
Abstract
Background
Prior to this study, relatively few strains of Francisella had been genome-sequenced. Previously published Francisella genome sequences were largely restricted to the zoonotic agent F. tularensis. Only limited data were available for other members of the Francisella genus, including F. philomiragia, an opportunistic pathogen of humans, F. noatunensis, a serious pathogen of farmed fish, and other less well described endosymbiotic species.
Results
We determined the phylogenetic relationships of all known Francisella species, including some for which the phylogenetic positions were previously uncertain. The genus Francisella could be divided into two main genetic clades: one included F. tularensis, F. novicida, F. hispaniensis and Wolbachia persica, and another included F. philomiragia and F. noatunensis.
Some Francisella species were found to have significant recombination frequencies. However, the fish pathogen F. noatunensis subsp. noatunensis was an exception due to it exhibiting a highly clonal population structure similar to the human pathogen F. tularensis.
Conclusions
The genus Francisella can be divided into two main genetic clades occupying both terrestrial and marine habitats. However, our analyses suggest that the ancestral Francisella species originated in a marine habitat. The observed genome to genome variation in gene content and IS elements of different species supports the view that similar evolutionary paths of host adaptation developed independently in F. tularensis (infecting mammals) and F. noatunensis subsp. noatunensis (infecting fish).
Similar content being viewed by others
Background
Tularemia is a zoonotic disease caused by the highly infectious, virulent, Gram-negative bacterium Francisella tularensis. Due to its infectious nature, ease of dissemination and associated high fatality rate (especially from respiratory infection), F. tularensis has been included in military biological weapons programs and is regarded as one of the top six agents which have a high potential for misuse in bioterrorism[1].
The known diversity of the genus Francisella has recently expanded substantially and the boundaries between taxonomic units within the genus is currently under debate[2, 3]. In mammals, including man, the disease tularemia is caused by two F. tularensis subspecies: subsp. tularensis (type A, endemic in North America) and subsp. holarctica (type B, found throughout the Northern hemisphere)[4]. Nearly all fatalities due to tularemia are caused by F. tularensis type A infections, for which mortality rates up to 30% have been reported[5]. In contrast, F. tularensis type B causes a milder disease, for which fatalities rarely occur. A third accepted subspecies, F. tularensis subsp. mediasiatica is found in central Asia. This subspecies has been found to display level of virulence comparable to strains of subspecies holarctica and very rarely cause human infections[6, 7]. Disease in humans has also been presented following opportunistic infection by F. novicida (in individuals with a weakened immune system) and F. philomiragia (in individuals with a weakened immune system exposed to sea water)[8–10].
A number of Francisella lineages also exist that do not appear to be associated with human disease. Strains closely related to F. philomiragia, which have recently been designated as Francisella noatunensis, have been identified as etiological agents of Francisellosis in fish[11–15]. F. noatunensis is further divided into two subspecies, subsp. noatunensis[16] and subsp. orientalis[17, 18]. Recent published studies have also identified several new related and as yet uncultured forms of Francisella from soil and water[19–23], as well as tick endosymbionts with high similarity to F. tularensis (Francisella-like endosymbionts)[24]. One isolated tick endosymbiont has inaccurately been designated Wolbachia persica[25], incorrectly placing it within the alpha- proteobacteria[26]. Further, a marine ciliate endosymbiont has been proposed to represent ‘Candidatus F. noatunensis subsp. endociliophora’[23] and a bacterium isolated from an air-conditioning system has been tentatively classified as Francisella cantonensis[27]. Thus, in recent years the existence of substantial sub-species diversity within the Francisella genus has become increasingly apparent. Members of Francisella appear to be associated with a multitude of different ecological niches, ranging from specialised endosymbionts and pathogens with different host spectrums, to generalists believed capable of a free-living existence.
There are several reasons for sequencing Francisella strains other than those belonging to F. tularensis. Analysis of gene content and genetic relationships may help to improve our understanding of the biology and evolution of these bacteria, including that of the human pathogen F. tularensis. Sequencing multiple strains of environmental Francisella may also reveal important sequence differences between environmental species and F. tularensis. This would allow the design of increasingly specific F. tularensis detection assays for use in anti-bioterrorism applications and epidemiological studies, particularly important since current methods frequently falsely detect F. tularensis in environmental samples[28].
The genome sequence data generated by the present study represents a considerable advancement of knowledge of the genus Francisella and includes genome sequences for 18 environmental Francisella strains, four F. tularensis and two distant but genetically related species Fangia hongkongensis[29] and Piscirickettsia salmonis[30]. Here we describe the population structure of the genus Francisella, suggest that ancestral Francisella strains originated in marine habitats, show that present-day strains can be divided into two main genetic clades occupying continental and aquatic habitats and provide evidence that similar evolutionary paths of host adaptations have occurred independently in F. tularensis (infecting mammals) and F. noatunensis subsp. noatunensis (infecting fish).
Results
Bacterial strains and classifications
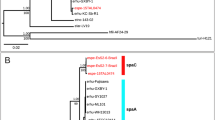
Here, we present 24 novel genome sequences representing all major subclades of the Francisella genus (Table 1), with the exception of the recently discovered F. cantonensis[27]. The data doubles the number of published Francisella genomes and significantly extends the known range of genetic diversity. The studied material includes seven previously unsequenced F. noatunensis isolates and a single representative from the tick endosymbiont group[31]. In addition, representatives of Fa. hongkongensis[29] and P. salmonis[32], both of which are related to the genus Francisella, were also sequenced. Interestingly, Francisella strain 3523[33, 34] appeared to belong to the F. hispaniensis clade and not the F. novicida clade as reported previously[34].
Overall, a major bifurcation into two genetic lineages was identified (Figure 1) with clade 1 comprising F. tularensis, F. novicida, F. hispaniensis and W. persica, and clade 2 containing F. noatunensis and F. philomiragia.

Whole-genome phylogeny. The phylogeny of 45 Francisella strains and two genetic relatives, Fangia hongkongensis and Piscirickettsia salmonis, based on single nucleotide polymorphism in the core genome. The Francisella genus is divided into two parts, clade 1 (F. tularensis and F. novicida) and clade 2 (F. noatunensis and F. philomiragia).
Francisella whole-genome phylogeny and average nucleotide identities
Whole-genome alignments were produced using progressiveMauve[35]. A phylogeny based on core genome single nucleotide polymorphisms is shown in Figure 1.
Analysis of the whole-genome phylogeny showed that strains of F. philomiragia and F. noatunensis subsp. noatunensis formed sister clades, while strains of F. noatunensis subsp. orientalis diverged at a more basal position, indicating the newly named species F. noatunensis is paraphyletic. This interpretation was supported by an analysis of average nucleotide identities (ANI): ANI for F. philomiragia compared to F. noatunensis subsp. noatunensis is higher (95%) than F. philomiragia compared to F. noatunensis subsp. orientalis (92–93%) Additional file1.
Phylogenetic inference using whole-genome data and 16 S ribosomal RNA genes resulted in incongruent topologies. The phylogenetic position of F. philomiragia (Figure 1) was different from previous publications of phylogenies calculated based on 16 S ribosomal RNA genes (Additional file2)[13, 14].
Analysis of Francisella pan and core genomes
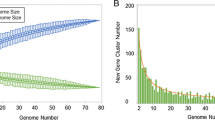
Based on the analysed gene orthologs, the core genome of the genus Francisella contained 803 genes (Figure 2A). Clade 1 shared 854 genes, while clade 2 (F. noatunensis and F. philomiragia) shared 1359 genes (Figure 2B).

Overview of core and pan genome structure for the Francisella genus. A. Accumulation curve (core genome) for the number of genes in common as a function of the number of genomes analysed for the Francisella genus. B. Accumulation curve for the total number of genes (pan genome) as a function of the number of genomes analysed for clade 1 (F. tularensis, W. persica and F. hispaniensis)(red) and clade 2 (F. philomiragia and F. noatunensis)(yellow). C. Cumulative accumulation of total number genes when genomes are added according to the structure in the SNP phylogeny. Ftt – F. tularensis subsp. tularensis, Fth – F. tularensis subsp. holarctica, Ftm – F. tularensis subsp. mediasiatica, Fn – F. novicida, Fh – F. hispaniensis, W – Wolbachia persica, Fno – F. noatunensis subsp. orientalis, Fnn – F. noatunensis subsp. noatunensis, Fp – F. Philomiragia.
The pan-genome of the genus Francisell a contained a total of 5396 genes (Figure 2C). Separate analysis of the pan-genome size within clade 1, excluding F. hispaniensis and W. persica, and clade 2 identified 3222 and 3535 genes, respectively (Figure 2B). The pan-genome of F. tularensis contained 2769 genes.
Clade-specific genes
An overview of the gene distribution in the genus Francisella is depicted in Figure 3 and the clade-specific genes are summarised in Table 2. The human pathogen, F. tularensis contained only seven unique genes (FTT0794, FTT0795, FTT0796, FTT1188, FTT1453c, FTT1454c and FTT1458), which grouped at three different locations in the genome. No unique genes were found in F. novicida genomes. F. hispaniensis, F. noatunensis and F. philomiragia exhibited seven, nine and ten unique genes, respectively. The complete clade 2 (F. noatunensis and F. philomiragia) genomes displayed 41 genes that were not present in clade 1. However, the clade 2 species F. noatunensis subsp. noatunensis and F. noatunensis subsp. orientalis shared 32 and 114 genes, respectively, which were not present in other genomes.

Whole-genome comparison of the pan-genome of 45 Francisella strains. Presence of a gene in a genome is indicated in black. The core genome of 803 genes, and the genes unique to clade 1 (F. tularensis, F. novicida, F. hispaniensis and W. persica) and clade 2 (F. noatunensis and F. philomiragia) are clustered together in the heat map.
IS elements
Notable differences in IS element content were identified between different species and clades. In clade 1, F. hispaniensis contained a few copies of ISFtu2, ISFtu3, ISFtu4, ISFpi1 and IS4502, while W. persica contained very few copies of ISFtu2 and ISFtu3. The isolates in clade 2 exhibited a different pattern of IS elements. Whereas F. noatunensis subsp. orientalis was almost completely lacking IS elements (only a potential trace of ISFpi1 was found), F. noatunensis subsp. noatunensis exhibited an expansion trend similar to that previously reported for F. tularensis[36]. The expansion was within the ISFpi1 group and included a handful of copies of the IS4502 element, and single copies of ISFtu1, ISFtu2 and ISFtu3 elements.
The more distant relatives studied, i.e. Fa. hongkongensis and P. salmonis, contained completely different sets of IS elements compared with Francisella. The genome of P. salmonis was enriched with ISPsa1 and ISPsa2[37], whereas only one copy of ISFpi1 was found in Fa. hongkongensis. The IS element distributions of F. tularensis, F. novicida and F. philomiragia have been reported previously[36, 38].
Recombination
Results of the recombination analyses are presented in Figure 4 and Additional file3. The proportion of genes within the Francisella core genome demonstrating evidence of recombination was 32%, although the presence of recombination was highly variable across the different Francisella lineages. For some species/subspecies, recombination was entirely absent (i.e. F. tularensis subsp. tularensis, F. tularensis subsp. holarctica and F. noatunensis subsp. noatunensis), whereas recombination in F. philomiragia and F. novicida occurred frequently.

Overview of the recombination. The proportion of genes showing evidence of recombination overlaid on the Francisella phylogeny. Recombination detection was performed using the PHI method with the number of significant tests (P < 0.01) reported as a percentage of recombinant genes within the Francisella genus.
Discussion
To date, sequenced Francisella genomes have mainly been of the human pathogenic species F. tularensis. Consequently, conclusions about evolutionary processes within the genus Francisella have largely been based on F. tularensis genomes[36, 38].
Moreover, a number of identification assays developed on the basis of F. tularensis genome sequences have been shown to give false-positive results. These have been attributed to the presence of the same sequence signatures as in other environmental Francisella lineages[39].
In the present study, we provide details of the genetic content of nearly all currently known members of the genus Francisella, with the exception of the recently published F. cantonensis[27]. Despite a generally high sequence similarity within the genus, we found that different genetic clades demonstrated different population structures (recombination frequencies). This suggests differences in lifestyle and host association.
F. tularensis has been found to be a highly clonal species, differentiating it from environmental lineages characterised by moderate recombination rates. We also found evidence that the fish pathogen F. noatunensis subsp. noatunensis also possesses a highly clonal lineage. The low recombination frequency in F. noatunensis subsp. noatunensis may indicate that a close host association reduced contact with other Francisella species, and thereby, lowered opportunities for gene acquisition by recombination, as previously hypothesised for F. tularensis[36]. The data of population structure and variability observed among F. novicida opposed to F. tularensis subspecies tularensis, holarctica and mediasiatica strains should be taken in consideration for the on-going discussion whether it should be a separate species or included as a subspecies in F. tularensis.
Whole genome phylogeny
Our analyses of 45 Francisella genomes showed that recognised species could be divided into two main genetic clades (Figure 1) and suggested that the pathogenic species in each clade emerged independently by similar evolutionary paths of host adaptation. F. tularensis, which can infect mammals, belonged to clade 1, whereas F. noatunensis subsp. noatunensis, which infects fish, belonged to clade 2. Our analyses based on the phylogeny of Francisella genomes and genetic relatives suggest that the ancestral Francisella species originated in a marine habitat.
Using whole-genome analysis, we found evidence that F. noatunensis subsp. noatunensis and F. noatunensis subsp. orientalis represent a paraphyletic group. F. philomiragia and F. noatunensis subsp. noatunensis together form a sister clade to F. noatunensis subsp. orientalis (Figure 1, Table 1). This result was not supported by 16 S RNA analysis, which suggested that the two subspecies F. noatunensis subsp. orientalis and F. noatunensis subsp. noatunensis form a clade. There are limited nucleotide differences in the 16 S region and more comprehensive core genome SNP analysis gives a better view of the real relationship. Identification of genetic factors coding for fish pathogenicity may therefore be possible via comparative analyses. Further phenotypic studies and experiments are needed to confirm this hypothesis. Since F. noatunensis subsp. noatunensis is isolated from cod and salmon, while F. noatunensis subsp. orientalis isolates are recovered from tilapia and three-line grunt, an alternative hypothesis is that these subspecies evolved their abilities for fish infection independently.
In agreement with other studies[26, 40], our analyses verified the phylogenetic position of the tick endosymbiont W. persica within the Francisella genus. Several lines of genomic evidence support the view that this organism adapted as a primary symbiont; its genome size was found to be significantly less (about 80–85%) than other Francisella genomes. Moreover, in W. persica, regulatory genes were found to be depleted, while biosynthesis pathways supplying amino acids to their insect host were retained, and there was a low abundance of IS elements. Our results confirm that W. persica and other Francisella-like endosymbionts (FLE)[19, 24, 41, 42] belong to the genus Francisella and that W. persica has undergone a significant reduction in its genome.
An important outcome of the present study was reclassification of the isolate 3523 from Australia. It was initially classified as an unusual F. novicida[33], a classification which remained despite whole genome sequencing[34]. It was clear from the present analyses that isolate 3523 belongs to the F. hispaniensis clade[43]. This result demonstrates the importance of broad-scale intra-genus sequencing for correct classification of new isolates.
Francisella pan- and core- genome
The overall core genome size of 799 genes for the genus Francisella was similar to that reported for the genus Bacillus (Figure 2A)[44]. Bacterial pan-genomes at the genus level have only recently been reported in the literature, and the number of gene families in these genera is expected to grow as more genomes are included. This approach may be valuable in revealing genes specific for niche commonalities between subspecies.
We detected different pan-genome sizes for clade 1 and clade 2 (Figure 2B), where clade 2 contain more genes compared to clade 1 and especially F. tularensis clade alone. As a result, F. tularensis may be fully sampled by sequencing smaller set of representative isolates, whereas more isolates of clade 2 would need to be fully sequenced to give a true pan-genome size.
The contribution of each genome to the complete pan-genome of the genus Francisella and related organisms is illustrated in Figure 2C. Most genomes contribute to the increase of the pan-genome, although the increase is less pronounced when similar genomes are added.
Clade specific genes
Genomes in the Francisella genus have, with the exception of W. persica, similar gene composition. W. persica has not retained genes included in the COG group ‘amino acid transport and metabolism’, which it does not require because of its endosymbiotic lifestyle.
An extensive list of 80 potential candidate virulence genes has previously been published, which are specific for the human pathogen F. tularensis[45]. Surprisingly, analysis of the more complete collection of Francisella genomes in the present study reduced this list to only seven F. tularensis specific genes. The general lack of specific genes agrees with previous observations that pathogens exhibit specialisation and host-adaption by gene loss rather than gene gain[46]. Thus, a pathogen such as F. tularensis should be defined not only by genes that are present but also by those that are lacking.
The seven specific genes in F. tularensis were located in three separate gene clusters. The genes in the first cluster (FTT0794, FTT0795, FTT0796) are organised in an operon and were predicted to be involved in exopolysaccharide synthesis. It has been suggested that an exopolysaccharide capsule plays a role in F. tularensis virulence[47].
The second gene cluster (FTT1453c, FTT1454c and FTT1458) is present within the wbt locus of F. tularensis. Genes in this locus encode proteins involved in lipopolysaccharide O-antigen synthesis. The O-antigens of F. tularensis contain an insertion element that differentiates it from F. novicida[48]. A similar insertion element structure has been reported to be essential for virulence in Shigella sonnei[49]. Finally, the last gene cluster contained a single gene, FTT1188, which encodes for a hypothetical membrane protein. This protein does not exhibit significant homology to other proteins in the NCBI non-redundant database and has not yet been characterised in depth. Identification of virulence genes and their role in the virulence of Francisella is not straight-forward. Our data suggest that F. tularensis virulence cannot be easily explained by a simplistic model which just considers the presence or absence of specific “virulence genes”[36].
Fa. hongkongensis and P. salmonis both contain many genes that are absent from the Francisella genus and are therefore likely to occupy distinctly different ecological niches.
IS elements
The two principal clades of the Francisella genus exhibit several similarities and differences in IS element content and composition. F. noatunensis subsp. noatunensis contains an expanded set of IS elements similar to those reported for F. tularensis. The expanded IS elements in F. tularensis are ISFtu1 and ISFtu2, whereas the expanded IS element in F. noatunensis is ISFpil. Interestingly, the two subspecies of F. noatunensis exhibited contrasting contents of IS elements. While F. noatunensis subsp. noatunensis contained the highest number of IS elements of all species in the genus, F. noatunensis subsp. orientalis contains very few IS elements. This may have arisen because of different evolutionary modes in these subspecies.
Recombination
Recombination is an important process for generating new genetic diversity within bacterial populations and is a known driver of evolution in many bacterial species[50]. The recombination analysis of 45 genomes in the present study confirmed that the level of recombination within the Francisella genus is highly variable, ranging from undetectable in F. tularensis to moderate in F. novicida[36].
We found that the subspecies of F. noatunensis exhibited apparently low rates of recombination at levels similar to the human pathogen group of F. tularensis. Otherwise, both clade 1 and II displayed moderate recombination rates, largely due to frequent recombination within F. novicida (clade 1) and F. philomiragia (clade 2). According to our findings, 33% of the core gene set provided evidence of past recombination events, similar in magnitude to estimates for Campylobacter[51] and Neisseria[52], but higher than previously reported for Francisella[36]. It was reported previously that 19.2% of the genes in the core genome of F. novicida and F. philomiragia have been recombined, but our data suggests that this is could be an underestimate due to a lack of genomic sequences available at that time.
Our findings are similar to reports for other bacterial genera that include highly specialized pathogens. Variable recombination rates have previously been reported for several pathogenic populations, (Yersina pestis[53], Bacillus anthracis[54] and Mycobacterium tuberculosis[55]). It should be noted that if closely related clones of pathogens with low recombination rates are over-represented, this can result in an under-estimation of recombination rates for the entire genus[50, 56]. By ensuring proper representation of all subspecies of the Francisella genus in the present work, such effects were minimised.
Conclusions
This work significantly expands the number of sequenced Francisella genomes, and therefore provides an important resource for assessing evolutionary and functional relationships within the genus. We identified the core genome in the Francisella genus and single nucleotide polymorphisms within these genes were used to infer a robust phylogeny. A high degree of clonality confirmed for F. tularensis type A and B, was also identified in F. noatunensis subsp. noatunensis. Despite infecting different species, it is apparent that these lineages also share certain important evolutionary properties.
Next-generation sequencing enables rapid generation of high quality data for the fast assembly of novel genomes[57]. Growing databases of genome sequences are of significant interest for improving the designs of specific detection assays, conducting epidemiological and ecological studies, microbial forensics[58] and classification of novel bacterial isolates. Comparative genomic approaches together with functional datasets will be critical for the development of assays that specifically target pathogenic Francisella strains.
Methods
Cultivation and DNA preparation
All strains (Table 1), except FSC845, were cultured on cysteine heart agar plates supplemented with 5–9% chocolatised sheep blood (CHAB) (BD Biosciences, Franklin Lakes, NJ USA), or Modified Thayer-Martin (McLeod) agar plates[59], at 25°C for the eight strains FSC769 to FSC776, and at 37°C for all other strains, in 5% CO2 for 5 days (F. tularensis strains) or up to ~30 days (non-F. tularensis strains).
A loopful of bacteria was suspended in 1 x TRIS-EDTA (TE) buffer to 109 CFU/ml and heat-killed at 95°C for 15 minutes. The bacteria were lysed using Buffer ATL (Qiagen, Hilden, Germany), or similar non-denaturing lysis buffer. DNA was prepared using phenol extraction (PE), or extraction with Biorobot EZ1 (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Sterility was verified by lack of growth on Modified Thayer-Martin (McLeod) plates[59] after incubation of 10 μl DNA (if PE was used) or lysate (if EZ1 was used) for 10 days.
The strain FSC845 (Wolbachia persica) was propagated in cells, and bacteria were collected by differential centrifugation. Bacterial cell pellets were suspended in phosphate-buffered saline (PBS) and DNA was prepared as above. For FSC772 and FDC178, DNA was prepared using the Gentra Puregene Cell kit (Qiagen, Hilden, Germany) and was re-suspended in a hydration solution supplied with the kit. DNA concentrations were measured using Nanodrop (Thermo Scientific, Wilmington, DE, USA).
Genome sequencing and assembly
DNA for genomic sequencing of 24 isolates from the Francisella genus and genetic relatives were prepared as described in Table 1. 19 genomes were sequenced at the National Bioforensic Analysis Center using a 454 FLX machine (454 Life Sequencing Inc., Branford, CT, USA) according to the manufacturer’s instructions. Additional sequencing was performed using the Illumina GS II sequencing 36 bp or 76 bp SE technology. Additional file4 provides summary data of the sequenced genomes.
Isolates sequenced with both technologies were first assembled using Newbler. Obtained contigs were corrected using the Illumina reads in Nesoni. Isolates sequenced only with the Illumina technology were assembled using Velvet.[60, 61].
All 24 isolates included in this study were submitted to IMG/ER[62] for functional annotation. All sequence reads were deposited in Genbank and are freely downloadable from the Genbank project number listed in Table 1.
16 S rRNA alignment
The Francisella 16 S ribosomal RNA data were obtained using the ribosomal database project[63, 64]. The key search words ‘Francisella’, ‘Fangia hongkongensis’ and ‘Piscirickettsia salmonis’ produced 171 hits in the database. After removing duplicates, small sequences and sequences of poor quality, the final dataset consisted of 69 sequences and 1557 bases. To infer the phylogenetic tree of the 16 S data, the software RAxML[65] was used, which is suitable for analysing ribosomal RNA data as covariation patterns are taken into account in the model. The secondary structure was analysed using the substitution model RNA6A by maximising the log-likelihood. The GTR and gamma model was chosen as the standard DNA substitution model. The number of bootstrap replicates was 1000.
Whole-genome alignment
Multiple genome alignments were computed by employing the progressiveMauve algorithm of the Mauve software[35] using default options. Sequences were extracted and concatenated from each of the 45 genomes to build a core genome of multiple alignments. Core genome SNPs were identified from this alignment as the loci where at least one sequence had a mutated base and it had no missing sequences from any genome included in this study. While it is possible that some of the SNP’s that were specific to a single sequence could have been incorrect due to sequencing errors, this was unlikely in the case of informative sites. Strain-specific SNP’s can have an effect on the estimated value of the mutation rate and the branch length of the phylogenetic tree. However, they do not affect the structure of the tree. The phylogenetic data is deposited in treeBASE[66, 67].
Whole-genome phylogenetic construction
An unrooted neighbour joining tree for the genus Francisella with the genetic relatives Fa. hongkongensis and P. salmonis as an outgroup was constructed using the MEGA5 software[68] based on the numbers of substitutions per nucleotide site in the genome sequences of all strains. The Complete Deletion Method was used for handling alignment gaps.
Estimation of evolutionary distance
Average nucleotide identity (ANI) was calculated by pairwise genome comparisons with the MUMmer and BLAST algorithms using JSpecies v1.2.1[69]. A threshold of >95% identity was used to classify genomes as the same species.
IS element finding
This study focuses on newly sequenced genomes of previously uncharacterized Francisella species and subspecies (Additional file5). Potential IS elements were identified using the BLASTN program (no low-complexity filtering), by querying genome sequences using the ISFinder tool[70] in the IS database and querying flanking regions of the draft genome contigs against the NCBI ‘nt’ database. Altogether, 41 IS elements were found to be present (e-value < 10-2) in the included genomes. Pairwise BLAST alignments of both left and right flanking regions, and full length IS sequence elements, were constructed for all genomes. The results were evaluated by e-value (<10-3), identity (90%), bit score (>40) alignment length and proximity to contig ends. Inherently, de novo assembly of next-generation sequence data implies problems in resolving repeat structures, resulting in an increased risk of underestimating the IS element copy number.
Protein orthologs
Forty-five genomes of Francisella and two genetic relatives were downloaded from IMG/ER (Table 1 and Additional file 6). Coding sequences were extracted from GenBank files and orthologs were determined using OrthoMCL with a BLAST e-value cut-off of 10-5 and an inflation parameter of 1.5. We used the OrthoMCL output to construct a table describing genome gene content (Additional file7) that were used for all pan- and core gene analysis.
Heat maps showing the presence of genes in the Francisella genus were generated using custom code in the statistical software R.
Analysis of recombination
To infer recombination events in the Francisella dataset, three recombination detection methods were used: pairwise homoplasy index (Phi)[71], neighbour similarity score (NSS)[72] and maximum chi-square (MaxChi)[73]. These methods are conveniently implemented in the PHI Package[71]. A subset of 742 genes (i.e. the core genome excluding multiple copies of genes) was tested for recombination detection on the entire data as well as on individual lineages in the phylogeny (subspecies populations). It should be mentioned that all recombination methods used here do not correct for multiple testing, which may result in an over-estimation of the presence of recombination, in particular for the full data analysis (comprising 45 isolates in total).
Disclaimer
The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies, either expressed or implied, of the U.S. Department of Homeland Security or the Swedish Civil Contingencies Agency.
Abbreviations
- ANI:
-
Average nucleotide identity
- FLE:
-
Francisella-like endosymbionts
- MaxChi:
-
Maximum chi-square
- NSS:
-
Neighbour similarity score
- Phi:
-
Pair wise homoplasy index.
References
Darling RG, Catlett CL, Huebner KD, Jarrett DG: Threats in bioterrorism. I: CDC category A agents. Emerg Med Clin North Am. 2002, 20: 273-309. 10.1016/S0733-8627(02)00005-6.
Johansson A, Celli J, Conlan W, Elkins KL, Forsman M, Keim PS, Larsson P, Manoil C, Nano FE, Petersen JM, Sjöstedt A: Objections to the transfer of Francisella novicida to the subspecies rank of Francisella tularensis. Int J Syst Evol Microbiol. 2010, 60: 1717-1718. 10.1099/ijs.0.022830-0. author reply 1718–20
Busse H-J, Huber B, Anda P, Escudero R, Scholz HC, Seibold E, Splettstoesser WD, Kämpfer P: Objections to the transfer of Francisella novicida to the subspecies rank of Francisella tularensis - response to Johansson et al. Int J Syst Evol Microbiol. 2010, 60: 1718-1720.
Keim P, Johansson A, Wagner DM: Molecular epidemiology, evolution, and ecology of Francisella. Ann N Y Acad Sci. 2007, 1105: 30-66. 10.1196/annals.1409.011.
Dienst FT: Tularemia: a perusal of three hundred thirty-nine cases. The Journal of the Louisiana State Medical Society: official organ of the Louisiana State Medical Society. 1963, 115: 114-127.
Sandström G, Sjöstedt A, Forsman M, Pavlovich NV, Mishankin BN: Characterization and classification of strains of Francisella tularensis isolated in the central Asian focus of the Soviet Union and in Japan. J Clin Microbiol. 1992, 30: 172-175.
Olsufiev NG, Meshcheriakova IS: Subspecific Taxonomy of Francisella tularensis McCoy and Chapin 1912. Int J Syst Bacteriol. 1983, 33: 872-874. 10.1099/00207713-33-4-872.
Hollis DG, Weaver RE, Steigerwalt AG, Wenger JD, Moss CW, Brenner DJ: Francisella philomiragia comb. nov. (formerly Yersinia philomiragia) and Francisella tularensis biogroup novicida (formerly Francisella novicida) associated with human disease. J Clin Microbiol. 1989, 27: 1601-1608.
Wenger JD, Hollis DG, Weaver RE, Baker CN, Brown GR, Brenner DJ, Broome CV: Infection caused by Francisella philomiragia (formerly Yersinia philomiragia). A newly recognized human pathogen. Ann Intern Med. 1989, 110: 888-892.
Mailman TL, Schmidt MH: Francisella philomiragia adenitis and pulmonary nodules in a child with chronic granulomatous disease. The Canadian journal of infectious diseases & medical microbiology. Journal canadien des maladies infectieuses et de la microbiologie médicale / AMMI Canada. 2005, 16: 245-248.
Brevik ØJ, Ottem KF, Nylund A: Multiple-locus, variable number of tandem repeat analysis (MLVA) of the fish-pathogen Francisella noatunensis. BMC Vet Res. 2011, 7: 5-10.1186/1746-6148-7-5.
Ottem KF, Nylund A, Karlsbakk E, Friis-Møller A, Kamaishi T: Elevation of Francisella philomiragia subsp. noatunensis Mikalsen et al. (2007) to Francisella noatunensis comb. nov. [syn. Francisella piscicida Ottem et al. (2008) syn. nov.] and characterization of Francisella noatunensis subsp. orientalis subsp. nov. J Appl Microbiol. 2009, 106: 1231-1243. 10.1111/j.1365-2672.2008.04092.x.
Colquhoun DJ, Duodu S: Francisella infections in farmed and wild aquatic organisms. Vet Res. 2011, 42: 47-10.1186/1297-9716-42-47.
Birkbeck T, Feist S, Verner-Jeffreys D: Francisella infections in fish and shellfish. J Fish Dis. 2011, 34: 173-187. 10.1111/j.1365-2761.2010.01226.x.
Olsen AB, Mikalsen J, Rode M, Alfjorden A, Hoel E, Straum-Lie K, Haldorsen R, Colquhoun DJ: A novel systemic granulomatous inflammatory disease in farmed Atlantic cod, Gadus morhua L., associated with a bacterium belonging to the genus Francisella. J Fish Dis. 2006, 29: 307-311. 10.1111/j.1365-2761.2006.00714.x.
Ottem KF, Nylund A, Karlsbakk E, Friis-Møller A, Krossøy B: Characterization of Francisella sp., GM2212, the first Francisella isolate from marine fish, Atlantic cod (Gadus morhua). Arch Microbiol. 2007, 187: 343-350. 10.1007/s00203-006-0198-1.
Kamaishi T, Fukuda Y, Nishiyama M, Kawakami H, Matsuyama T, Yoshinaga T, Oseko N: Identification and pathogenicity of intracellular Francisella bacterium in three-line grunt Parapristipoma trilineatum. Fish Pathology. 2005, 40: 67-72. 10.3147/jsfp.40.67.
Birkbeck TH, Bordevik M, Frøystad MK, Baklien A: Identification of Francisella sp. from Atlantic salmon, Salmo salar L., in Chile. J Fish Dis. 2007, 30: 505-507. 10.1111/j.1365-2761.2007.00837.x.
Barns SM, Grow CC, Okinaka RT, Keim P, Kuske CR: Detection of diverse new Francisella-like bacteria in environmental samples. Appl Environ Microbiol. 2005, 71: 5494-5500. 10.1128/AEM.71.9.5494-5500.2005.
Dillon J, McMath L, Trout A: Seasonal changes in bacterial diversity in the Salton Sea. Hydrobiologia. 2009, 632: 49-64. 10.1007/s10750-009-9827-4.
Kugeler KJ, Mead PS, McGowan KL, Burnham JM, Hogarty MD, Ruchelli E, Pollard K, Husband B, Conley C, Rivera T, Kelesidis T, Lee WM, Mabey W, Winchell JM, Stang HL, Staples JE, Chalcraft LJ, Petersen JM: Isolation and characterization of a novel Francisella sp. from human cerebrospinal fluid and blood. J Clin Microbiol. 2008, 46: 2428-2431. 10.1128/JCM.00698-08.
Petersen JM, Carlson J, Yockey B, Pillai S, Kuske C, Garbalena G, Pottumarthy S, Chalcraft L: Direct isolation of Francisella spp. from environmental samples. Lett Appl Microbiol. 2009, 48: 663-667.
Schrallhammer M, Schweikert M, Vallesi A, Verni F, Petroni G: Detection of a novel subspecies of Francisella noatunensis as endosymbiont of the ciliate Euplotes raikovi. Microb Ecol. 2011, 61: 455-464. 10.1007/s00248-010-9772-9.
Scoles GA: Phylogenetic analysis of the Francisella-like endosymbionts of Dermacentor ticks. J Med Entomol. 2004, 41: 277-286. 10.1603/0022-2585-41.3.277.
Weiss E, Myers WF, Suitor EC, Neptune EM: Respiration of a Rickettsialike Microorganism, Wolbachia Persica. J Infect Dis. 1962, 110: 155-164. 10.1093/infdis/110.2.155.
Noda H, Munderloh UG, Kurtti TJ: Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl Environ Microbiol. 1997, 63: 3926-3932.
Qu P, Deng X, Zhang J, Chen J, Zhang J, Zhang Q, Xiao Y, Chen S: Identification and characterization of the Francisella sp. strain 08HL01032 isolated in air condition systems. Wei sheng wu xue bao = Acta microbiologica sinica. 2009, 49: 1003-1110.
Kugeler KJ, Gurfield N, Creek JG, Mahoney KS, Versage JL, Petersen JM: Discrimination between Francisella tularensis and Francisella-like endosymbionts when screening ticks by PCR. Appl Environ Microbiol. 2005, 71: 7594-7597. 10.1128/AEM.71.11.7594-7597.2005.
Lau KWK, Ren J, Fung M-C, Woo PCY, Yuen K-Y, Chan KKM, Qian P-Y, Wong P-K, Wu M: Fangia hongkongensis gen. nov., sp. nov., a novel gammaproteobacterium of the order Thiotrichales isolated from coastal seawater of Hong Kong. Int J Syst Evol Microbiol. 2007, 57: 2665-2669. 10.1099/ijs.0.65156-0.
Fryer JL, Lannan CN, Giovannoni SJ, Wood ND: Piscirickettsia salmonis gen. nov., sp. nov., the causative agent of an epizootic disease in salmonid fishes. Int J Syst Bacteriol. 1992, 42: 120-126. 10.1099/00207713-42-1-120.
Suitor EC, Weiss E: Isolation of a Rickettsialike Microorganism (Wolbachia persica, n. sp.) from Argas persicus (Oken). J Infect Dis. 1961, 108: 95-106. 10.1093/infdis/108.1.95.
Fryer JL, Lannan CN LHG, Larenas JJ, Smith PA: Isolation of a rickettsiales-like organism from diseased coho salmon Oncorhynchus kisutch in Chile. Fish pathology. 1990, 25: 107-114. 10.3147/jsfp.25.107.
Whipp MJ, Davis JM, Lum G, de Boer J, Zhou Y, Bearden SW, Petersen JM, Chu MC, Hogg G: Characterization of a novicida-like subspecies of Francisella tularensis isolated in Australia. J Med Microbiol. 2003, 52: 839-842. 10.1099/jmm.0.05245-0.
Siddaramappa S, Challacombe JF, Petersen JM, Pillai S, Hogg G, Kuske CR: Common ancestry and novel genetic traits of Francisella novicida-like isolates from North America and Australia as revealed by comparative genomic analyses. Appl Environ Microbiol. 2011, 77: 5110-5122. 10.1128/AEM.00337-11.
Darling AE, Mau B, Perna NT: progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010, 5: e11147-10.1371/journal.pone.0011147.
Larsson P, Elfsmark D, Svensson K, Wikström P, Forsman M, Brettin T, Keim P, Johansson A: Molecular evolutionary consequences of niche restriction in Francisella tularensis, a facultative intracellular pathogen. PLoS Pathog. 2009, 5: e1000472-10.1371/journal.ppat.1000472.
Marshall SH, Henríquez V, Gómez FA, Cárdenas C: ISPsa2, the first mobile genetic element to be described and characterized in the bacterial facultative intracellular pathogen Piscirickettsia salmonis. FEMS Microbiol Lett. 2011, 314: 18-24. 10.1111/j.1574-6968.2010.02132.x.
Champion MD, Zeng Q, Nix EB, Nano FE, Keim P, Kodira CD, Borowsky M, Young S, Koehrsen M, Engels R, Pearson M, Howarth C, Larson L, White J, Alvarado L, Forsman M, Bearden SW, Sjöstedt A, Titball R, Michell SL, Birren B, Galagan J: Comparative genomic characterization of Francisella tularensis strains belonging to low and high virulence subspecies. PLoS Pathog. 2009, 5: e1000459-10.1371/journal.ppat.1000459.
Nübel U, Reissbrodt R, Weller A, Grunow R, Porsch-Ozcürümez M, Tomaso H, Hofer E, Splettstoesser W, Finke E-J, Tschäpe H, Witte W: Population structure of Francisella tularensis. J Bacteriol. 2006, 188: 5319-5324. 10.1128/JB.01662-05.
Forsman M, Sandström G, Sjöstedt A: Analysis of 16 S ribosomal DNA sequences of Francisella strains and utilization for determination of the phylogeny of the genus and for identification of strains by PCR. Int J Syst Bacteriol. 1994, 44: 38-46. 10.1099/00207713-44-1-38.
Escudero R, Toledo A, Gil H, Kovácsová K, Rodríguez-Vargas M, Jado I, García-Amil C, Lobo B, Bhide M, Anda P: Molecular method for discrimination between Francisella tularensis and Francisella-like endosymbionts. J Clin Microbiol. 2008, 46: 3139-3143. 10.1128/JCM.00275-08.
Berrada ZL, Telford SR: Diversity of Francisella species in environmental samples from Martha’s Vineyard, Massachusetts. Microb Ecol. 2010, 59: 277-283. 10.1007/s00248-009-9568-y.
Huber B, Escudero R, Busse H-J, Seibold E, Scholz HC, Anda P, Kämpfer P, Splettstoesser WD: Description of Francisella hispaniensis sp. nov., isolated from human blood, reclassification of Francisella novicida (Larson et al. 1955) Olsufiev et al. 1959 as Francisella tularensis subsp. novicida comb. nov. and emended description of the genus Franc. Int J Syst Evol Microbiol. 2010, 60: 1887-1896. 10.1099/ijs.0.015941-0.
Alcaraz LDD, Moreno-Hagelsieb G, Eguiarte LE, Souza V, Herrera-Estrella L, Olmedo G: Understanding the evolutionary relationships and major traits of Bacillus through comparative genomics. BMC Genomics. 2010, 11: 332-10.1186/1471-2164-11-332.
Rohmer L, Fong C, Abmayr S, Wasnick M, Larson Freeman TJ, Radey M, Guina T, Svensson K, Hayden HS, Jacobs M, Gallagher LA, Manoil C, Ernst RK, Drees B, Buckley D, Haugen E, Bovee D, Zhou Y, Chang J, Levy R, Lim R, Gillett W, Guenthener D, Kang A, Shaffer SA, Taylor G, Chen J, Gallis B, D’Argenio DA, Forsman M, Olson MV, Goodlett DR, Kaul R, Miller SI, Brittnacher MJ: Comparison of Francisella tularensis genomes reveals evolutionary events associated with the emergence of human pathogenic strains. Genome Biol. 2007, 8: R102-10.1186/gb-2007-8-6-r102.
Georgiades K, Raoult D: Genomes of the most dangerous epidemic bacteria have a virulence repertoire characterized by fewer genes but more toxin-antitoxin modules. PLoS One. 2011, 6: e17962-10.1371/journal.pone.0017962.
Hood AM: Virulence factors of Francisella tularensis. J Hyg. 1977, 79: 47-60. 10.1017/S0022172400052840.
McLendon MK, Apicella MA, Allen L-AA: Francisella tularensis: taxonomy, genetics, and Immunopathogenesis of a potential agent of biowarfare. Annu Rev Microbiol. 2006, 60: 167-185. 10.1146/annurev.micro.60.080805.142126.
Prior JL, Prior RG, Hitchen PG, Diaper H, Griffin KF, Morris HR, Dell A, Titball RW: Characterization of the O antigen gene cluster and structural analysis of the O antigen of Francisella tularensis subsp. tularensis. J Med Microbiol. 2003, 52: 845-851. 10.1099/jmm.0.05184-0.
Didelot X, Maiden MCJ: Impact of recombination on bacterial evolution. Trends Microbiol. 2010, 18: 315-322. 10.1016/j.tim.2010.04.002.
Lefébure T, Bitar PDP, Suzuki H, Stanhope MJ: Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol Evol. 2010, 2: 646-655. 10.1093/gbe/evq048.
Joseph B, Schwarz RF, Linke B, Blom J, Becker A, Claus H, Goesmann A, Frosch M, Müller T, Vogel U, Schoen C: Virulence evolution of the human pathogen Neisseria meningitidis by recombination in the core and accessory genome. PLoS One. 2011, 6: e18441-10.1371/journal.pone.0018441.
Achtman M, Morelli G, Zhu P, Wirth T, Diehl I, Kusecek B, Vogler AJ, Wagner DM, Allender CJ, Easterday WR, Chenal-Francisque V, Worsham P, Thomson NR, Parkhill J, Lindler LE, Carniel E, Keim P: Microevolution and history of the plague bacillus, Yersinia pestis. Proc Natl Acad Sci USA. 2004, 101: 17837-17842. 10.1073/pnas.0408026101.
Van Ert MN, Easterday WR, Huynh LY, Okinaka RT, Hugh-Jones ME, Ravel J, Zanecki SR, Pearson T, Simonson TS, U’Ren JM, Kachur SM, Leadem-Dougherty RR, Rhoton SD, Zinser G, Farlow J, Coker PR, Smith KL, Wang B, Kenefic LJ, Fraser-Liggett CM, Wagner DM, Keim P: Global genetic population structure of Bacillus anthracis. PLoS One. 2007, 2: e461-10.1371/journal.pone.0000461.
Liu X, Gutacker MM, Musser JM, Fu Y-X: Evidence for recombination in Mycobacterium tuberculosis. J Bacteriol. 2006, 188: 8169-8177. 10.1128/JB.01062-06.
Smith JM, Smith NH, O’Rourke M, Spratt BG: How clonal are bacteria?. Proc Natl Acad Sci USA. 1993, 90: 4384-4388. 10.1073/pnas.90.10.4384.
Ahmed N: A flood of microbial genomes-do we need more?. PLoS One. 2009, 4: e5831-10.1371/journal.pone.0005831.
Enserink M: Anthrax investigation. Full-genome sequencing paved the way from spores to a suspect. Science. 2008, 321: 898-899. 10.1126/science.321.5891.898. New York, N.Y
Sandström G, Tärnvik A, Wolf-Watz H, Löfgren S: Antigen from Francisella tularensis: nonidentity between determinants participating in cell-mediated and humoral reactions. Infect Immun. 1984, 45: 101-106.
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18: 821-829. 10.1101/gr.074492.107.
Zerbino DR, McEwen GK, Margulies EH, Birney E: Pebble and rock band: heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PLoS One. 2009, 4: e8407-10.1371/journal.pone.0008407.
Markowitz VM, Mavromatis K, Ivanova NN, Chen I-MA, Chu K, Kyrpides NC: IMG ER: A system for microbial genome annotation expert review and curation. Bioinformatics Oxford England. 2009, 25: 2271-2278. 10.1093/bioinformatics/btp393.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM: The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37: D141-D145. 10.1093/nar/gkn879.
Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, Bandela AM, Cardenas E, Garrity GM, Tiedje JM: The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007, 35: D169-D172. 10.1093/nar/gkl889.
Stamatakis A: RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics (Oxford, England). 2006, 22: 2688-2690. 10.1093/bioinformatics/btl446.
Sjödin A, Svensson K, Ohrman C, Ahlinder J, Lindgren P, Duodu S, Hnath J, Burans JP, Johansson A, Colquhoun DJ, Larsson P, Forsman M: Genome characterisation of the genus Francisella. TreeBASE,http://purl.org/phylo/treebase/phylows/study/TB2:S12723,
Anwar N, Hunt E: Improved data retrieval from TreeBASE via taxonomic and linguistic data enrichment. BMC Evol Biol. 2009, 9: 93-10.1186/1471-2148-9-93.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011, 11: 2731-2739.
Richter M, Rosselló-Móra R: Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 2009, 106: 19126-19131. 10.1073/pnas.0906412106.
Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M: ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34: D32-D36. 10.1093/nar/gkj014.
Bruen TC, Philippe H, Bryant D: A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006, 172: 2665-2681.
Jakobsen IB, Easteal S: A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Computer applications in the biosciences: CABIOS. 1996, 12: 291-295.
Smith J: Analyzing the mosaic structure of genes. J Mol Evol. 1992, 34: 126-129.
Acknowledgments
Research for this publication was funded by the U.S. Department of Homeland Security and the Swedish Civil Contingencies Agency pursuant to the Agreement between the Government of the United States of America and the Government of the Kingdom of Sweden on Cooperation in Science and Technology for Homeland Security Matters.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
No competing interests for any of the authors exist.
Authors’ contributions
AS assembled all genomes and performed analysis of genome and gene content. JA performed the 16 S rRNA alignment, analysis of over-represented COGs and recombination. CO performed the analysis of average nucleotide identity, IS elements and clade specific genes. SD and DJC provided Francisella isolates that infect fish and knowledge of fish pathogenic Francisella, and contributed to completion of the manuscript. PLA and MF conceived the study. AS, KS, JA, PLI, AJ, PLA, MF drafted the manuscript. All authors read and approved the manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sjödin, A., Svensson, K., Öhrman, C. et al. Genome characterisation of the genus Francisella reveals insight into similar evolutionary paths in pathogens of mammals and fish. BMC Genomics 13, 268 (2012). https://doi.org/10.1186/1471-2164-13-268
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-268