting with MAX to form heterodimers, which compete for binding to target sites for transcriptional regulation. Whereas the MYCN-MAX complexes induce transcriptional activation, the MXI1-MAX heterodimers repress transcription [73]. Thus, the balance among the different TFs determines the proliferation and tissue growth rate. Also, the CDCA7 gene codes for another TF which regulates cell proliferation being a target of MYC-dependent transcriptional regulation [74]. The GRHL3 transcription factor is involved in development and migration of endothelial cells being considered an angiogenic factor [75], and participates in the regulation of actin cytoskeleton organization. Interestingly, the Iroquois Homeobox 3 gene (IRX3) is identified in our work as a potential regulator for the gene expression differences observed between genetic types. This gene has very recently been proposed as a novel determinant of body mass and composition [76].
Also, RF specifically related to adipogenesis and lipid metabolism were predicted to regulate the expression changes. Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1 β (PPARGC1B) is involved in fat oxidation, non-oxidative glucose metabolism, mitochondrial biogenesis, and the regulation of energy expenditure [77]. It stimulates lipogenic gene expression by activating SREBP transcription factor family. Consistently, SREBF2, a known gene with role on cholesterol homeostasis and control of lipid levels, is also predicted to regulate the set of DE genes. CCAAT/enhancer binding protein delta (CEBPD) belongs to the CEBP family of adipogenic TF and plays a crucial role in mitotic clonal expansion in the early stages of adipocyte differentiation [78]. Retinoic acid receptor related orphan receptor alpha (RORα) is an orphan member of the nuclear receptor superfamily of TF related to lipid homeostasis. It influences genes associated with lipid and carbohydrate metabolism, fatty acid oxidation, insulin signalling, LXR nuclear receptor signalling, and Akt and AMPK signalling in mouse skeletal muscle [79]. Also it influences the expression of SREBP and PPARGC1B TF. Hence, these RF with extreme RIF z-scores are important metabolic regulators which could be especially relevant for the differences in adipocyte differentiation pattern, lipid metabolism, energy balance regulation and fat deposition between muscles of both genetic types.
The identification of different RF corresponding to the same pathways (MAX-MXI-MYCN-CDCA7, MSTN-GATA3, EYA2-SIX4, PPARGC1B-CEBPD-GATA3-SREBF2) is an indication of the reliability of the results and the involvement of the whole pathways in the expression differences found between the compared genetic types.
The RIF approach also gives novel findings regarding functional relationships not previously reported in muscle. Not much is known about ZHX2, LHX6, PAX2, PER3, IRF2, ZNF27, ELK3 or ZFP36L1 regulatory factors in relation to muscle structure or function, although some of them have known roles on developmental processes in other tissues. Also, other ones show key roles in the regulation of cell proliferation and differentiation and are associated with tumour development as MTA3, MN1 or DACH1.
In order to reinforce the reliability of the RIF study, the potential compatibility of the RIF-predicted RF and DE genes was also explored at the DNA sequence level, to analyze the presence of transcription factor binding sites (TFBS) in the DE genes promoters. This study was applied to the DNA-binding and sequence-specific transcription factors, which are the ones analyzed by the Genomatix software. On average, we detected TFBS for the RIF-predicted DNA-binding TF in a 60% of the DE genes promoters. This software also allowed the identification of TF for which the number of TFBS in the DE genes was significantly enriched. The transcription factors MXI1, MAX, MYCN, ELK3, GRHL3, SIX4, PAX2, SREBF2 and KLF11 showed significantly higher number of matches with the promoter sequences than the value expected by chance (Benjamini-corrected P values =6×10-16 for MXI1, MAX and MYCN; 0.001 for ELK3, 0.0008 for GRHL3, 0.037 for SIX4, 3.5×10-8 for PAX2, 0.0008 for SREBF2 and 5.7×10-25 for KLF11).
b) Prediction of regulatory factors for the main networks of DE genes detected with IPA
Several gene networks detected by IPA were selected because of its potential role on the phenotypic differences between genetic types, and RIF metrics were also calculated for the DE genes included in each one of these three main networks.
For the first NW (Development, growth, proliferation), we detected 31 potential regulators (Additional file 3). Out of them, sixteen were coincident with RF previously identified in the whole dataset study and fifteen RF were new ones. Among these, several RF with known roles in the functions affected by this NW can be identified, as MYF6 which is a main regulator of muscle cell differentiation [80], KLF10 which inhibits growth, acting as negative regulator of cellular proliferation [81], or ETS2, which regulates genes related with development and adipogenesis [82]. Moreover, some of the network-specific identified regulators have a role in cell differentiation or proliferation (KLF5, MAFF or SRF).
The relationships found among RF and DE genes were separately analyzed for each NW employing IPA and GENEMANIA [83] tools. Both softwares allow finding relationships in a set of input genes, using a very large set of functional association data, including protein and genetic interactions, pathways, co-expression, co-localization and protein domain similarity. For NW1, all the RF detected with RIF algorithm were predicted to be biologically related with the molecules included in the NW, except LHX6. According to GENEMANIA each RF was found to be related to a mean of nine molecules in the NW. The genes CEBPD, ZHX2 and RORA were the most connected ones.
For the genes included in the second NW (cellular assembly), 27 RF were detected with extreme RIF scores (Additional file 3). Most of them (21) coincide with previously identified ones. Among the 6 new ones, FOSB gene may be highlighted as a regulator of cell proliferation, differentiation and transformation, known to regulate cell-matrix adhesion [84]. As for the first NW, most relationships between RF and DE genes were confirmed to have a biological support. This NW showed and average number of eight relationships among RFs and DE genes, and several predicted RF were connected to many DE genes, suggesting a more important regulatory role (FOSB, CEBPD, SOX4, RORA, ZHX2, ZFP36L1 and specially the complex MXI-MAX-MYC).
For the third NW (proteolysis), which includes only genes upregulated in IB, we detected 29 RF. Out of them, 12 genes were common with the global study and a few more were detected previously in the other networks, but 13 RF were specific for this NW (Additional file 3). Among these, we can highlight some interesting genes, as ETV5, potentially related to energy balance [85], and the HOXA7, HOXA9, HOXB7 and KLF4 genes involved in cellular proliferation and differentiation, cell fate determination and adipogenesis [86–88]. On the other hand, the identification of MSTN and STAT5B as regulators of this NW is interesting, as GH effects on muscle growth are known to act via STAT5B, which regulates the abundance of mature myostatin by proteolytic cleavage [89]. Also, several TF related to lipid metabolism are identified (RXRB, PER2). For NW3 (proteolysis), all the detected RF were predicted to be significantly related with DE genes in this NW. However, most of them showed scarce known connections with the DE genes, with a mean number of five connections. Only the KLF4, ZHX2 and EYA2 genes were related to a higher number of DE genes according to the available information.
The potential relationship of the RIF-predicted RF and DE genes was also explored at the DNA sequence level using the Genomatix software. In each NW, we detected TFBS for the RIF-predicted DNA-binding transcription factors in a mean of 58% of the DE genes’ promoters (54, 58 and 63% for networks 1, 2 and 3, respectively). The results allowed the identification of TF for which the number of TFBS in the DE genes was significantly higher than the number expected by chance. For NW1, the transcription factors MXI1, KLF5, KLF10, KLF11 and SREBF2 showed significantly higher number of matches than expected (Benjamini-corrected P values =0.003 for the first four genes and 0.005 for SREBF2). Transcription factor binding sites for KLF11 were significantly overrepresented in the set of DE genes included in NW2 (Benjamini-corrected P value =0.03). At last, for NW3 the transcription factors MXI1, MAX, KLF4 and KLF11 were highlighted (Benjamini-corrected P values =0.006, 0.006, 0.03 and 0.03, respectively). Thus, although this tool is only applicable to sequence-specific DNA-binding TF, these results reinforce the previous evidences, highlighting the transcription factors MXI1, MAX and KLFs as the most solid findings according all the available information (RIF z-scores, biological relationship and sequence information).
On the other hand, among the RF predicted in the global study of DE genes and in each functional NW, we found RF which consistently appear with elevated RIF z-scores in all analyses (MSTN, MXI1, SIX4, EYA2, IRX3 and ZHX2), which may be responsible of a large part of the gene expression differences detected in muscles from crossbred and purebred Iberian piglets. A regulatory role of these RF on the main phenotypic differences between the compared genetic types can be thus suggested. Also, RF with potential role in specific functions can be identified, as the RF detected exclusively for the proteolysis network, most of which have poorly known roles.
c) Differential correlation among RF and DE genes in the two genetic types
The correlation structure of gene expression conveys important biological information far beyond the marginal measures of differential expression. In fact, co-expression measures can be used to uncover significant features of cellular control and may help in the determination of gene function [59]. RIF’s scores measure differential co-expression (differential connectivity) between two conditions, which integrates three sources of information: the amount of differential expression of DE genes; the abundance of DE genes and the change in correlation existing between the RF and the DE genes [60, 61]. In order to better understand the relationships of the RF predicted with the DE genes, we studied the correlations among their expression values in each one of the genetic types. These correlations were calculated for the three functional networks (Additional file 4). For the first NW, gene expression correlations were calculated between 31 RF and 29 DE genes (899 correlations in each genetic type). Among these, 28 significantly differed between genetic types (FDR <0.10). For the second NW, we calculated in each genetic type the 594 correlations between 27 RF and 22 DE genes, with 80 resulting statistically different between genetic types (FDR < 0.10). Finally, for the third NW, 29 RF and 20 DE genes were employed for the correlation study (580 correlations for each group). In this case 33 correlations were significantly different (FDR < 0.10). For NW 2, those 80 correlations significantly different between genetic types were represented employing Cytoscape 2.8.0 [90] (Additional file 5).
For the first NW, a higher number of correlations significantly differing from zero were observed in IB piglets’ data (Additional file 4). This would suggest a higher activity of transcriptional regulation in purebred animals (either induction or repression), in relation to the biological functions affected by this NW. This would be in agreement with the enriched biological function of transcriptional regulation observed in the IB group in the GO study with DAVID tool (Table 3 and additional file 2). For NW2, a higher proportion of negative correlations was observed in IB dataset (Additional file 5). Several RF involved in the regulation of muscle growth, as MSTN, RORA, SIX4, KLF11 or EYA2, are negatively correlated with the DE genes (which are mostly upregulated in crossbred animals in this NW) in IB dataset, but the correlations are not significantly different from zero in DU×IB piglets. This suggests a transcriptional repression in IB animals of genes involved in ECM development and function. In DU×IB piglets, some RF correlate with the DE genes (LHX6, MAX, MTA3), with most correlations being positive. In NW3, the differential correlation allows to highlight several RF which show opposite behavior in both genetic types. The genes ZHX2 and EYA2 are correlated with several of the DE genes in IB but not in DU×IB, while LHX6 and MAX show the opposite results. These RF may have a role in the expression differences observed between genetic types regarding proteolytic genes.
Conclusions
In this study, purebred and crossbred Iberian piglets differing in muscle traits were compared at the transcriptome level, and a remarkable number of DE probes were detected. The study of DE genes allowed us the identification of biological functions and pathways with relevant role in the differences in development of muscle and intramuscular adipose tissues between the studied genetic types. Muscle development and ECM components are clearly upregulated in crossbred piglets, and the results indicate its main role in the differentiation and development of muscular, adipose and connective tissues, and thus on many growth and meat quality parameters. Over-expression of lipid metabolism genes in purebred Iberian muscle agrees with an earlier adipocyte development in purebred Iberian pigs. In addition, proteolysis pathways were upregulated in purebred Iberian muscle, with potential negative consequences on protein deposition and lean growth.
Apart from measuring differential expression across genetic types, we studied differential co-expression with regulatory factors, thus improving the understanding of the gene expression data and increasing the biological knowledge generated from the experiment. Several RF were identified which could be responsible of the transcriptional regulation of muscles of both genetic types. Some of them have known roles on myogenesis and cell proliferation (as MSTN, MYF6, SIX4, EYA2, MXI1, MAX, MYCN, KLF11, IRX3) or adipogenesis (as PPARGC1, SREBF2 or CEBPD). Also transcriptional regulators for the specific affected functions were identified, which deserve further attention, as ETS2, FOSB, KLF4 or PAX2. At last, RF prediction and correlation study suggest a transcriptional repression of genes involved in muscle growth and ECM function in muscles of purebred piglets.
Results provide candidate genes (DE genes and putative regulatory factors) to explain the phenotypic differences that characterize the genetic types compared. The identification of polymorphisms responsible for these expression changes would be the following step for the practical application of these findings to improve meat quality.
Methods
Animal material
The current study was carried out at the facilities of the CIA Deheson del Encinar (Toledo, Spain). Animal manipulations were performed in compliance with the regulations of the Spanish Policy for Animal Protection RD1201/05, which meets the European Union Directive 86/609 about the protection of animals used in experimentation. The experiment was specifically assessed and approved (report CEEA 2010/003) by the Spanish National Institute for Agricultural and Food Research and Technology (INIA) Committee of Ethics in Animal Research. Two groups of Iberian sows were either mated with Iberian boars or inseminated with Duroc semen. Thirteen contemporary litters were generated (ten of purebred and three of crossbred animals). At weaning (28 days) at least one male piglet was randomly chosen from each litter. A total number of 14 piglets of each genetic type were slaughtered and eviscerated. Loin tissue samples were collected from the carcasses at the level of the last rib and stored at -80°C. Samples were employed for composition and gene expression studies.
Tissue composition analyses
Intramuscular fat was obtained as proposed by Marmer and Maxwell [91]. Longissimus dorsi muscle fat extracts were methylated in the presence of sulphuric acid and analysed by gas chromatography. Fatty acid methyl esters (FAMEs) were identified by gas chromatography as described elsewhere [92] using a Hewlett Packard HP-6890 (Avondale, PA, USA) gas chromatograph equipped with an automatic injector, a flame ionization detector and a capillary column (HP-Innowax, 30 m × 0.32 mm i.d. and 0.25 μm polyethylene glycol-film thickness) (Agilent Technologies Gmbh, Germany). A split ratio of 1:50 and a temperature program of 170 to 245°C were used. The injector and detector were maintained at 250°C. The carrier gas (helium) flow rate was 2 ml/min. Results were expressed as grams per 100 grams of detected FAMEs.
Microarray study
RNA isolation and microarray hybridization and analysis
Loin muscle RNA from 28 animals (14 of each genetic type), was isolated using RiboPure RNA isolation kit (Ambion) following the manufacturer’s recommendations. RNA obtained was quantified using a NanoDrop equipment (NanoDrop Technologies, Wilmington, USA) and RNA quality was assessed with an Agilent bioanalyzer device (Agilent Technologies, Palo Alto, USA). The RNA Integrity Number (RIN) values obtained showed an average of 8.5 ± 0.4, thus assuring their homogeneity and high quality. A non-competitive hybridization with the GeneChip® Porcine Genome Array (Affymetrix, Santa Clara, CA, USA) was performed in two successive series of 16 and 12 samples. This microarray contains 24,123 probe sets that interrogate around 23,250 transcripts from 20,201 Sus scrofa genes. The RNA samples were transferred to the Institut de Recerca Hospital Universitari Vall d’Hebron (Barcelona, Spain) for reverse transcription, fluorescent labeling, hybridization on chips and scanning. Briefly, for each sample 5 μg of total RNA was reverse-transcribed into cDNA molecules, labeled with biotin and hybridized to the high density oligonucleotide chip. Hybridizations were done according to Affymetrix standard protocols and expression data were generated with GeneChip Operating Software (GCOS). All protocols followed the MIAME recommendations [93] developed by the Microarray Gene expression Database Group (http://www.fged.org/). The data sets supporting the results and discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus repository [94] and are accessible through GEO Series accession number GSE53029 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE53029).
Quality control, normalization and filtering of expression data
Microarray data quality evaluation was carried out with the “Affy” and “Sympleaffy” packages of Bioconductor software (http://www.bioconductor.org/) [95]. All the 28 hybridizations performed overcame the quality control and were used for statistical analysis. Normalization was conducted to reduce technical variation between chips. GCRMA normalization was carried out with BRB-Array Tools (v. 3.7.1) (http://linus.nci.nih.gov/BRB-ArrayTools.html) [96]. A filtering step was performed to exclude from the analyses the genes showing minimal variation across the set of arrays. This filtering has been shown to improve the power to detect differential expression [97]. Only genes displaying more than 20% of expression values over ± 1.5 times the median expression of all arrays were used for further analysis. From the total 24,123 probe sets of the array, 5,226 spots overcame these filtering conditions and were used in the statistical analysis of differential expression.
Statistical analysis of microarray data
Normalized microarray expression data (background corrected and base-2 logarithmic-transformed) were analyzed through Bayesian inference using the GEAMM v.1.6 software [98]. The following model was used for searching the effects on expression data of both genetic types:
where y (pq × 1 elements) is the vector of gene expression data sorted by successfully hybridized array (q = 28) and probe within array (p = 5,226), and influenced by the overall effect of each array (a) as well as discrete (Di) within-probe effects (genetic type and hybridization series), both with dimensions 1 × p. All the unknowns in the model were sampled from their joint posterior distribution by Gibbs sampling [99]. Additional details of the performed Bayesian procedure are reported by Casellas et al. [98].
Inferences were made on the probe-specific difference between Di levels from the appropriate posterior distributions summarized by its mean, standard deviation and posterior probability (PP) above (negative mean) or below (positive mean) zero. When the magnitude of PP values is very small, it provides substantial evidence on the differential expression of a given probe. In this study these posterior probabilities were treated as P-values for calculating their maximum value under multiple testing within the false discovery rate (FDR) approach of Benjamini and Hochberg [100]. Because the probe expression values are correlated, the effective number of independent probes (Meff = 4,068) was calculated according to Moskvina and Schmidt [101] and used in the calculation of FDR value.
GeneChip porcine genome array reannotation
Probes were annotated using the latest Affymetrix annotation file available (http://www.affymetrix.com/catalog/131488/AFFY/Porcine-Genome-Array#1_1). Owing to the possibility of missannotation, gene annotation of the DE genes was confirmed from the available sequence used by Affymetrix to design the probes in the array. Each sequence was analyzed by BLAST [102] to confirm the gene annotation, based on homology with other genomes such as human, mouse or bovine, among others.
Gene ontology and functional annotation
To study the functionality of the DE genes we used Gene Ontology (GO) information. The biological interpretation of the data was carried out using the DAVID 2008 database tool [29] which provides batch enrichment analyses to highlight the most relevant GO terms associated to a gene list. This tool detects overrepresented functional gene categories in the gene list compared with a background genome, which in our case was the set of genes present on the filtered array. Significance levels are calculated following a modification of Fisher’s exact test (also named EASE score). Functional terms with P-values lower than 0.05 are usually considered enriched in the annotation categories. A multiple testing-corrected P-value was also calculated using Benjamini and Hochberg algorithm [100], and GO terms with Benjamini-corrected P < 0.10 were considered significant (Additional file 2). Functional annotation clustering was also performed by employing biological process GO terms. Usually, an enrichment score of 1.3, which is equivalent to non-log scale P-value of 0.05, is employed as threshold for cluster significance [29]. Further, we calculated the geometric mean of the Benjamini-corrected P values of the GO terms included in each functional cluster, and retained those clusters with values lower than 0.10 (Tables 3 and 4).
As a complementary approach, Ingenuity Pathway Analysis, Ingenuity Systems (http://www.ingenuity.com) bioinformatics tools were employed to identify and characterize biological functions, gene networks and canonical pathways affected by the treatment. These tools integrate the Ingenuity Knowledge Base of gene-to-gene or protein-to-protein interaction information and the annotated data from the gene expression experiment to generate biological relevant gene regulatory networks. Networks are collections of interconnected molecules assembled by a NW algorithm. Each connection represents known relationships between the molecules, found in the Ingenuity Knowledge Base. Networks are created from “seed” molecules. IPA searches the Ingenuity Knowledge Base for molecules that are known to biologically interact with the seed molecules and makes connections based on the findings. The most highly connected molecules in the DE list and the knowledge base are consolidated into Networks. Network analysis returns a score that ranks networks according to their degree of connectivity and relevance to the network eligible molecules in the dataset [103]. The network score is based on the hypergeometric distribution and is calculated with the right-tailed Fisher’s exact test. The score is the negative log of this P-value. Molecules that demonstrate direct and indirect relationships to other genes, or proteins were integrated into the analysis. The IPA Canonical Pathways Analysis identified the pathways from the Ingenuity Pathways Analysis library of canonical pathways that were most significant in our dataset. The significance of the association between the dataset and the canonical pathway was measured with Fischer’s exact test, to calculate a P‒value determining the probability that the association between the genes in the dataset and the canonical pathway is explained by chance alone.
The global list of 256 DE genes affected by genetic type and the two partial lists of 102 and 154 genes upregulated in IB and DU×IB, respectively, were explored using both the DAVID tool and the core analysis function included in IPA Analysis.
Search of regulatory factors with RIF metrics
RIF1 and RIF2 metrics [60, 61] were calculated for the whole set of DE genes obtained conditional on genetic type (256 genes), and also for the genes included in the most interesting networks identified by IPA software. A manually curated census of 1072 regulatory factors was obtained from Perez-Montarelo et al. [104]. This list was elaborated from previous publications [105, 106] and transcription factor databases (http://www.bioguo.org/AnimalTFDB/; http://www.hprd.org/). Out of those 1072 RF, 310 had probes present in the filtered Affymetrix array, and were used for the RIF analysis (Additional file 6). This list includes sequence-specific DNA-binding transcription factors, but also other transcription factors and cofactors. The MSTN gene was included in the list despite not being a transcription factor, due to its relevant and widely known involvement in muscle growth regulation in mammals. The RIF1 and RIF2 values were computed for the ith RF as follows:
and
where n de is de number of DE genes, and the estimated average expression and differential expression of the jth DE gene, r1 ij and r2 ij the co-expression correlation between the ith RF and the jth DE gene in each one of the genetic types and being e1 j and e2 j the expression of the jth gene in each genetic type [59]. Both RIF measures for each analyzed RF were transformed to standardized z-scores by substracting the mean and dividing by its standard deviation. We identified relevant RF as those with extreme RIF z-scores according to the corresponding confidence intervals (CI) calculated by bootstrap. In each iteration of bootstrapping, a set of nde = 256 genes (nde = 29, 22 and 20 genes for the networks 1, 2 and 3) were randomly selected from the 5,226 probes of the filtered array, and the RIF1 and RIF2 z-scores of the 310 RF were calculated. The procedure was repeated 10,000 times for each scenario to obtain the corresponding 95 and 99% CI intervals of both z-scores.
Different softwares were employed to analyze the compatibility of RIF-predicted RF with the DE genes. IPA (IPA, http://www.ingenuity.com) and Genemania (http://genemania.org) [83] softwares were employed to check for available information about biological relationships between RF and DE genes, and for the construction of networks joining both types of molecules. Genomatix software suite (http://www.genomatix.de/) was employed for the verification of the presence of TFBS in the promoter sequences of the DE genes, which can bind the transcription factors predicted with RIF metrics. The Gene2promoter tool was employed to retrieve promoter sequences for the DE genes from Eldorado database. The selected promoters were subjected to the search of TFBS with the CommonTFs tool, which searches multiple sequences for common TFBSs. For this search, user-defined matrix subsets were created including the matrix families corresponding to the set of RF identified in each step (whole study with the 256 DE genes and each one of the networks), obtained from MatBase. Matrix families were available for a subset of the RIF-predicted RF (Additional file 6). The CommonTFs tool returns the number of sequence matches found between transcription factors and gene promoters included in the analysis and also a P value, which denotes the probability to obtain an equal or greater number of sequences with a match in a randomly drawn sample of the same size as the input sequence set. A multiple testing-corrected P-value was also calculated using Benjamini and Hochberg algorithm [100].
Pearson correlations between the gene expression values of RIF-predicted RF and DE genes in each one of the three IPA networks were calculated. The correlations were calculated for the whole set of available animals, and also separately for IB and DU×IB animals. Statistically significant differences in the correlations between both genetic types were identified for each network (Benjamini-corrected P values < 0.10). Because the probe expression values are correlated, the effective numbers of independent RF and DE genes were calculated according to Moskvina and Schmidt [101] and used in the calculation of Benjamini-corrected P values. Correlations which differ between genetic types were graphically represented employing Cytoscape 2.8.0 [90].
Validation of DE by quantitative PCR (qPCR)
The RNA obtained from loin of the 28 animals under study was employed to perform the technical validation of the differential expression of some probes and also to assess the expression of one candidate gene absent in the microarray data (DLK1). First-strand cDNA synthesis was carried out with Superscript II (Invitrogen, Life Technologies, Paisley, UK) and random hexamers in a total volume of 20 μl containing 1 μg of total RNA and following the supplier’s instructions.
The expression of 19 genes (eight upregulated in DU×IB, eight upregulated in IB, two non-changed ones and one absent in the Affymetrix microarray) was quantified by qPCR. Primer pairs used for quantification were designed using Primer Select software (DNASTAR, Wisconsin, USA) from the available GENBANK and/or ENSEMBL sequences, covering different exons in order to assure the amplification of the cDNA. Sequence of primers and amplicon lengths are indicated in Table 6. Standard PCRs on cDNA were carried out to verify amplicon sizes. Transcript quantification was performed using SYBR Green mix (Roche, Basel, Switzerland) in a LightCycler480 (Roche, Basel, Switzerland). The qPCR reactions were prepared in a total volume of 20 μl containing 2.5 μl of cDNA (1/20 dilution), 10 μl of SYBR Green mix (2X) and 0.15 μM of both forward and reverse primers. As negative controls, mixes without cDNA were used. Cycling conditions were 95°C for 10 min, followed by 45 cycles of 95°C (15 s) and 60°C (1 min) where the fluorescence was acquired. Finally, a dissociation curve to test PCR specificity was generated by one cycle at 95°C (15 s) followed by 60°C (20s) and ramp up to 95°C with acquired fluorescence during the ramp to 0.01°C/s. Data were analysed with LyghtCycler480 SW1.5 software (Roche, Basel, Switzerland). All points and samples were run in triplets as technical replicates and dissociation curves were carried out for each individual replicate. Single peaks in the dissociation curves confirmed the specific amplification of the genes. For each gene, PCR efficiency was estimated by standard curve calculation using four points of cDNA serial dilutions. Values of PCR efficiency are indicated in Table 6. Mean Cp values were employed for the statistical analyses of differential expression. Data normalization was carried out using the two most stable endogenous genes selected out of: GAPDH, B2M, TBP and ACTB. Endogenous genes stability measures were calculated from Genorm software [107]. The GAPDH and ACTB genes were finally employed.
Statistical analyses of tissue composition and qPCR gene expression data
The influence of genetic type on FA composition was separately analyzed for each fatty acid with a linear model fitting as systematic effects the mean and genetic type, and as random the full-sib family and residual effects. For the analysis of qPCR expression data Cp values were transformed to relative quantities using the comparative Cp method. This method is a relative quantification method in which relative gene expression quantities are calculated from Ct/Cp values by employing the specific PCR efficiency values previously calculated for each gene and making the values relative to the sample with higher expression, which is employed as calibrator (Qty = 10-ΔCp/slope) [108]. The qPCR expression data normalization was performed using normalization factors calculated with Genorm software (http://medgen.ugent.be/~jvdesomp/genorm/) from the GAPDH and the ACTB expression values. Relative quantities were divided by the normalization factors which were the geometric means of the two reference genes quantities. Normalized gene expression relative measures obtained were analyzed conditional on genetic type with a t-test. All the analyses were performed using the GLM procedure of SAS 9.1 (SAS Institute Inc., Cary, NC, USA).
The concordance correlation coefficient (CCC) between the fold-change values estimated in loin from microarrays and qPCR expression measures for the 18 genes was calculated to validate the global microarrays results [28].
Availability of supporting data
The data sets supporting the results and discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus repository [94] and are accessible through GEO accession number GSE53029 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE53029).
Abbreviations
- CCC:
-
Concordance correlation coefficient
- CI:
-
Confidence interval
- DAVID:
-
Database for annotation, visualization and integrated discovery
- DE:
-
Differentially expressed
- DUxIB:
-
Duroc x Iberian
- FDR:
-
False discovery rate
- GEO:
-
Gene expression omnibus
- IB:
-
Iberian
- IMF:
-
Intramuscular fat
- ECM:
-
Extracellular matrix
- FA:
-
Fatty acid
- FAME:
-
Fatty acid methyl esters
- GO:
-
Gene ontology
- IPA:
-
Ingenuity pathway analysis
- MUFA:
-
Monounsaturated fatty acid
- NW:
-
Network
- PP :
-
Posterior probability
- qPCR:
-
Real-time quantitative PCR
- RF:
-
Regulatory factor
- RIF:
-
Regulatory Impact Factor
- TF:
-
Transcription factor
- TFBS:
-
Transcription factor binding site.
References
Verbeke W, Van Oeckel MJ, Warnants N, Viaene J, Boucque CV: Consumer perceptions, facts and possibilities to improve acceptability of health and sensory characteristics of pork. Meat Sci. 1999, 53: 77-99. 10.1016/S0309-1740(99)00036-4.
Wood JD, Enser M, Fisher AV, Nute GR, Sheard PR, Richardson RI, Hughes SI, Whittington FM: Fat deposition, fatty acid composition and meat quality: a review. Meat Sci. 2008, 78 (4): 343-358. 10.1016/j.meatsci.2007.07.019.
Hausman GJ, Dodson MV, Ajuwon K, Azain M, Barnes KM, Guan LL, Jiang Z, Poulos SP, Sainz RD, Smith S, Spurlock M, Novakofski J, Fernyhough ME, Bergen WG: The biology and regulation of preadipocytes and adipocytes in meat animals. J Anim Sci. 2009, 87: 1218-1246. 10.2527/jas.2008-1427.
Hocquette JF, Gondret F, Baéza E, Médale F, Jurie C, Pethick DW: Intramuscular fat content in meat-producing animals: development, genetic and nutritional control, and identification of putative markers. Animal. 2010, 4 (2): 303-319. 10.1017/S1751731109991091.
Zheng GS, Mei ZS: Physiology, affecting factors and strategies for control of Pig meat intramuscular Fat. Recent Pat Food Nutr Agric. 2009, 1: 59-74.
Lee YB, Kauffman RG: Cellular and enzymatic changes with animal growth in porcine intramuscular adipose tissue. J Anim Sci. 1974, 38 (3): 532-537.
Cianzio DS, Topel DG, Whitehurst GB, Beitz DC, Self HL: Adipose tissue growth and cellularity: changes in bovine adipocyte size and number. J Anim Sci. 1985, 60 (4): 970-976.
Hood RL, Allen CE: Cellularity of porcine adipose tissue: effects of growth and adiposity. J Lipid Res. 1977, 18: 275-284.
Li WZ, Zhao SM, Huang Y, Yang MH, Pan HB, Zhang X, Ge CR, Gao SZ: Expression of lipogenic genes during porcine intramuscular preadipocyte differentiation. Res Vet Sci. 2012, 93 (3): 1190-1194. 10.1016/j.rvsc.2012.06.006.
Pena RN, Quintanilla R, Manunza A, Gallardo D, Casellas J, Amills M: Application of the microarray technology to the transcriptional analysis of muscle phenotypes in pigs. Anim Genet. 2014, Published online: 27 MAR 2014. doi: 10.1111/age.12146
Wimmers K, Murani E, te Pas MF, Chang KC, Davoli R, Merks JW, Henne H, Muraniova M, da Costa N, Harlizius B, Schellander K, Böll I, Braglia S, de Wit AA, Cagnazzo M, Fontanesi L, Prins D, Ponsuksili S: Associations of functional candidate genes derived from gene-expression profiles of prenatal porcine muscle tissue with meat quality and muscle deposition. Anim Genet. 2007, 38: 474-484. 10.1111/j.1365-2052.2007.01639.x.
Kim NK, Park HR, Lee HC, Yoon D, Son ES, Kim YS, Kim SR, Kim OH, Lee CS: Comparative studies of skeletal muscle proteome and transcriptome profilings between pig breeds. Mamm Genome. 2010, 21: 307-319. 10.1007/s00335-010-9264-8.
Murgiano L, D’Alessandro A, Egidi MG, Crisa A, Prosperini G, Timperio AM, Valentini A, Zolla L: Proteomics and transcriptomics investigation on longissimus muscles in Large White and Casertana pig breeds. J Proteome Res. 2010, 9: 6450-6466. 10.1021/pr100693h.
Zhao X, Mo D, Li A, Gong W, Xiao S, Zhang Y, Qin L, Niu Y, Guo Y, Liu X, Cong P, He Z, Wang C, Li J, Chen Y: Comparative analyses by sequencing of transcriptomes during skeletal muscle development between pig breeds differing in muscle growth rate and fatness. PLoS One. 2011, 6: e19774-10.1371/journal.pone.0019774.
Damon M, Wyszynska-Koko J, Vincent A, Herault F, Lebret B: Comparison of muscle transcriptome between pigs with divergent meat quality phenotypes identifies genes related to muscle metabolism and structure. PLoS One. 2012, 7 (3): e33763-10.1371/journal.pone.0033763.
Yu K, Shu G, Yuan F, Zhu X, Gao P, Wang S, Wang L, Xi Q, Zhang S, Zhang Y, Li Y, Wu T, Yuan L, Jiang Q: Fatty acid and transcriptome profiling of longissimus dorsi muscles between pig breeds differing in meat quality. Int J Biol Sci. 2013, 9 (1): 108-118. 10.7150/ijbs.5306.
Cagnazzo M, te Pas MF, Priem J, de Wit AA, Pool MH, Davoli R, Russo V: Comparison of prenatal muscle tissue expression profiles of two pig breeds differing in muscle characteristics. J Anim Sci. 2006, 84 (1): 1-10.
Davoli R, Braglia S, Russo V, Varona L, te Pas MF: Expression profiling of functional genes in prenatal skeletal muscle tissue in Duroc and Pietrain pigs. J Anim Breed Genet. 2011, 128 (1): 15-27. 10.1111/j.1439-0388.2010.00867.x.
Lin CS, Hsu CW: Differentially transcribed genes in skeletal muscle of Duroc and Taoyuan pigs. J Anim Sci. 2005, 83: 2075-2086.
Bentzinger CF, Wang YX, Rudnicki MA: Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol. 2012, 4: a008342-
Rodríguez-López JM, Pardo C, Bee G: Hyperplastic muscle growth occurs from birth to weaning in pigs. J Anim Sci. 2010, 88 (Suppl. 2): 364-
López-Bote C: Sustained utilization of Iberian pig breed. Meat Sci. 1998, 49: S17-S27.
Ventanas S, Ventanas J, Jurado A, Estévez M: Quality traits in muscle biceps femoris and back-fat from purebred Iberian and reciprocal Iberian x Duroc crossbred pigs. Meat Sci. 2006, 73: 651-659. 10.1016/j.meatsci.2006.03.009.
Serrano MP, Valencia DG, Nieto M, Lazaro R, Mateos GG: Influence of sex and terminal sire line on performance and carcass and meat quality of Iberian pigs reared under intensive production systems. Meat Sci. 2008, 78: 420-428. 10.1016/j.meatsci.2007.07.006.
Ferraz ALJ, Ojeda A, López-Béjar M, Fernandes LT, Castelló A, Folch JM, Pérez-Enciso M: Transcriptome architecture across tissues in the pig. BMC Genomics. 2008, 9: 173-10.1186/1471-2164-9-173.
Pérez-Enciso M, Ferraz ALJ, Ojeda A, López-Béjar M: Impact of breed and sex on porcine endocrine transcriptome: a bayesian biometrical analysis. BMC Genomics. 2009, 10: 89-10.1186/1471-2164-10-89.
Tsai S, Mir B, Martin AC, Estrada JL, Bischoff SR, Hsieh WP, Cassady JP, Freking BA, Nonneman DJ, Rohrer GA, Piedrahita JA: Detection of transcriptional difference of porcine imprinted genes using different microarray platforms. BMC Genomics. 2006, 7: 328-10.1186/1471-2164-7-328.
Miron M, Woody OZ, Marcil A, Murie C, Sladek R, Nadon R: A methodology for global validation of microarray experiments. BMC Bioinforma. 2006, 7: 333-10.1186/1471-2105-7-333.
Huang DW, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc. 2009, 4: 44-57.
Van Laere AS, Nguyen M, Braunschweig M, Nezer C, Collette C, Moreau L, Archibald AL, Haley CS, Buys N, Tally M, Andersson G, Georges M, Andersson L: A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature. 2003, 425 (6960): 832-836. 10.1038/nature02064.
Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Löwer A, Langer A, Merdes G, Paro R, Masters CL, Müller U, Kins S, Beyreuther K: Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005, 24 (20): 3624-3634. 10.1038/sj.emboj.7600824.
Thinakaran G, Koo EH: Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008, 283: 29615-29619. 10.1074/jbc.R800019200.
Zhang H, Apfelroth SD, Hu W, Davis EC, Sanguineti C, Bonadio J, Mecham RP, Ramirez F: Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices. J Cell Biol. 1994, 5: 855-863.
Bílek K, Knoll A, Stratil A, Svobodová K, Horák P, Bechyňová R, Van Poucke M, Peelman LJ: Analysis of mRNA expression of CNN3, DCN, FBN2, POSTN, SPARC and YWHAQ genes in porcine foetal and adult skeletal muscles K. Czech J Anim Sci. 2008, 53 (5): 181-186.
Zhang L, Reidy SP, Nicholson TE, Lee HJ, Majdalawieh A, Webber C, Stewart BR, Dolphin P, Ro HS: The role of AEBP1 in Sex-specific diet-induced obesity. Mol Med. 2005, 11 (1–12): 39-47.
Majdalawieh A, Ro HS: PPARγ1 and LXRα face a new regulator of macrophage cholesterol homeostasis and inflammatory responsiveness, AEBP1. Nucl Recept Signal. 2010, 8: e004-
Gagnon A, Abaiian KJ, Crapper T, Layne MD, Sorisky A: Down-regulation of aortic carboxypeptidase-like protein during the early phase of 3 T3-L1 adipogenesis. Endocrinology. 2002, 143 (7): 2478-2485. 10.1210/endo.143.7.8875.
Velleman SG: Extracellular matrix regulation of skeletal muscle formation. J Anim Sci. 2012, 90: 936-941. 10.2527/jas.2011-4497.
Clause KC, Barke TH: Extracellular matrix signaling in morphogenesis and repair. Curr Opin Biotechnol. 2013, 24 (5): 830-833. 10.1016/j.copbio.2013.04.011.
Boudreau NJ, Jones PL: Extracellular matrix and integrin signalling: the shape of things to come. Biochem J. 1999, 339: 481-488. 10.1042/0264-6021:3390481.
Watt F, Huck WTS: Role of the extracellular matrix in regulating stem cell fate. Nat Rev Mol Cell Biol. 2013, 14: 467-473. 10.1038/nrm3620.
Du M, Carlin KM: Extracellular matrix in skeletal muscle development and meat quality. J Anim Sci. 2012, 90: 922-923. 10.2527/jas.2011-4937.
Stupack DG, Cheresh DA: Integrins and angiogenesis. Curr Top Dev Biol. 2004, 64: 207-238.
Fukumura D, Ushiyama A, Duda DG, Xu L, Tam J, Krishna V, Chatterjee K, Garkavtsev I, Jain RK: Paracrine regulation of angiogenesis and adipocyte differentiation during in vivo adipogenesis. Circ Res. 2003, 93 (9): e88-e97. 10.1161/01.RES.0000099243.20096.FA.
Karunaratne JF, Ashton CJ, Stickland NC: Fetal programming of fat and collagen in porcine skeletal muscles. J Anat. 2005, 207: 763-768. 10.1111/j.1469-7580.2005.00494.x.
Karunaratne JP, Bayol SA, Ashton CJ, Simbi BH, Stickland NC: Potential molecular mechanisms for the prenatal compartmentalisation of muscle and connective tissue in pigs. Differentiation. 2009, 77: 290-297. 10.1016/j.diff.2008.10.002.
Hausman GJ, Richardson RL, Simmen FA: Expression of insulin-like growth factor binding proteins (IGFBPs) before and during the hormone sensitive period of adipose tissue development in the fetal pig. Growth Dev Aging. 2000, 64: 51-67.
Hausman GJ, Dodson MV: Stromal vascular cells and adipogenesis: cells within adipose depots regulate adipogenesis. J Genomics. 2012, 1: 56-66.
Purslow PP, Archile-Contreras AC, Cha MC: Manipulating meat tenderness by increasing the turnover of intramuscular connective tissue. J Anim Sci. 2012, 90: 950-959. 10.2527/jas.2011-4448.
Hausman GJ: Meat science and muscle biology symposium: the influence of extracellular matrix on intramuscular and extramuscular adipogenesis. J Anim Sci. 2012, 90 (3): 942-949. 10.2527/jas.2011-4616.
Pena RN, Noguera JL, Casellas J, Díaz I, Fernández AI, Folch JM, Ibáñez-Escriche N: Transcriptional analysis of intramuscular fatty acid composition in the longissimus thoracis muscle of Iberian × Landrace back-crossed pigs. Anim Genet. 2013, 44 (6): 648-660. 10.1111/age.12066.
Hamill RM, Aslan O, Mullen AM, O’Doherty JV, McBryan J, Morris DG, Sweeney T: Transcriptome analysis of porcine M. semimembranosus divergent in intramuscular fat as a consequence of dietary protein restriction. BMC Genomics. 2013, 14: 453-10.1186/1471-2164-14-453.
Lähdesmäki H, Shmulevich I, Dunmire V, Yli-Harja O, Zhang W: In silico microdissection of microarray data from heterogeneous cell populations. BMC Bioinforma. 2005, 6: 54-10.1186/1471-2105-6-54.
Urs S, Smith C, Campbell B, Saxton AM, Taylor J, Zhang B, Snoddy J, Jones Voy B, Moustaid-Moussa N: Gene expression profiling in human preadipocytes and adipocytes by microarray analysis. J Nutr. 2004, 134 (4): 762-770.
Wang Y, Hudak C, Sul HS: Role of preadipocyte factor 1 in adipocyte differentiation. Clin Lipidol. 2010, 5 (1): 109-115. 10.2217/clp.09.80.
Glickman MH, Ciechanover A: The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002, 82 (2): 373-428.
Rivera-Ferre MG, Aguilera JF, Nieto R: Muscle fractional protein synthesis is higher in Iberian than in Landrace growing pigs fed adequate or lysine-deficient diets. J Nutr. 2005, 135 (3): 469-478.
Rivera-Ferre MG, Aguilera JF, Nieto R: Differences in whole-body protein turnover between Iberian and Landrace pigs fed adequate or lysine-deficient diets. J Anim Sci. 2006, 84 (12): 3346-3355. 10.2527/jas.2005-405.
Almudevar A, Klebanov LB, Qiu X, Salzman P, Yakovlev AY: Utility of correlation measures in analysis of gene expression. NeuroRx. 2006, 3 (3): 384-395. 10.1016/j.nurx.2006.05.037.
Reverter A, Hudson NJ, Nagaraj SH, Pérez-Enciso M, Dalrymple BP: Regulatory impact factors: unraveling the transcriptional regulation of complex traits from expression data. Bioinformatics. 2010, 26 (7): 896-904. 10.1093/bioinformatics/btq051.
Hudson NJ, Dalrymple BP, Reverter T: Beyond differential expression: the quest for causal mutations and effector molecules. BMC Genomics. 2012, 13: 356-10.1186/1471-2164-13-356.
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P: Genesis, Modulation, and Regeneration of Skeletal Muscle. Molecular Biology of the Cell. 2002, New York: Garland Science, Available from: http://www.ncbi.nlm.nih.gov/books/NBK26853/, 4
Heldin CH, Miyazono K, ten Dijke P: TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997, 390 (6659): 465-471. 10.1038/37284.
Liu T, Feng XH: Regulation of TGF-β Signaling by Protein Phosphatases. Biochem J. 2010, 430 (2): 191-198. 10.1042/BJ20100427.
Hudson N, Reverter A, Dalrymple B: A differential wiring analysis of expression data correctly identifies the gene containing the causal mutation. PLoS Comput Biol. 2009, 5: e1000382-10.1371/journal.pcbi.1000382.
Tong Q, Tsai J, Tan G, Dalgin G, Hotamisligil GS: Interaction between GATA and the C/EBP family of transcription factors is critical in GATA-mediated suppression of adipocyte differentiation. Mol Cell Biol. 2005, 25 (2): 706-715. 10.1128/MCB.25.2.706-715.2005.
Sun J, He H, Pillai S, Xiong Y, Challa S, Xu L, Chellappan S, Yang S: GATA3 transcription factor abrogates Smad4 transcription factor-mediated fascin overexpression, invadopodium formation, and breast cancer cell invasion. J Biol Chem. 2013, 288 (52): 36971-36982. 10.1074/jbc.M113.506535.
Richard AF, Demignon J, Sakakibara I, Pujol J, Favier M, Strochlic L, Le Grand F, Sgarioto N, Guernec A, Schmitt A, Cagnard N, Huang R, Legay C, Guillet-Deniau I, Maire P: Genesis of muscle fiber-type diversity during mouse embryogenesis relies on Six1 and Six4 gene expression. Dev Biol. 2011, 359 (2): 303-320. 10.1016/j.ydbio.2011.08.010.
Grifone R, Demignon J, Giordani J, Niro C, Souil E, Bertin F, Laclef C, Xu PX, Maire P: Eya1 and Eya2 proteins are required for hypaxial somitic myogenesis in the mouse embryo. Dev Biol. 2007, 302 (2): 602-616. 10.1016/j.ydbio.2006.08.059.
Kaczynski J, Cook T, Urrutia R: Sp1- and Krüppel-like transcription factors. Genome Biol. 2003, 4: 206-10.1186/gb-2003-4-2-206.
Jang SM, Kim JW, Kim D, Kim CH, An JH, Choi KH, Rhee S: Sox4-mediated caldesmon expression facilitates skeletal myoblast differentiation. J Cell Sci. 2013, doi:10.1242/jcs.131581
Mal A, Sturniolo M, Schiltz RL, Ghosh MK, Harter ML: A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. EMBO J. 2001, 20: 1739-1753. 10.1093/emboj/20.7.1739.
Lee T, Ziff EB: Mxi1 is a repressor of the c-myc promoter and reverses activation by USF. J Biol Chem. 1999, 274: 595-606. 10.1074/jbc.274.2.595.
Gill RM, Gabor TV, Couzens AL, Schei MP: The MYC-associated protein CDCA7 is phosphorylated by AKT to regulate MYC-dependent apoptosis and transformation. Mol Cell Biol. 2013, 33 (3): 498-513. 10.1128/MCB.00276-12.
Guardiola-Serrano F, Haendeler J, Lukosz M, Sturm K, Melchner H, Altschmied J: Gene trapping identifies a putative tumor suppressor and a new inducer of cell migration. Biochem Biophys Res Commun. 2008, 376 (4): 748-752. 10.1016/j.bbrc.2008.09.070.
Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, Lee JH, Puviindran V, Tam D, Shen M, Son JE, Vakili NA, Sung HK, Naranjo S, Acemel RD, Manzanares M, Nagy A, Cox NJ, Hui CC, Gomez-Skarmeta JL, Nóbrega MA: Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014, 507: 371-375. 10.1038/nature13138.
Handschin C, Spiegelman BM: Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006, 27: 728-735. 10.1210/er.2006-0037.
Hishida T, Nishizuka M, Osada S, Imagawa M: The role of C/EBPdelta in the early stages of adipogenesis. Biochimie. 2009, 91 (5): 654-657. 10.1016/j.biochi.2009.02.002.
Raichur S, Fitzsimmons RL, Myers SA, Pearen MA, Lau P, Eriksson N, Wang SM, Muscat GEO: Identification and validation of the pathways and functions regulated by the orphan nuclear receptor, ROR alpha1, in skeletal muscle. Nucleic Acids Res. 2010, 38 (13): 4296-4312. 10.1093/nar/gkq180.
Fan H, Cinar MU, Phatsara C, Tesfaye D, Tholen E, Looft C, Schellander K: Molecular mechanism underlying the differential MYF6 expression in postnatal skeletal muscle of Duroc and Pietrain breeds. Gene. 2011, 486 (1–2): 8-14.
Parakati R, DiMario JX: Repression of myoblast proliferation and fibroblast growth factor receptor 1 promoter activity by KLF10 protein. J Biol Chem. 2013, 288 (19): 13876-13884. 10.1074/jbc.M113.457648.
Birsoy K, Berry R, Wang T, Ceyhan O, Tavazoie S, Friedman JM, Rodeheffer MS: Analysis of gene networks in white adipose tissue development reveals a role for ETS2 in adipogenesis. Development. 2011, 138 (21): 4709-4719. 10.1242/dev.067710.
Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q: GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008, 9 (1): S4-10.1186/gb-2008-9-1-r4. doi:10.1186/gb-2008-9-s1-s4
Ohnishi YN, Sakumi K, Yamazaki K, Ohnishi YH, Miura T, Tominaga Y, Nakabeppu Y: Antagonistic regulation of cell-matrix adhesion by FosB and DeltaFosB/Delta2DeltaFosB encoded by alternatively spliced forms of fosB transcripts. Mol Biol Cell. 2008, 19 (11): 4717-4729. 10.1091/mbc.E07-08-0768.
Speakman JR: Functional analysis of seven genes linked to body mass index and adiposity by genome-wide association studies: a review. Hum Hered. 2013, 75 (2–4): 57-79.
Cantile M, Schiavo G, Terracciano L, Cillo C: Homeobox genes in normal and abnormal vasculogenesis. Nutr Metab Cardiovasc Dis. 2008, 18: 651-658. 10.1016/j.numecd.2008.08.001.
Bernot D, Barruet E, Poggi M, Bonardo B, Alessi MC, Peiretti F: Down-regulation of tissue inhibitor of metalloproteinase-3 (TIMP-3) expression is necessary for adipocyte differentiation. J Biol Chem. 2010, 285 (9): 6508-6514. 10.1074/jbc.M109.078444.
Saulnier N, Puglisi MA, Lattanzi W, Castellini L, Pani G, Leone G, Alfieri S, Michetti F, Piscaglia AC, Gasbarrini A: Gene profiling of bone marrow- and adipose tissue-derived stromal cells: a key role of Kruppel-like factor 4 in cell fate regulation. Cytotherapy. 2011, 13 (3): 329-340. 10.3109/14653249.2010.515576.
Oldham JM, Osepchook CC, Jeanplong F, Falconer SJ, Matthews KG, Conaglen JV, Gerrard DF, Smith HK, Wilkins RJ, Bass JJ, McMahon CD: The decrease in mature myostatin protein in male skeletal muscle is developmentally regulated by growth hormone. J Physiol. 2009, 587: 669-677. 10.1113/jphysiol.2008.161521.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T: Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13: 2498-2504. 10.1101/gr.1239303.
Marmer WN, Maxwell RJ: Dry column method for the quantitative extraction and simultaneous class separation of lipids from muscle tissue. Lipids. 1981, 16: 365-371. 10.1007/BF02534964.
López-Bote CJ, Rey A, Ruiz J, Isabel B, Sanz Arias R: Effects of feeding diets high in monounsaturated fatty acids and α-tocopheryl acetate to rabbits on resulting carcass fatty acid profile and lipid oxidation. Anim Sci. 1997, 64: 177-186. 10.1017/S1357729800015691.
Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FCP, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze-Kremer S, Stewart J, Taylor R, Vilo J, Vingron M: Minimum information about a microarray experiment (MIAME)—toward standards for microarray data. Nat Genet. 2001, 29: 365-371. 10.1038/ng1201-365.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A: NCBI GEO: archive for functional genomics data sets—update. Nucl Acids Res. 2013, 41 (D1): D991-D995. 10.1093/nar/gks1193. http://www.ncbi.nlm.nih.gov/geo/,
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J: Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5: R80-10.1186/gb-2004-5-10-r80.
Xu X, Zhao Y, Simon R: Gene Set Expression Comparison kit for BRB-ArrayTools. Bioinformatics. 2008, 24: 137-139. 10.1093/bioinformatics/btm541.
Hackstadt AJ, Hess AM: Filtering for increased power for microarray data analysis. BMC Bioinforma. 2009, 10: 11-10.1186/1471-2105-10-11. doi:10.1186/1471-2105-10-11
Casellas J, Ibáñez-Escriche N, Martínez-Giner M, Varona L: GEAMM v. 1.4: a versatile program for mixed model analysis of gene expression data. Anim Genet. 2008, 39: 89-90. 10.1111/j.1365-2052.2007.01670.x.
Gelfand AE, Smith AFM: Sampling-based approaches to calculating marginal densities. J Am Stat Assoc. 1990, 85: 398-409. 10.1080/01621459.1990.10476213.
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc Ser B. 1995, 57: 289-300.
Moskvina V, Schmidt KM: On multiple-testing correction in genome-wide association studies. Genet Epidemiol. 2008, 32 (6): 567-573. 10.1002/gepi.20331.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410. 10.1016/S0022-2836(05)80360-2.
Calvano SE, Wenzhong X, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF: A network-based analysis of systemic inflammation in humans. Nature. 2005, 437: 1032-1037. 10.1038/nature03985.
Perez-Montarelo D, Hudson NJ, Fernandez AI, Ramayo-Caldas Y, Dalrymple BP: Porcine tissue-specific regulatory networks derived from meta-analysis of the transcriptome. PLoS One. 2012, 7 (9): e46159-10.1371/journal.pone.0046159. doi:10.1371/journal.pone.0046159
Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM: A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009, 10 (4): 252-263. 10.1038/nrg2538.
Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, Akalin A, Schmeier S, Kanamori-Katayama M, Bertin N, Carninci P, Daub CO, Forrest AR, Gough J, Grimmond S, Han JH, Hashimoto T, Hide W, Hofmann O, Kamburov A, Kaur M, Kawaji H, Kubosaki A, Lassmann T, van Nimwegen E, MacPherson CR, Ogawa C, Radovanovic A, Schwartz A, Teasdale RD: An atlas of combinatorial transcriptional regulation in mouse and Man. Cell. 2010, 141: 369-369. 10.1016/j.cell.2010.04.004.
Vandesompele J, De Preter K, Pattyn F, Pappe B, Van Roy N, De Paepe A, Speleman F: Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3: 7-
Pfaffl MW: A new mathematical model for relative quantification in real-time RT-PCR. Nucl Acids Res. 2001, 29: 2002-2007.
Acknowledgements
The research was supported by INIA grant RTA2007-00075-00-00, CAM grant P2009-AGR1704 (Comunidad de Madrid) and AGL2010-21991-C03-02. We are grateful to Jaime Rodrigañez, Pedro Martín-Palomino, and all the staff of “El Dehesón del Encinar” for their invaluable help.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CO and LS participated in experimental design, gene expression data analysis and interpretation; and drafting the manuscript. RB, MA and YN participated in animal sampling, RNA isolation and qPCR analysis. AF, AIF and CR participated in data analysis and interpretation. BI and AR carried out meat samples composition analysis. CLB participated in experimental design and coordination; and editing of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_6121_MOESM1_ESM.xlsx
Additional file 1: Annotated list of probes showing differential expression between genetic types. List and annotation of the 271 probes differentially expressed between l.dorsi muscle of purebred Iberian piglets and crossbred with Duroc (FDR < 0.10), at 28 d of age. Posterior probabilities (PP) for the genetic type effect and expression ratios (calculated from mean log2 intensities) are shown. (XLSX 35 KB)
12864_2013_6121_MOESM2_ESM.xlsx
Additional file 2: Gene ontology terms. List of significantly overrepresented GO terms (distributed over the three main categories) and KEGG pathways in the three analysed gene lists: the 256 DE genes between genetic types, the 154 genes upregulated in DUxIB and the 102 genes upregulated in IB. Parametric P-values are indicated along with Benjamini multiple test corrections (FDR < 0.10). (XLSX 41 KB)
12864_2013_6121_MOESM3_ESM.xlsx
Additional file 3: Prediction of RF for the three functional networks. RIF metrics were employed for the identification of RF potentially involved in the three functional gene networks detected with IPA software. Annotation and RIF z-scores are provided for the RFs identified in each network. Bootstrap 99% and 95% confidence intervals for RIF1 and RIF2 z-scores are indicated for each network, as inserted comments in the corresponding columns. (XLSX 28 KB)
12864_2013_6121_MOESM4_ESM.xlsx
Additional file 4: Gene expression correlations between RF and DE genes. Correlations were calculated among the expression values of RF and DE genes, for the three functional IPA networks, separately for the animals of each genetic type. For each network, magnitude and significance of the correlation in each genetic type and significance of the difference in the correlation between genetic types are shown (nominal P-values). Shaded lines are correlations significantly different in both genetic types (FDR < 0.10). (XLSX 229 KB)
12864_2013_6121_MOESM5_ESM.docx
Additional file 5: Graphical representation of the correlations RF-DE genes in network 2 (Connective Tissue Disorders, Dermatological Diseases and Conditions, Cellular Assembly and Organization). Gene expression correlations between RIF-predicted regulatory factors and DE genes, which were significantly different in both genetic types, were graphically represented with Cytoscape 2.8.0, for each genetic type. Light circle nodes represent regulatory factors. Squared nodes are DE genes, red ones are upregulated in IB while green ones are upregulated in DUxIB. Solid lines are significant correlations and dotted lines are non-significantly different from cero (in each genetic type). Blue lines are positive correlations and pink ones are negative correlations. (DOCX 785 KB)
12864_2013_6121_MOESM6_ESM.xlsx
Additional file 6: Regulatory factors employed for the RIF analysis. Annotated list of 310 regulatory factors with probes available in the filtered array and employed for the RIF analysis. The list includes sequence-specific DNA-binding transcription factors, other transcription factors and cofactors. Also MSTN is included,despite not being a TF, because of its known involvement in muscle growth regulation. Gene ontology and Genomatix annotation information is provided. (XLSX 469 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Óvilo, C., Benítez, R., Fernández, A. et al. Longissimus dorsi transcriptome analysis of purebred and crossbred Iberian pigs differing in muscle characteristics. BMC Genomics 15, 413 (2014). https://doi.org/10.1186/1471-2164-15-413
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-413