
Abstract
Background
Overexpression of the Cut homeobox 1 gene, CUX1, inverselycorrelates with patient survival in breast cancers. Cell-based assays andmolecular studies have revealed that transcriptional regulation byCUX1 involves mostly the proteolytically processed p110isoform. As there is no antibody specific to p110 CUX1 only, an alternatestrategy must be employed to identify its targets.
Results
We expressed physiological levels of a tagged-p110 CUX1 protein and performedchromatin affinity purification followed by hybridization on ENCODE andpromoter arrays. Targets were validated by chromatin immunoprecipitation andtranscriptional regulation by CUX1 was analyzed in expression profiling andRT-qPCR assays following CUX1 knockdown or p110 CUX1 overexpression.Approximately 47% and 14% of CUX1 binding sites were respectively mappedless than 4 Kbp, or more than 40 Kbp, away from a transcription start site.More genes exhibited changes in expression following CUX1 knockdown thanp110 CUX1 overexpression. CUX1 directly activated or repressed 7.4% and 8.4%of putative targets identified on the ENCODE and promoter arraysrespectively. This proportion increased to 11.2% for targets with 2 bindingsites or more. Transcriptional repression was observed in a slightly higherproportion of target genes. The CUX1 consensus binding motif, ATCRAT, wasfound at 47.2% of the CUX1 binding sites, yet only 8.3% of the CUX1consensus motifs present on the array were bound in vivo. Thepresence of a consensus binding motif did not have an impact on whether atarget gene was repressed or activated. Interestingly, the distance betweena binding site and a transcription start site did not significantly reducedthe ability of CUX1 to regulate a target gene. Moreover, CUX1 not only wasable to regulate the next adjacent gene, but also regulated the gene locatedbeyond this one as well as the gene located further away in the oppositedirection.
Conclusion
Our results demonstrate that p110 CUX1 can activate or repress transcriptionwhen bound at a distance and can regulate more than one gene on certaingenomic loci.
Similar content being viewed by others


Background
The last decade has seen significant advances in the field of transcription. Thediscovery of nuclear histone acetyltransferases (HATs) in the mid nineties hasliterally opened a new field of investigation into post-translational modificationsthat target histones and modulate the chromatin state either locally or over largegenomic loci. The multiple types of modifications that take place on specifichistone residues and the regulatory cascades that can be triggered in this mannerled investigators to propose that a "histone code" regulates gene expression in amanner reminiscent of the genetic code translating nucleic acid coding sequencesinto protein sequences [1, 2]. In parallel, a number of novel experimental approaches have contributedto move the transcription field from a gene-by-gene approach focused on corepromoters to a genome-wide non-biased approach that enables us to study largenumbers of transcriptional targets as well as the mechanisms by which these targetsare regulated [3]. Recent tools in our arsenal include the increasing availability ofgenomic microarrays [4], siRNA-mediated gene knockdown [5], more efficient virus-based gene delivery systems [6, 7], and high-throughput sequencing [8]. Importantly, the "rediscovery" of chromatin immunoprecipitation combinedwith the development of microchip arrays containing large numbers of genomicsequences has opened new horizons. Indeed, chromatin immunoprecipitation was firstdescribed in the mid eighties by the group of John T. Lis who used this assay toshow that RNA polymerase II molecules were already present at the 5' end of thehsp70 gene in uninduced cells and that heat shock somehow enabled transcriptionelongation to take place [9]. Curiously, the method was not applied to specific transcription factorsbefore another decade [10]. Interestingly, the genomic microarray that was designed as part of theENCODE project provided a sampling of the human genome that can be interrogated todefine the distribution types of transcriptional regulation of specifictranscription factors [11]. The information thus gathered has forced us to reconsider our originalunderstanding of basic mechanisms of transcriptional regulation [12]. For example, a common belief was that a specific transcription factorcould bind to a few dozen genes whose core promoters contain its consensus bindingsite as defined in vitro, and once recruited to a promoter could almostsingle-handedly regulate transcription [13]. We now know that c-MYC binds to approximately 20% of gene promoters andis also capable of regulating genes at a distance [14–16]. Another major conceptual advance concerns the criteria to define atranscriptional target. Experimental evidence typically included the presence of aconsensus binding motif within a core promoter, in vitro binding assays andluciferase reporter assays. While these assays are still employed, it is clear thatthey cannot provide definitive evidence that a transcription factor regulates aspecific gene. Additional evidence must also include chromatin immunoprecipitationassays to demonstrate "in vivo" DNA binding, and change in expression of theendogenous gene target in response to the knockdown and/or overexpression of thetranscription factor.
Cut homeobox 1 (CUX1) has previously been called CCAAT-displacement protein(CDP), CDP/Cut and Cut-like 1 (CUTL1). CUX1 encodes two mainisoforms that exhibit different DNA binding and transcriptional properties (reviewedin [17]). The full-length protein, p200 CUX1, is a very abundant protein thatbinds DNA with extremely fast kinetics [18]. In mid-G1 phase, 1% to 10% of p200 CUX1 is proteolytically processed bya nuclear isoform of cathepsin L to produce the p110 CUX1 isoform [19, 20]. This shorter isoform can stably interact with DNA and, depending onpromoter-context, can function as transcriptional repressor or activator [21, 22]. The expression and activity of p110 CUX1 are tightly regulated in a cellcycle-dependent manner, mostly through phosphorylation-dephosphorylation by cyclinA/Cdk2, cyclin A/Cdk1 cyclin B/Cdk1, and Cdc25A, as well as proteolytic processingby nuclear cathepsin L and a caspase-like protease [19, 20, 23–27]. These post-translational modifications circumscribe the transcriptionalactivity of p110 CUX1 to the period between mid-G1 to sometimes in G2. In contrastto p110 CUX1, the DNA binding activity of p200 CUX1 is constant throughout the cellcycle [19]. Its transcriptional activity, if any, would be limited to the"CAATT-displacement activity", a mechanism of passive repression involvingcompetition for binding site occupancy [18].
Homozygous inactivation of Cux1 in mice causes perinatal lethality in alarge proportion of animals due to delayed lung development and associatedrespiratory failure [28]. Surviving mice are usually male and exhibit growth retardation,disrupted hair follicle morphogenesis, purulent rhinitis, infertility, cachexia, andreduction of B and T cell content in bone marrow and thymus, respectively [28–30]. In transgenic mouse models, overexpression of CUX1 generated variouscancer-associated disorders depending on the specific isoform and tissue typeexpression. These include multi-organ organomegaly, glomerulosclerosis andpolycystic kidneys, pre-cancerous lesions in the liver,myeloproliferative-disease-like myeloid leukemias and mammary tumors sometimesassociated with lung metastasis [31–36]. Cell-based assays demonstrated a role for CUX1 in cell cycle progressionand cell proliferation [27, 37], strengthening of the spindle assembly checkpoint [38], cell migration and invasion [22, 39–41], resistance to apoptotic signals [42], and dendrite branching and spine development in cortical neurons [43]. Which CUX1 isoform(s) is active in these processes cannot be determinedfrom siRNA or shRNA-mediated knockdown approaches, however, in overexpressionstudies the p110 CUX1 isoform was shown to regulate transcription of genes involvedin cell cycle progression, DNA damage response, spindle assembly checkpoint and cellmotility.
Many specific transcription factors are able bind to genomic sites that are far awayfrom TSS. These studies also revealed that only about up to 10% of putativetranscriptional targets showed evidence of regulation in response to changes intranscription factor concentrations [44–46]. Whether CUX1 binds preferentially to core promoter sequences, like E2F1,or whether it can also bind at a distance from TSS, like c-Myc, has not beendetermined [14, 15]. Also, what proportion of all CUX1 targets is regulated in response tooverexpression or silencing of CUX1 is not known. To begin to address thesequestions, we have performed ChAP-chip using ENCODE and promoter microarrays.Putative targets were validated in independent ChIP followed by q-PCR, whileregulatory effects were measured in expression profiling experiments and confirmedby RT-qPCR. The results show that CUX1 binds to a large number of genomic sites thatare located far away from a TSS and can regulate genes at a distance even whenanother gene is located in the intervening region.
Results
Strategy to identify p110 CUX1 binding sites
The overall goal of the present study was to define the modes of transcriptionalregulation by CUX1 and, in particular, determine whether CUX1 can regulate genesat a distance. As detailed in the introduction, previous transcriptional studiesand cell-based assays have implicated the p110 CUX1 isoform in transcriptionalactivation and repression of target genes. Since p110 CUX1 is generated byproteolytic processing, its primary sequence is included in the full-length CUX1protein sequence. Consequently, all available antibodies that bind to p110 CUX1also recognize p200 CUX1. Our strategy to identify in vivo binding sites forp110 CUX1 was to isolate chromatin by two different methods. First, we purifiedchromatin by tandem affinity purification (TAP) using a population of Hs578tcells stably expressing moderate levels of a p110 CUX1 protein with two epitopetags at its C-terminus, p110-Tag2 (Figure 1A and B). Chromatin isolated in this manner as well as totalchromatin (input) were used in hybridizations on the NimbleGen HG17 ENCODE highdensity oligonucleotide tiling array. Secondly, binding sites identified in themicroarray were then validated by performing independent ChIP in the parentalHs578t cells using CUX1 antibodies, 861 and 1300 (Figure 1A). Importantly, these cells express endogenous CUX1 proteins only.The strategy of chromatin affinity purification (ChAP) followed by microarrayanalysis (ChAP-chip) has previously been validated [47], and described in detail [48].

Expression of CUX1 Recombinant Proteins. (A) Schematicrepresentation of CUX1 proteins with some of the functional domains: ID,inhibitory domain; CC, coiled-coil; CR1, CR2 and CR3, Cut repeat 1, 2and 3; HD homeodomain; CBD, calmodulin binding domain; Prot A, proteinA. The regions recognized by the 861 and 1300 antibodies are shown.(B) Hs578t cells were infected with a retroviral vector toestablish a population of cells stably expressing a recombinant p110CUX1 protein with two tags at its C-terminus, p110 CUX1-Tag2.A population stably carrying the empty vector was used as a control.Nuclear extracts were prepared from each population of cells andanalyzed by Western blot using the 861 and 1300 CUX1 antibodies.
Distribution of CUX1 binding sites on the ENCODE array
Using a stringent false discovery rate (FDR = 0.05), 513 CUX1 bindingsites were identified on the ENCODE array (Table 1).The recruitment of CUX1 to 23 out of 25 genomic sites (92%) was validated inquantitative-PCR assays using chromatin that was independently obtained fromHs578t cells by immunoprecipitation with CUX1 antibodies (Table 1). 79.6% of probes on the ENCODE array derive fromtranscribed genomic regions. 70.9% of CUX1 binding sites were located withintranscribed regions, indicating a 1.6-fold enrichment in non-transcribedregions. In comparison, data obtained from ChIP on the ENCODE platform [14] for c-MYC reveals a 1.56 fold enrichment in non-transcribed regionswhile E2F1 showed a strong enrichment for transcribed regions (Table 2).
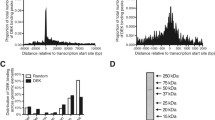
Mapping of CUX1 binding sites relative to transcription start sites (TSS)generated a bell-shaped curve of low height around TSS (Figure 2A). 14.2% of all binding sites overlapped a TSS, and anadditional 17% and 16% of binding sites were respectively located in the 4 Kbpregion upstream and downstream of a TSS. The number of binding sites graduallydeclined with increasing distance. Yet, over 6% and 8% of binding sites weresituated at more than 40,000 bp upstream or downstream, respectively, fromthe closest TSS. 53% of CUX1 binding sites are located more than 4,000 bpaway from a TSS and approximately 14% of all CUX1 binding sites are situated atmore than 40,000 bp from a TSS.

Distribution of CUX1, C-Myc and E2F1 Binding Sites Relative toTranscription Start Sites. (A) Percentage of CUX1 bindingsites located at various distances from the closest transcription startsite. The "0" column indicates genes where the CUX1 binding siteoverlaps the start site. (B) Location of C-Myc binding sites asper A. (C) Location of E2F1 binding sites as perA.
We compared the distribution of CUX1 binding sites with those of 3 randomlygenerated sets of binding sites, as well as those of c-Myc and E2F1 using thedata of Bieda et al., 2006 [14] (Figure 2B and C). We note that thedistributions of randomly generated sets of binding sites exhibited flatterbell-shaped curves around TSS (Additional file 1:Figure S1). We conclude that the higher frequency of CUX1 binding sites close toTSS reflects the preferential recruitment of CUX1 to promoter regions. The samecannot be said regarding the binding sites that are located at more than 40 Kbpfrom TSS, since the same proportions of randomly generated binding sites werelocated in these regions.
In contrast to CUX1 and c-Myc, the E2F1 transcription factor was found to bindalmost exclusively to the region immediately adjacent to TSS. The preference ofE2F1 to core promoter regions led the authors to posit that E2F1 is recruitedvia protein interactions with components of the general transcription machinery [14]. The wider distribution of binding sites observed for CUX1 and c-Mycis also observed for other transcription factors [15, 49, 50] (Additional file 2: Figure S2A-C), whileother factors show a preference for TSS similarly to E2F1 (Additional file2: Figure S2D-E). Yet other factors show differentpatterns of binding, such as Pax8, which exhibits preference for non-promoterCpG islands and a tendency to bind in the 10–100 Kbp range rather thanclose to the TSS of genes [51].
Binding of CUX1 to distant regulatory elements
We compared the location of CUX1 binding sites that are more than 4 Kb from thenearest TSS to DNAse hypersensitivity mappings and ChromHMM data in humanmammary epithelial cells from published datasets. DNAse hypersensitivity siteshave been used as markers of regulatory DNA elements such as enhancers,silencers, insulators and locus control regions [52–55]. ChromHMM is a computational method that compiles data from histonemodification mappings and integrates them to predict genomic elements such asenhancers [56]. This analysis revealed that respectively 19.2% and 22.1% ofdistantly located CUX1 binding sites are present within 1 kb of a DNAsehypersensitivity site and of an enhancer predicted (Table 3). Both of these proportions are greater than what is seen forrandomly distributed binding sites. However, there was no enrichment of CUX1binding sites in proximity of insulator elements (Table 3). These results are in agreement with the notion that CUX1 canperform some regulatory functions when binding at a distance from transcriptionstart sites.
Detection of CUX1 binding sites and consensus binding motif on promoterarrays
Promoter microarrays are useful because they enable one to interrogate easilyover 30,000 gene promoters. A limitation is that only a limited amount ofpromoter sequences can be included for each gene, precluding the detection offar away binding sites that could play a role in transcriptional regulation.Based on the localization of CUX1 binding sites on the ENCODE array, wecalculated that between 17.2% to 26.6% of CUX1 binding sites would be identifiedon commercially available promoter arrays (Table 4).However, since for many distant CUX1 binding sites another binding site is alsopresent close to the transcription start site, we estimated that between 44.6%to 58.5% of gene targets would be identified on distinct promoter arrays(Table 5). In contrast, as E2F1 is targeted totranscription start sites, between 80.4% to 85.8% of E2F1 binding sites would beexpected to be identified on a promoter array.
We verified these predictions by performing a ChAP-chip experiment using theNimblegen promoter microarray. Total chromatin (input) as well as purifiedchromatin from Hs578t cells expressing p110 CUX1-Tag2 were used in hybridizationon the promoter array of NimbleGen. Using a stringent false discovery rate(FDR = 0.05), 5828 CUX1 binding sites were identified on 4706 genepromoters (Table 6). The recruitment of CUX1 to 25out of 25 genomic sites (100%) was validated in quantitative-PCR assays usingchromatin that was independently obtained from Hs578t cells byimmunoprecipitation with CUX1 antibodies (Table 6).The vast majority of target genes (83.7%) contained only one CUX1 binding site,yet a sizable fraction contained 2 or more binding sites (Table 6).
According to the predictions shown in Table 5, 44.6%of CUX1 target genes should be identified on the promoter array from Nimblegen.We calculated the proportion of ENCODE genes with a CUX1 binding site that werealso identified as putative targets of CUX1 in the promoter array. When weconsidered all 513 CUX1 binding sites and 445 adjacent ENCODE genes, we foundthat 92 genes (21%) were identified in the promoter array (Table 7, third column). When we considered only the 85 ENCODEgenes that were regulated in response to changes in CUX1 levels (see below), wefound that 27 genes (32%) were identified as putative target of CUX1 in thepromoter array (Table 7, third column).
The CUX1 consensus binding site, ATCRAT (where R = C or A), was foundto be present at 47.2% of the 5828 bound genomic sites (Table 8). This frequency was judged to be significant as the CUX1consensus binding site was found to be present in only 17.5% of 5828 randomlychosen regions of equal size. Notably, the GC content between bound and unboundregions is practically identical, and thus cannot account for the difference inbinding site occurrence (Table 8). Yet, only 8.3%(3633/43778) of the CUX1 consensus sites present on the array were bound invivo. We conclude that the CUX1 consensus binding site plays a role inthe recruitment of CUX1 at specific genomic locations, but the presence of aconsensus site is not sufficient.
Identification of binding motifs in genomic regions bound by CUX1
We envisioned that interactions with other transcription factors play animportant role in recruiting CUX1 to specific locations. In agreement with thisnotion, functional analysis revealed distinct sets of cellular functions amonggene targets that contain an ATCRAT consensus and those that do not(Tables 9 and 10). Tofurther test the possibility that CUX1 may interact with other factors, weinvestigated the presence of binding motifs other than that of CUX1 using theMEME suite of analysis tools (meme.nbcr.net/). We first tested the reliabilityof the tool by using it to find motifs in the sequences of CUX1 BS in which wehad independently determined that they contained the established ATCRATconsensus. As expected, it identified the ATCRAT consensus as the most enrichedmotif in the set of sequences, by a vast margin (Table 11, entry 1). We then analyzed binding motifs in the two sets ofCUX1 binding sites: those that contained the ATCRAT motif and those that didnot. While the size of bound regions varied from 149 to 1107 bp, theaverage size was 532 and 477 bp, respectively. Interestingly, only onecommon binding motif was found in the two sets, while the rest of the bindingmotifs were unique to each set (Tables 11 and 12). Extending the search to the 500 bp regions oneither side of bound regions did not highlight other differences between the twosets or reveal additional contributing factors (Tables 13 and 14). These findings support the notionthat targeting of CUX1 to specific genomic sites is influenced byprotein-protein interactions with other DNA binding proteins.
Regulatory effects of CUX1 on putative targets
To verify the effect of CUX1 on putative targets, we performed expressionprofiling on three Hs578t cell populations: cells that had been infected with aretrovirus expressing an shRNA against CUX1, cells infected with a retrovirusexpressing p110 CUX1, or cells infected with an empty retrovirus. In each case,replicate microarray hybridizations were carried out such that a p value couldbe calculated for each difference in gene expression. Results from expressionprofiling were validated by repeating the infections and performing RT-qPCRanalysis on 20 genes whose expression went up or down in response to onetreatment or the other (Figure 3). All genes testedin this manner displayed changes in gene expression in the same direction asthat observed in the microarray hybridization: genes that were repressed inexpression profiling were also repressed when mRNA levels were measured byRT-qPCR. Similar observations were made for genes that were activated. We note,however, that the fold activation or repression calculated by RT-qPCR were notnecessarily proportional to the changes observed in microarray hybridization.For example, EEF1A1 and C20ORF44 mRNA were increased respectively 7.3 and 1.8fold when measured by RT-qPCR, but were increased 1.7 and 1.4 fold in microarrayanalyses. Some of these differences could be due to the fact that measurementsby the two methods were made with RNA prepared from independent experiments.Notwithstanding the differences in magnitude, the effects of CUX1 on geneexpression was confirmed for all tested genes.

Overexpression and Knockdown of CUX1 and Expression ProfilingValidation. (A) Hs578t cells were infected with alentiviral vector expressing p110 CUX1 or nothing (vector). RNA andproteins were purified 48 hours post-infection. CUX1 expression wasanalyzed by RT-qPCR and immunoblotting. (B) Hs578t cells wereinfected with a lentiviral vector expressing CUX1 shRNA or a scrambledRNA. RNA and proteins were purified 5 days afterinfection. CUX1 expression was analyzed by and RT-qPCR andimmunoblotting. (C) RNA levels of the indicated genes weremeasured by RT-qPCR in cells treated as in A. Expected up or downindicates regulation that was observed by expression profiling.(D) RNA levels of the indicated genes were measured byRT-qPCR in cells treated as in B. Expected up or down indicatesregulation that was observed by expression profiling. * p<0.05, ***p<0.001 on a Student's T test.
A total of 445 genes are present on the ENCODE array, and all have a CUX1 bindingsite located within 213 Kbp of their TSS. Expression profiling results could bematched for 327 of these genes. Using a cut-off of 50% either up ordown-regulated and a p value below 0.05, we observed differences in theexpression of 26 target genes (7.4%), following changes in CUX1 levels(Table 15). 20 genes responded to CUX1knockdown, and 6 genes, to p110 CUX1 overexpression (Table 15). Among the 26 regulated target genes, 10 genes (38%) wereactivated and 16 genes (62%) were repressed by CUX1 (Table 15). Similar proportions of activated and repressed genes were foundwhen a cut-off of 25% change in gene expression was employed (Table 16). These findings confirm that p110 CUX1 can participatein transcriptional activation or repression depending on promoter context.
Similar results were obtained when we analyzed the expression of putative targetsidentified on the promoter array. A total of 347 genes, 8.4% of all putativetargets for which expression profiling results could be matched, were regulatedby CUX1. 287 and 85 genes exhibited regulation in response to CUX1 knockdown orp110 CUX1 overexpression, respectively. 181 (52%) were up-regulated by CUX1while 167 (48%) were down-regulated by CUX1.
Effect of distance on transcriptional regulation by CUX1
We noted that CUX1 regulated 7.4% and 8.4% of putative targets from the ENCODEand the promoter arrays, respectively. We next investigated the relationshipbetween the position of a CUX1 binding site relative to a transcription startsite and the probability of a gene to be regulated in response to changes inCUX1 levels. When genes were classified according to the distance between theCUX1 binding site and the transcription start site, we did not observesignificant difference in the fraction of targets that were regulated by CUX1(Figure 4A and B). However, we observed muchvariability in the fraction of regulated genes because the number of geneswithin some distance intervals were very small. Therefore, to increase thesample size, we repeated the analysis this time using a cut-off of 25% either upor down and a p value below 0.05 (Figure 4C and D).We observed differences in the expression of 62 and 36 genes in response to CUX1shRNA and CUX1 overexpression, respectively (Table 16). Again, more genes were found to be regulated by CUX1 using theshRNA approach. Among genes that exhibited regulation by CUX1, 35 genes (41%)were activated by CUX1, and 50 genes (59%) were repressed by CUX1(Table 16). The histogram presenting thepercentage of regulated genes versus the distance of CUX1 binding sites to TSSshows that essentially the same proportion of genes are regulated whether CUX1binds close or far away from the TSS (Figure 4C).Indeed, no statistical difference was observed between genes bound at the TSSand those bound more than 40 Kbp away. We conclude that CUX1 can activate orrepress transcription when bound at a distance from a transcription startsite.

Effect of Distance on Regulation by CUX1. (A) Genes fromthe ENCODE array have been organized according to the distance betweentheir transcription start site (TSS) and the closest CUX1 binding site.The "0" column indicates genes where the CUX1 binding site overlaps thestart site. The histogram shows, for each interval of distance, thepercentage of genes that exhibit a 1.5 fold change in expressionfollowing p110 CUX1 overexpression or CUX1 knockdown. The total numberof genes within each interval is indicated within each column.(B) As in A, except that the regulation by CUX1 isexpressed as either activation by CUX1 or repression by CUX1. (C)As in A, but with a threshold of 1.25 fold change in expression.(D) As in B, but with a threshold of 1.25 fold changein expression.
Effect of multiple CUX1 sites
The presence of multiple CUX1 binding sites has a modest, yet significant, impacton the probability that a gene is regulated by CUX1. CUX1 regulated 7.9%, 11.2%of genes that contain respectively one or two CUX1 binding sites, respectively(Table 17).
Effect of gene position on transcriptional regulation by CUX1
Intuitively, one would assume that a transcription factor is more likely toregulate the closest promoter. Yet, some enhancers will exhibit an effect on apromoter situated on one side, but no effect on the promoter that is on theother side on the map. This sort of selectivity between an enhancer and apromoter has been explained by the presence of boundary or insulator elements orby specific interactions between proteins bound at the enhancer and theregulated promoter. Previous studies on CUX1 have all focused on genes thatcontain a CUX1 binding site within the immediate promoter. To begin toinvestigate the rules that govern the action of CUX1, we calculated the fractionof different types of CUX1 targets that were regulated in response to changes inCUX1 levels. Three types of genes were analyzed: 1, genes that are the closestto the CUX1 binding site; 2, genes that are further away and in the otherdirection from the CUX1 binding site; 3, genes that are located further away andare separated by another gene from the CUX1 binding site. For each category, wecalculated the percentage of genes that exhibit a 1.25 or 1.5-fold change inexpression following p110 CUX1 overexpression or CUX1 knockdown. Strikingly,essentially similar fractions of genes were regulated whether they were closestto the CUX1 binding site or were located further away in the other direction(Figure 5, compare 1 and 2). Moreover, theproportion of regulated genes was not significantly lower among genes thatbelong to the third category (Figure 5, type 3genes). We conclude that CUX1 is capable of regulating genes at a distance.Moreover, CUX1 can regulate more than one gene on certain genomic loci.

Relationship Between Gene Position and Regulation by CUX1. Threetypes of situations are depicted in the diagram. 1, genes that are theclosest to the CUX1 binding site; 2, genes that are further away and inthe other direction from the CUX1 binding site; 3, genes that arelocated further away and are separated by another gene from the CUX1binding site. For each category, the table shows the percentage of genesthat exhibit a 1.25 or 1.5 change in expression following p110 CUX1overexpression or CUX1 knockdown.
Discussion
Genome-wide location analysis on the ENCODE array revealed that ~47% of CUX1 bindingsites are located in the 4-Kbp region upstream and downstream of a TSS, while morethan 14% of CUX1 binding sites are situated at more than 40 Kbp from a TSS(Figure 2). Overall, 7.4% and 8.4% of putativetargets on the ENCODE and promoter arrays respectively, exhibited a 1.5-fold changein expression following CUX1 knockdown or p110 CUX1 overexpression(Tables 15, 16, 17, 18 and 19).This proportion is within the 1-10% range of potential targets that have beenreported to be regulated by other transcription factors [44–46].
Importantly, analysis of the percentage of regulated genes versus the distance ofCUX1 binding sites to TSS showed that essentially the same proportion of genes areregulated whether CUX1 binds close or far away from the TSS (Figure 4A and B). In other words, the probability that a gene isregulated by CUX1 is not affected by the distance between the CUX1 binding site andthe TSS. In addition, our results indicate that the position of genes relative to aCUX1 binding site do not determine whether these genes are regulated by CUX1. CUX1regulated similar percentages of genes whether they were closest to the CUX1 bindingsite or were located further away in the other direction (Figure 5, compare 1 and 2). Moreover, CUX1 regulated a surprisingly highproportion (5.4%) of genes that were separated from their binding site by anothergene (Figure 5). Altogether these results demonstratethat CUX1 can regulate genes at a distance and can regulate more than one gene oncertain genomic loci.
The proportion of target genes that were found to be activated or repressed by CUX1,respectively 52% and 48% (Tables 18), is significantlydifferent from what we reported in previous studies on target genes involved in cellcycle progression, cell motility, or the DNA damage response [21, 22, 57]. In each case, a vast majority of genes were found to be activated byp110 CUX1, whether we performed siRNA-mediated knockdown or overexpression of p110CUX1. One factor that may explain this could be the functional classes of genes thatwere studied previously. The functional class of “cell cycle” genesincludes mostly genes that stimulate cell cycle progression. Out of 25 cell cyclegene targets identified by ChIP-chip, 22 were activated and 2 were repressed by CUX1(while only one was not affected) [21]. One of the two repressed genes, p21WAF1/CKI1, code for aCDK-inhibitor that blocks cell cycle progression, while the other, CCNH, is involvedin transcription and DNA repair. All target genes that were activated play apositive role in cell cycle progression. Similarly, among 19 targets that play arole in DNA damage response, 18 were activated and one was repressed [57]. The repressed gene again was p21WAF1/CKI1. Overall, theseresults are consistent with the notion that CUX1 establishes a transcriptionalprogram that promotes cell cycle progression and at the same time ensures themaintenance of genetic integrity.
We employed two experimental approaches to examine the transcriptional regulation ofgenes by CUX1. Expression profiling was performed following shRNA-mediated knockdownof CUX1 or p110 CUX1 overexpression. Among targets identified on the promoter array,287 genes exhibited a 1.5-fold change in expression following CUX1 knockdown, while85 genes were regulated in response to p110 CUX1 overexpression. Therefore, moregenes were found to be regulated by CUX1 using the shRNA approach. This result canbe interpreted to mean that CUX1 is required for optimal expression of many targetgenes, however, increasing CUX1 expression is not sufficient to modulate theexpression of some target genes.
The CUX1 consensus binding site, ATCRAT (where R = C or A), was found tobe present at 47.2% of the 5828 bound genomic sites (Table 8). We conclude that the presence of a CUX1 consensus binding sitecontributes to, but is not sufficient for, the recruitment of CUX1 to specificgenomic locations. We envision that interactions with other transcription factorsplay an important role in recruiting CUX1 to specific locations. In agreement withthis notion, functional analysis revealed distinct sets of cellular functions amonggene targets that contain an ATCRAT consensus and those that do not(Tables 9 and 10). We notethat functional classes involved in cell cycle were over-represented among targetgenes that do not contain a consensus CUX1 binding site (Table 10). In previous studies, CUX1 was shown to interact with E2F factorsand cooperate with these factors in the regulation of several cell cycle genes [58, 59]. It is likely that protein-protein interaction with E2F factors reducesthe requirement for the presence of a high-affinity binding site for the recruitmentof CUX1 on this class of genes.
CUX1 can be purified efficiently by immunoprecipitation or affinity chromatography.Following cross-linking, however, the yield of purification is drastically reducedsuch that we need 500 million cells to perform chromatin immunoprecipitation oraffinity purification (ChIP or ChAP) for CUX1. This caveat has limited our abilityto perform ChIP-sequencing and therefore our study relied on microarrayhybridizations. While sequence coverage is admittedly smaller on microarrays, datacollected from both ENCODE and promoter arrays have enabled us to define theimportance of the CUX1 consensus binding site in the recruitment of CUX1 to genomiclocations and determine whether CUX1 can regulate genes at a distance.
Conclusions
Our results demonstrate that p110 CUX1 can mediate transcriptional repression oractivation of specific genes when bound at variable distances from the transcriptionstart site. Although the CUX1 consensus binding motif, ATCRAT, plays a role in therecruitment of CUX1 to specific genomic sites, protein-protein interactions mustcontribute to its transcriptional activity.
Methods
Cell culture
Hs578T is a human mammary carcinoma cell line [60]. Previous studies have documented changes in gene expression inresponse both to CUX1 knockdown and overexpression [21, 33]. Hs578T cells were maintained in Dulbecco’s modified minimumessential medium (DMEM)(Wisent) supplemented with penicillin-streptomycin, and5% fetal bovine serum (FBS) (Gibco).
Retroviral infection and stable cell lines
Retroviruses were produced by transfecting 293VSV cells with the pREV/TRE vectoreither empty or encoding p110 CUX1-Tag2 (CUX1 a.a. 612–1336with protein A and CBP tags inserted at the C-terminus) (Clontech). Preparationof the retroviruses and stable cell lines was done as previously described [37].
Chromatin Affinity Purification (ChAP)
The method of chromatin affinity purification (ChAP) has previously beenvalidated [47], and described in detail [48]. To ensure that the recombinant p110-Tag2 protein would beexpressed at moderate level, we employed the pRevTRE retroviral vector(Clontech), which contains the minimal CMV promoter with a tetracyclineresponsive element. Importantly, no tetracycline was added to the medium.Moreover, the Hs578T breast tumor cells do not express a tetracycline-responsivetransactivator. Basal expression from the pRevTRE vector was previously shown tobe very low [61–63]. ChAP was performed on 5 x108 Hs578T. The cell nuclei werepurified as described in [64], then lysed in RIPA-M buffer (10 mM Tris–HCl pH8,1 mM EDTA, 0.5 mM EGTA, 150 mM NaCl, 1% Triton X-100, 0.5% DOC,0.1% SDS, 1 mM PMSF, protease inhibitors) and sonicated on ice to obtain250- to 800-bp-long DNA fragments. Stably expressed recombinantp110-Tag2 protein was purified by the Taptag purification methodwith some modifications [65]. The IgG matrix bound p110-Tag2/DNA were washed in washbuffer I (20 mM Tris–HCl pH8, 2 mM EDTA, 2 mM EGTA,150 mM NaCl, 1% NP-40, 0.5% DOC, 0.2% SDS), wash buffer II (20 mMTris–HCl pH9, 2 mM EDTA, 2 mM EGTA, 500 mM NaCl, 1% NP-40,0.5% DOC, 0.1% SDS), wash buffer III (50 mM Tris–HCl pH7.5, 2 mMEDTA, 1 mM EGTA, 0.5 M LiCl, 1% NP-40, 0.7% DOC,) and then TEV buffer(10 mM Tris–HCl (pH8.0), 100mMNaCl, 0.1% TX-100, 0.5 mM EDTA,10% glycerol, 1 mM DTT). After TEV protease digestion, the releasedprotein/DNA complexes were purified by affinity chromatography on calmodulinbeads in the presence of calcium and then eluted with EGTA. Afterde-crosslinking, samples were treated with RNase A and Proteinase K. Un-enrichedinput chromatin was put aside as a control.
Preparation of ChAP purified DNA for hybridization
ChAP purified chromatin was amplified by the method of Ligation-Mediated PCR asdetailed previously [66]. Briefly, ChAPed DNAs and input DNA were blunted, ligated to aunidirectional linker and amplified by PCR for 24 cycles to generate asufficient amount of DNA. Amplified DNA samples were Cy5 labeled and amplifiedinput controls were Cy3 labeled using Nimblegen's Dual-Color DNA Labeling Kitaccording to the manufacturer's instructions.
DNA microarray hybridization
Labeled samples were hybridized to either NimbleGen's HG17 ENCODE or their HG18Human Promoter Array Set high density oligonucleotide tiling array (385 kprobe format) and then washed according to the manufacturer's instructions.Arrays were scanned on an Agilent 5 μm scanner model G2505B usingcustomized scan area settings (X: 28, Y:6, Width: 20, Height: 14, values inmm).
ChAP-microarray result analysis
For both array platforms (Encode and promoter array), grid alignment, raw signalextraction, peak identification and peak mapping were carried out using theNimblescan v8.0 software according to the company's instructions. Identifiedpeaks were considered significant with a false discovery rate (FDR) below 0.05,which is considered highly confident. Further analysis of identified bindingsites was carried our using either the R platform for statistical computing(http://www.R-project.org) or scripts written in PERL (PracticalExtraction and Report Language, http://www.perl.org). All peaksidentified in ChAP-Chip experiments on the Encode Array and the Human PromoterArray Set are provided (Additional files 3 and 4, respectively).
ChAP-microarray result validation
Independent ChIP experiments using antibodies specific for endogenous CUX1 werecarried out in Hs578t, as previously described [21]. Real-time PCR was used to measure the level of enrichment of genomictarget regions in ChIP DNA vs. the un-enriched input DNA. We selected 25 genesfrom both the targets identified on the ENCODE array and on the Promoter arrayset and designed primers specific for the corresponding regions where CUX1 wasputatively identified as binding.
ENCODE binding sites for c-MYC and E2F1
We used ChIP-chip binding sites for E2F1 and c-Myc downloaded from the website ofDr. Peggy Farnham laboratory at<http://genomics.ucdavis.edu/farnham/suppdata.html>. Thisdataset contains the binding sites predicted for E2F1, c-MYC and POLR2A (RNApolymerase II) in the ENCODE regions classified by 4 criteria: L1(P < 0.0001 and 98th percentile), L2(P < 0.0001 and 95th percentile), L3(P < 0.05 and 98th percentile) and L4(P < 0.05 and 95th percentile) [14]. Based on the validation of 29 binding sites, Bieda et al. concludethat L1 binding sites are highly reliable, L2 and L3 binding sites are alsoreliable however based on sparser testing and L4 binding sites are usuallyartifacts. Binding sites identified with the L1 criteria were used for ouranalyses. The chromosomal intervals for binding sites predicted for E2F1 andPOLR2A belonged to genomic coordinates using hg16, whereas c-MYC binding siteswere in hg17. Therefore, the lift-over program found on the online GALAXYplatform [67–69] was used and random results were verified using UCSC genome browserto convert hg16 coordinates to those of hg17. There were 1 and 2 binding sitesfor E2F1 at L1 and L3 respectively (hg16), which could not be mapped tohg17.
DHS and ChromHMM data analysis
Data tracks were downloaded from the UCSC's Encode data portal(http://genome.ucsc.edu/ENCODE/). Genomic locations were comparedto those of the CUX1 binding sites using scripts written in R. UCSC Accessionnumbers of the tracks used are: wgEncodeEH000503 (GEO accessions GSM736552 andGSM736634) for the DHS data and wgEncodeEH000786 for the ChromHMM data.
Consensus sequence analysis
Genomic sequences corresponding to regions of interest (binding sites or other)were obtained using the online GALAXY platform. Scripts written in R were usedto identify the ATCRAT consensus motif within regions of interest.
De novo binding motif identification
De Novo motif discovery was performed using the DREME (Discriminative DNA MotifDiscovery) motif discovery tools form the MEME suite of tools. Comparison withknown DNA binding motifs was performed using the TOMTOM algorithm using theJASPAR CORE database as a reference for comparison. (meme.nbcr.net/) [70–72].
Functional overrepresentation analysis
Identification of overrepresented gene functions was carried out using the onlineannotation tool DAVID. Genes that were bound by CUX1 (Targets) were comparedwith all genes present on the microarray (Background). Overrepresentation of afunction depends on the increase in the proportion of genes involved in a givenfunction between CUX1 targets and the background. The P-value is determinedusing an improved Fisher’s exact test from the DAVID software [73, 74].
p110 CUX1 overexpression and CUX1 shRNA
For overexpression, Hs578t cells were infected with a lentiviral vectorexpressing p110 CUX1. Duplicate infections were carried out in parallel andcells were harvested after 24 hours. For CUX1 knockdown, a stable Hs578tcell line containing a doxycycline inducible shRNA was established by retroviralinfection. Doxycycline was applied to the cells for 6 days before harvestwith control cells left untreated. Knockdown experiments were carried out inbiological duplicates in parallel.
Expression profiling sample preparation and hybridization
Total RNA was isolated from cells using the Arcturus Picopure RNA isolation kit.2 μg of RNA was then amplified using the Arcturus RiboAmp PLUS RNAamplification kit according to the manufacturer's instructions for a singleround of amplification. Amplified mRNA (aRNA) was labeled using the ArcturusTurbo Labelling Cy5 and Cy3 kits using the manufacturer's instructions with amodification: The labeling reaction was carried out using 5 μg of aRNAin a 20 μl volume instead of 50 μl to increase the dyeincorporation rate. Labelled aRNA was hybridized to Agilent's Whole Human GenomeMicroarry (G4112F) according to the manufacturer's instructions, washed andscanned on a 5 μm Agilent scanner. Hybridizations of the biologicalduplicates of each experiment were carried out in technical duplicates using dyeswaps (Cy3 and Cy5), for a total of 4 replicates for each of the overexpressionand downregulation experiment.
Expression profiling data analysis
Raw signal and background intensities were extracted from the scanned images ofexpression arrays using the Feature Extraction software from Agilent. Raw datawas processed and normalized using the R platform and the LIMMA package [75]. Processed expression profiling results are provided (Additional file5).
Expression profiling result validation
Independent p110 CUX1 overexpression and CUX1 shRNA knockdown experiments werecarried out in Hs578t cells using retroviral vectors. 10 genes were selectedfrom each experiment and real-time PCR was used to confirm the changes inexpression seen in expression profiling.
Abbreviations
- ChAP:
-
Chromatin Affinity Purification
- ChIP:
-
Chromatin Immuno Precipitation
- DHS:
-
DNAse hypersensitivity site
- ENCODE:
-
ENCyclopedia of DNA Elements
- FDR:
-
Falsediscovery rate
- TAP:
-
Tandem affinity purification
- TSS:
-
Transcription startsite.
References
Jenuwein T, Allis CD: Translating the histone code. Science. 2001, 293 (5532): 1074-1080. 10.1126/science.1063127.
Turner BM: Cellular memory and the histone code. Cell. 2002, 111 (3): 285-291. 10.1016/S0092-8674(02)01080-2.
MacQuarrie KL, Fong AP, Morse RH, Tapscott SJ: Genome-wide transcription factor binding: beyond direct target regulation. Trends Genet. 2011, 27 (4): 141-148. 10.1016/j.tig.2011.01.001.
Mantripragada KK, Buckley PG, Diaz de Stahl T, Dumanski JP: Genomic microarrays in the spotlight. Trends Genet. 2004, 20 (2): 87-94. 10.1016/j.tig.2003.12.008.
Bosher JM, Labouesse M: RNA interference: genetic wand and genetic watchdog. Nat Cell Biol. 2000, 2 (2): E31-E36. 10.1038/35000102.
Hofmann A, Nolan GP, Blau HM: Rapid retroviral delivery of tetracycline-inducible genes in a singleautoregulatory cassette [see comments]. Proc Natl Acad Sci U S A. 1996, 93 (11): 5185-5190. 10.1073/pnas.93.11.5185.
Paulus W, Baur I, Boyce FM, Breakefield XO, Reeves SA: Self-contained, tetracycline-regulated retroviral vector system for genedelivery to mammalian cells. J Virol. 1996, 70 (1): 62-67.
Meyerson M, Gabriel S, Getz G: Advances in understanding cancer genomes through second-generationsequencing. Nat Rev Genet. 2010, 11 (10): 685-696. 10.1038/nrg2841.
Gilmour DS, Lis JT: In vivo interactions of RNA polymerase II with genes of Drosophilamelanogaster. Mol Cell Biol. 1985, 5 (8): 2009-2018.
Marzio G, Tyagi M, Gutierrez MI, Giacca M: HIV-1 tat transactivator recruits p300 and CREB-binding protein histoneacetyltransferases to the viral promoter. Proc Natl Acad Sci U S A. 1998, 95 (23): 13519-13524. 10.1073/pnas.95.23.13519.
Consortium EP: The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004, 306 (5696): 636-640.
Farnham PJ: Insights from genomic profiling of transcription factors. Nat Rev Genet. 2009, 10 (9): 605-616. 10.1038/nrg2636.
Amati B, Land H: Myc-Max-Mad: a transcription factor network controlling cell cycleprogression, differentiation and death. Curr Op Genet Dev. 1994, 4 (1): 102-108. 10.1016/0959-437X(94)90098-1.
Bieda M, Xu X, Singer MA, Green R, Farnham PJ: Unbiased location analysis of E2F1-binding sites suggests a widespread rolefor E2F1 in the human genome. Genome Res. 2006, 16 (5): 595-605. 10.1101/gr.4887606.
Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ: Unbiased mapping of transcription factor binding sites along humanchromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004, 116 (4): 499-509. 10.1016/S0092-8674(04)00127-8.
Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA: Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell. 2012, 151 (1): 56-67. 10.1016/j.cell.2012.08.026.
Sansregret L, Nepveu A: The multiple roles of CUX1: Insights from mouse models and cell-basedassays. Gene. 2008, 412 (1–2): 84-94.
Moon NS, Berube G, Nepveu A: CCAAT displacement activity involves Cut repeats 1 and 2, not the Cuthomeodomain. J Biol Chem. 2000, 275 (40): 31325-31334.
Moon NS, Premdas P, Truscott M, Leduy L, Berube G, Nepveu A: S Phase-Specific Proteolytic Cleavage Is Required to Activate Stable DNABinding by the CDP/Cut Homeodomain Protein. Mol Cell Biol. 2001, 21: 6332-6345. 10.1128/MCB.21.18.6332-6345.2001.
Goulet B, Baruch A, Moon NS, Poirier M, Sansregret LL, Erickson A, Bogyo M, Nepveu A: A Cathepsin L Isoform that Is Devoid of a Signal Peptide Localizes to theNucleus in S Phase and Processes the CDP/Cux Transcription Factor. Mol Cell. 2004, 14 (2): 207-219. 10.1016/S1097-2765(04)00209-6.
Harada R, Vadnais C, Sansregret L, Leduy L, Berube G, Robert F, Nepveu A: Genome-wide location analysis and expression studies reveal a role for p110CUX1 in the activation of DNA replication genes. Nucleic Acids Res. 2008, 36 (1): 189-202.
Kedinger V, Sansregret L, Harada R, Vadnais C, Cadieux C, Fathers K, Park M, Nepveu A: p110 CUX1 homeodomain protein stimulates cell migration and invasion in partthrough a regulatory cascade culminating in the repression of E-cadherin andoccludin. J Biol Chem. 2009, 284 (40): 27701-27711. 10.1074/jbc.M109.031849.
Coqueret O, Berube G, Nepveu A: The mammalian Cut homeodomain protein functions as a cell-cycle-dependenttranscriptional repressor which downmodulates p21WAF1/CIP1/SDI1 in Sphase. EMBO J. 1998, 17 (16): 4680-4694. 10.1093/emboj/17.16.4680.
Santaguida M, Ding Q, Berube G, Truscott M, Whyte P, Nepveu A: Phosphorylation of the CCAAT displacement protein (CDP)/Cux transcriptionfactor by cyclin A-Cdk1 modulates its DNA binding activity in G(2). J Biol Chem. 2001, 276 (49): 45780-45790. 10.1074/jbc.M107978200.
Santaguida M, Nepveu A: Differential regulation of CDP/Cux p110 by cyclin A/Cdk2 and cyclinA/Cdk1. J Biol Chem. 2005, 280 (38): 32712-32721. 10.1074/jbc.M505417200.
Sansregret L, Gallo D, Santaguida M, Leduy L, Harada R, Nepveu A: Hyperphosphorylation by cyclin B/CDK1 in mitosis resets CUX1 DNA bindingclock at each cell cycle. J Biol Chem. 2010, 285 (43): 32834-32843. 10.1074/jbc.M110.156406.
Truscott M, Denault JB, Goulet B, Leduy L, Salvesen GS, Nepveu A: Carboxyl-terminal proteolytic processing of CUX1 by a caspase enablestranscriptional activation in proliferating cells. J Biol Chem. 2007, 282 (41): 30216-30226. 10.1074/jbc.M702328200.
Ellis T, Gambardella L, Horcher M, Tschanz S, Capol J, Bertram P, Jochum W, Barrandon Y, Busslinger M: The transcriptional repressor CDP (Cutl1) is essential for epithelial celldifferentiation of the lung and the hair follicle. Genes Dev. 2001, 15 (17): 2307-2319. 10.1101/gad.200101.
Sinclair AM, Lee JA, Goldstein A, Xing D, Liu S, Ju R, Tucker PW, Neufeld EJ, Scheuermann RH: Lymphoid apoptosis and myeloid hyperplasia in CCAAT displacement proteinmutant mice. Blood. 2001, 98 (13): 3658-3667. 10.1182/blood.V98.13.3658.
Luong MX, van der Meijden CM, Xing D, Hesselton R, Monuki ES, Jones SN, Lian JB, Stein JL, Stein GS, Neufeld EJ: Genetic ablation of the CDP/Cux protein C terminus results in hair cycledefects and reduced male fertility. Mol Cell Biol. 2002, 22 (5): 1424-1437. 10.1128/MCB.22.5.1424-1437.2002.
Ledford AW, Brantley JG, Kemeny G, Foreman TL, Quaggin SE, Igarashi P, Oberhaus SM, Rodova M, Calvet JP, Vanden Heuvel GB: Deregulated Expression of the Homeobox Gene Cux-1 in Transgenic Mice Resultsin Downregulation of p27(kip1) Expression during Nephrogenesis, GlomerularAbnormalities, and Multiorgan Hyperplasia. Dev Biol. 2002, 245 (1): 157-171. 10.1006/dbio.2002.0636.
Brantley JG, Sharma M, Alcalay NI, Heuvel GBV: Cux-1 transgenic mice develop glomerulosclerosis and interstitialfibrosis. Kidney Int. 2003, 63 (4): 1240-1248. 10.1046/j.1523-1755.2003.00889.x.
Cadieux C, Kedinger V, Yao L, Vadnais C, Drossos M, Paquet M, Nepveu A: Mouse mammary tumor virus p75 and p110 CUX1 transgenic mice develop mammarytumors of various histologic types. Cancer Res. 2009, 69 (18): 7188-7197. 10.1158/0008-5472.CAN-08-4899.
Cadieux C, Harada R, Paquet M, Cote O, Trudel M, Nepveu A, Bouchard M: Polycystic kidneys caused by sustained expression of Cux1 isoform p75. J Biol Chem. 2008, 283 (20): 13817-13824. 10.1074/jbc.M709332200.
Cadieux C, Fournier S, Peterson AC, Bedard C, Bedell BJ, Nepveu A: Transgenic mice expressing the p75 CCAAT-displacement protein/Cut homeoboxisoform develop a myeloproliferative disease-like myeloid leukemia. Cancer Res. 2006, 66 (19): 9492-9501. 10.1158/0008-5472.CAN-05-4230.
Heuvel GBV, Bodmer R, McConnell KR, Nagami GT, Igarashi P: Expression Of a Cut-Related Homeobox Gene In Developing and Polycystic MouseKidney. Kidney Int. 1996, 50 (2): 453-461. 10.1038/ki.1996.336.
Sansregret L, Goulet B, Harada R, Wilson B, Leduy L, Bertoglio J, Nepveu A: The p110 isoform of the CDP/Cux transcription factor accelerates entry into Sphase. Mol Cell Biol. 2006, 26 (6): 2441-2455. 10.1128/MCB.26.6.2441-2455.2006.
Sansregret L, Vadnais C, Livingstone J, Kwiatkowski N, Awan A, Cadieux C, Leduy L, Hallett MT, Nepveu A: Cut homeobox 1 causes chromosomal instability by promoting bipolar divisionafter cytokinesis failure. Proc Natl Acad Sci U S A. 2011, 108 (5): 1949-1954. 10.1073/pnas.1008403108.
Michl P, Ramjaun AR, Pardo OE, Warne PH, Wagner M, Poulsom R, D'Arrigo C, Ryder K, Menke A, Gress T: CUTL1 is a target of TGF(beta) signaling that enhances cancer cell motilityand invasiveness. Cancer Cell. 2005, 7 (6): 521-532. 10.1016/j.ccr.2005.05.018.
Michl P, Downward J: CUTL1: a key mediator of TGFbeta-induced tumor invasion. Cell Cycle. 2006, 5 (2): 132-134. 10.4161/cc.5.2.2311.
Aleksic T, Bechtel M, Krndija D, von Wichert G, Knobel B, Giehl K, Gress TM, Michl P: CUTL1 promotes tumor cell migration by decreasing proteasome-mediated Srcdegradation. Oncogene. 2007, 26 (40): 5939-5949. 10.1038/sj.onc.1210398.
Ripka S, Neesse A, Riedel J, Bug E, Aigner A, Poulsom R, Fulda S, Neoptolemos J, Greenhalf W, Barth P: CUX1: target of Akt signalling and mediator of resistance to apoptosis inpancreatic cancer. Gut. 2010, 59 (8): 1101-1110. 10.1136/gut.2009.189720.
Cubelos B, Sebastian-Serrano A, Beccari L, Calcagnotto ME, Cisneros E, Kim S, Dopazo A, Alvarez-Dolado M, Redondo JM, Bovolenta P: Cux1 and Cux2 regulate dendritic branching, spine morphology, and synapses ofthe upper layer neurons of the cortex. Neuron. 2010, 66 (4): 523-535. 10.1016/j.neuron.2010.04.038.
Scacheri PC, Davis S, Odom DT, Crawford GE, Perkins S, Halawi MJ, Agarwal SK, Marx SJ, Spiegel AM, Meltzer PS: Genome-wide analysis of menin binding provides insights into MEN1tumorigenesis. PLoS Genet. 2006, 2 (4): e51-10.1371/journal.pgen.0020051.
Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, Gingeras TR, Struhl K: Relationships between p63 binding, DNA sequence, transcription activity, andbiological function in human cells. Mol Cell. 2006, 24 (4): 593-602. 10.1016/j.molcel.2006.10.018.
Krig SR, Jin VX, Bieda MC, O'Geen H, Yaswen P, Green R, Farnham PJ: Identification of genes directly regulated by the oncogene ZNF217 usingchromatin immunoprecipitation (ChIP)-chip assays. J Biol Chem. 2007, 282 (13): 9703-9712. 10.1074/jbc.M611752200.
Harada R, Berube G, Tamplin OJ, Denis-Larose C, Nepveu A: DNA-binding specificity of the cut repeats from the human cut-likeprotein. Mol Cell Biol. 1995, 15 (1): 129-140.
Harada R, Nepveu A: Chromatin affinity purification. Meth Mol Biol (Clifton, NJ). 2012, 809 (22113280): 237-253.
Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B: A high-resolution map of active promoters in the human genome. Nature. 2005, 436 (7052): 876-880. 10.1038/nature03877.
Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR: Chromosome-wide mapping of estrogen receptor binding reveals long-rangeregulation requiring the forkhead protein FoxA1. Cell. 2005, 122 (1): 33-43. 10.1016/j.cell.2005.05.008.
Ruiz-Llorente S, de Pau EC, Sastre-Perona A, Montero-Conde C, Gomez-Lopez G, Fagin JA, Valencia A, Pisano DG, Santisteban P: Genome-wide analysis of Pax8 binding provides new insights into thyroidfunctions. BMC Genomics. 2012, 13: 147-10.1186/1471-2164-13-147.
Li Q, Harju S, Peterson KR: Locus control regions: coming of age at a decade plus. Trends Genet. 1999, 15 (10): 403-408. 10.1016/S0168-9525(99)01780-1.
Gaszner M, Felsenfeld G: Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet. 2006, 7 (9): 703-713. 10.1038/nrg1925.
Gross DS, Garrard WT: Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988, 57: 159-197. 10.1146/annurev.bi.57.070188.001111.
Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B: The accessible chromatin landscape of the human genome. Nature. 2012, 489 (7414): 75-82. 10.1038/nature11232.
Ernst J, Kellis M: ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012, 9 (3): 215-216. 10.1038/nmeth.1906.
Vadnais C, Davoudi S, Afshin M, Harada R, Dudley R, Clermont PL, Drobetsky E, Nepveu A: CUX1 transcription factor is required for optimal ATM/ATR-mediated responsesto DNA damage. Nucleic Acids Res. 2012, 40 (10): 4483-4495. 10.1093/nar/gks041.
Seguin L, Liot C, Mzali R, Harada R, Siret A, Nepveu A, Bertoglio J: CUX1 and E2F1 regulate coordinated expression of the mitotic complex genesEct2, MgcRacGAP, and MKLP1 in S phase. Mol Cell Biol. 2009, 29 (2): 570-581. 10.1128/MCB.01275-08.
Truscott M, Harada R, Vadnais C, Robert F, Nepveu A: p110 CUX1 cooperates with E2F transcription factors in the transcriptionalactivation of cell cycle-regulated genes. Mol Cell Biol. 2008, 28 (10): 3127-3138. 10.1128/MCB.02089-07.
Kraus MH, Yuasa Y, Aaronson SA: A position 12-activated H-ras oncogene in all HS578T mammary carcinosarcomacells but not normal mammary cells of the same patient. Proc Natl Acad Sci U S A. 1984, 81 (17): 5384-5388. 10.1073/pnas.81.17.5384.
Geck P, Maffini MV, Szelei J, Sonnenschein C, Soto AM: Androgen-induced proliferative quiescence in prostate cancer cells: the roleof AS3 as its mediator. Proc Natl Acad Sci U S A. 2000, 97 (18): 10185-10190. 10.1073/pnas.97.18.10185.
Saqr HE, Omran O, Dasgupta S, Yu RK, Oblinger JL, Yates AJ: Endogenous GD3 ganglioside induces apoptosis in U-1242 MG glioma cells. J Neurochem. 2006, 96 (5): 1301-1314. 10.1111/j.1471-4159.2005.03640.x.
Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E: Early transcriptional profiles in huntingtin-inducible striatal cells bymicroarray analyses. Hum Mol Genet. 2002, 11 (17): 1953-1965. 10.1093/hmg/11.17.1953.
Weinmann AS, Farnham PJ: Identification of unknown target genes of human transcription factors usingchromatin immunoprecipitation. Methods. 2002, 26 (1): 37-47. 10.1016/S1046-2023(02)00006-3.
Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B: The tandem affinity purification (TAP) method: a general procedure of proteincomplex purification. Methods. 2001, 24 (3): 218-229. 10.1006/meth.2001.1183.
Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD: E2F integrates cell cycle progression with DNA repair, replication, andG(2)/M checkpoints. Genes Dev. 2002, 16 (2): 245-256. 10.1101/gad.949802.
Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J: Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010, Chapter 19: Unit 19 10 11–21-
Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J: Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005, 15 (10): 1451-1455. 10.1101/gr.4086505.
Goecks J, Nekrutenko A, Taylor J: Galaxy: a comprehensive approach for supporting accessible, reproducible, andtransparent computational research in the life sciences. Genome Biol. 2010, 11 (8): R86-10.1186/gb-2010-11-8-r86.
Bailey TL: DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011, 27 (12): 1653-1659. 10.1093/bioinformatics/btr261.
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS: MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009, 37 (Web Server issue): W202-W208.
Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS: Quantifying similarity between motifs. Genome Biol. 2007, 8 (2): R24-10.1186/gb-2007-8-2-r24.
da Huang W, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVIDbioinformatics resources. Nat Protoc. 2009, 4 (1): 44-57.
da Huang W, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functionalanalysis of large gene lists. Nucleic Acids Res. 2009, 37 (1): 1-13. 10.1093/nar/gkn923.
Smyth GK: Linear models and empirical bayes methods for assessing differentialexpression in microarray experiments. Stat Appl Genet Mol Biol. 2004, 3: Article3-
Acknowledgements
C.V. is the recipient of a scholarship from the Fonds de la Recherche enSanté du Québec. This research was supported by grant # MOP-98010 fromthe Canadian Institute of Health Research of Canada and grant #019389 from theCanadian Cancer Society to AN. We acknowledge infrastructure support andtechnical assistance from the Breast Cancer Functional Genomics Group, which issupported by funds from the Terry Fox Foundation and CIHR.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AN, CV and AA conceived the study. CV, AA, RH, PLC, LL, and GB performed theexperiments. CV and AA performed data analysis. AN and CV wrote the manuscript. Allauthors read and approved the final manuscript.
Electronic supplementary material
12864_2012_4981_MOESM1_ESM.ps
Additional file 1: Figure S1: Distribution of 3 random sets of binding sites relative to transcriptionstart sites. (PS 343 KB)
12864_2012_4981_MOESM2_ESM.ps
Additional file 2: Figure S2: Distribution of binding sites relative to transcription start sites for 6transcription factors. (PS 445 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Vadnais, C., Awan, A.A., Harada, R. et al. Long-range transcriptional regulation by the p110 CUX1 homeodomain protein on theENCODE array. BMC Genomics 14, 258 (2013). https://doi.org/10.1186/1471-2164-14-258
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-258