
Abstract
Background
Scanning the genome with high density SNP markers has become a standard approach for identifying regions of the genome showing substantial between-population genetic differentiation, and thus evidence of diversifying selection. Such regions may contain genes of large phenotypic effect. However, few studies have attempted to address the power or efficacy of such an approach.
Results
In this study, the patterns of allele frequency differences between two cattle breeds based on the Bovine HapMap study were compared with statistical evidence for QTL based on a linkage mapping study of an experimental population formed by a cross between the same breeds. Concordance between the two datasets was seen for chromosomes carrying QTL with strong statistical support, such as BTA5 and BTA18, which carry genes associated with coat color. For these chromosomes, there was a correspondence between the strength of the QTL signal along the chromosome and the degree of genetic differentiation between breeds. However, such an association was not seen in a broader comparison that also included chromosomes carrying QTL with lower significance levels. In addition, other chromosomal regions with substantial QTL effects did not include markers showing extreme between-breed genetic differentiation. Furthermore, the overall consistency between the two studies was weak, with low genome-wide correlation between the statistical values obtained in the linkage mapping study and between-breed genetic differentiation from the HapMap study.
Conclusions
These results suggest that genomic diversity scans are capable of detecting regions associated with qualitative traits but may be limited in their power to detect regions associated with quantitative phenotypic differences between populations, which may depend on the marker resolution of the study and the level of LD in the populations under investigation.
Similar content being viewed by others

Background
With the development of dense genome-wide marker panels for many species, it is becoming common to use these markers to characterize genetic diversity across the genome. Such genomic scans are designed to identify regions where selection has acted and which therefore, may contain genes of large phenotypic effect. The rationale is that, even without phenotypic information, one can use patterns of genetic variation to highlight genomic regions under selection. However, the power and reliability of these studies has not been assessed because usually there is no independent data set against which the results can be compared. The current study was designed to address this point by examining the concordance between a genomic diversity dataset comprising genome-wide SNP data from two cattle breeds and a set of linkage mapping results from a study in an F2/Backcross population bred from founders of these same breeds.
Using these datasets, an overall concordance between level of genetic differentiation and linkage mapping signal was evaluated. In addition, the more specific question to be addressed is whether regions with large allele frequency differences between the breeds are more likely to contain genes controlling phenotypes that differ between the breeds than regions with small allele frequency differences. However, because not all of the traits that might distinguish the breeds can be measured, the question was addressed in the converse direction in this study, i.e. whether regions of the genome where QTL have been identified are characterized by larger SNP allele frequency differences compared with other genomic regions.
Methods
Interval mapping experiment
Source population
A three-generation Charolais x Holstein cattle herd was bred from seven founder Charolais sires, which were mated with Holstein cows to produce 137 F1 animals. From these, a total of 501 second generation cross-bred animals were produced, 315 were F2 individuals from crossing eight F1 sires with F1 cows, and 186 were reciprocal backcrosses of the F1 animals with the founder breeds (88 Charolais backcrosses, CB1, and 98 Holstein backcrosses, HB1). The second generation animals were measured for a wide variety of traits [1–5] (results for dairy traits have not yet been published). For the analysis presented here, a subset of traits related to growth, dairy and meat production and coat colour were selected, as these traits were expected to have been under the strongest divergent selection in the two founder breeds. Specific traits were selected that were representative of these trait groups but were not strongly correlated with each other. These included size and growth rate, coat color, carcass conformation characteristics, detailed carcass measurements, meat quality traits (as assessed by a trained taste panel), chemical composition of meat and milk, milk yield and udder characteristics (Additional File 1, Table S1). Other trait groups were not included (e.g. behavioral and immunological traits). All work in this study involving the use of animals was designed under guidance of and carried out under a United Kingdom Government Home Office animal experimentation license, under the supervision of the local Home Office inspector, and all procedures were formally inspected annually.
Microsatellite marker data
A panel of 165 microsatellite markers were genotyped in the population; 139 of these were selected at random from across the autosomes and the other 26 were added later in regions on nine chromosomes where QTL were detected following initial analysis [4]. Linkage maps were constructed using CRIMAP 2.4 [6]. The maps obtained were compared with the latest published version of the bovine linkage map [7] and found to be consistent.
Microsatellite markers used to derive the linkage maps were then mapped against the Baylor bovine genome assembly Btau_4.0 [8] in order to derive genomic sequence coordinates, either by extracting pre-computed locations from Ensembl (http://www.ensembl.org/Bos_taurus) (88 markers) or by megaBLAST [9] sequence similarity searches using flanking sequence (64 markers). The sequence positions of the remaining 13 markers, for which a definitive megaBLAST-based location could not be identified within the expected genomic location, were assigned estimated positions based on their proximity to flanking markers on the genetic linkage map.
Interval mapping analysis
The QTL analysis was performed using the QTL Express software [10], which implements the linear regression method of Knott and Haley [11] assuming the founder lines to be fixed for alternative alleles at the QTL loci. A single QTL model analysis with additive and dominance effects was fitted at intervals of 100,000 basepairs along each chromosome according to the maps described above. Fixed effects considered in the linear model and the numbers of phenotypes analyzed are given in Additional file 1, Table S1. Fixed effects included genetic composition (F2, CB1, and HB1) for all traits as well as sex, birth cohort, dairy cohort, taste panel grouping, feeding regime and background coat colour, as detailed in Additional file 1, Table S1. The F-ratio for each combination of trait and chromosomal position was recorded. The maximum of F-ratios (Fmax) over all traits at each chromosomal position was used as the test statistic for the interval mapping results.
Bovine HapMap study
Genomic diversity data
Genome-wide SNP data was obtained from the Bovine HapMap Consortium [12] and is subsequently referred to as HapMap. Details of the HapMap samples, SNP markers, genotypes and quality control have been described previously [12]. Briefly, DNA samples for individuals from Bos taurus breeds (as unrelated as possible, based on pedigree), plus sire-dam-offspring trios for data quality assessment, were collected and genotyped for 33,851 biallelic SNPs distributed across the genome (encompassing 3.2 Gb for Assembly Btau_4.0). The majority of SNPs were discovered by comparing shotgun genome sequence reads for cows from six breeds (Angus, Brahman, Holstein, Jersey, Limousin and Norwegian Red) to the Hereford reference genome sequence. SNP density was fairly uniform across the genome except that higher densities of markers were positioned on three chromosomes (BTA6, 14 and 25: average: ~27 per Mbp compared to ~10 per Mbp for the other 26 autosomes).
Only the genotype data for 49 Holstein and 20 Charolais individuals included in the HapMap project was considered (excluding calves that formed part of family trios and one Charolais that was missing a majority of genotypes). Loci with low minor allele frequencies, high error rates, evidence of deviation from Hardy-Weinberg Equilibrium or excessive missing data were removed from the data set (as in the HapMap analysis, [12]), as were loci that could not be positioned on Assembly Btau_4.0 or were found on the X chromosome. This left 31,312 SNPs covering the autosomes. SNP positions along the genome were based on Assembly Btau_4.0.
Measures of marker differentiation
To calculate genetic differentiation from the HapMap data, the following protocol was followed. The frequency of one allele was calculated for each SNP for each of the two breeds. Two statistics were then used to characterize the differences between the two breeds. First the absolute value of the difference in allele frequencies between the Holstein and Charolais breed groups (δ) was calculated at each SNP position (equivalent to δ c for two alleles [13]). Secondly, estimates of pairwise FST (θ) were calculated for each SNP [14, 15], adjusting for different sample sizes as in Ref. [16]. If FST estimates were negative, they were set to 0 [16]. The dataset was restricted to those markers for which FST was defined (i.e. the average allele frequency across the two breeds was not 0 or 1). Sequence positions for each marker were rounded to the nearest 100,000 bp and an average value of δ (or FST) was calculated for all markers with the same rounded position. Two smoothed statistics, MA_δ and MA_ FST, were calculated as moving averages of 11 δ or FST values, respectively, centered on each rounded position, i.e. including that position and its 10 flanking positions (excluding the first and last 5 positions of the chromosome).
Data Analysis
Correlation coefficients were calculated between Fmax values from interval mapping and each of the four measures of SNP breed differences from the HapMap study (δ, FST, MA_δ and MA_ FST) over the entire genome and for each chromosome. Then chromosomal positions were grouped according to whether their Fmax value was above (highF) or below (lowF) a given threshold. Three groupings were defined using the approximate cut-offs for chromosome-wise and genome-wide significance previously calculated by permutation testing for this data (chromosome-wise, 0.05, F = 5; chromosome-wise, 0.01, F = 7; genome-wide, 0.01, F = 10; [1–4]). For each of these three thresholds, the mean δ and FST values (and their respective moving averages) were calculated for the highF and lowF groups on each chromosome. Numbers of chromosomes for which any Fmax values exceeded the thresholds decreased with threshold values (27 chromosomes for F = 5, 13 chromosomes for F = 7 and 6 chromosomes for F = 10). Wilcoxon signed rank tests were used to test whether the mean values of δ and FST (and their respective moving averages) for each chromosome differed for the highF and lowF groups, for each of the three F-thresholds. A one-tailed test was applied, i.e. the alternative hypothesis was that the highF group had greater δ or FST values than the lowF group.
Results
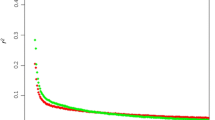
The genome-wide correlation between FST and δ was high (0.8764) and results for both measures were generally similar. Thus, results are presented only for δ (and MA_δ) except where qualitative differences were seen between results for δ and FST. Visual inspection did not show a consistent trend between Fmax and δ across the genome, although such a trend was seen for some chromosomes. Figure 1 shows Fmax and MA_δ across the chromosomes with the largest QTL in the study, on BTA5, 6 and 18 (all chromosomes are shown in Additional file 2, Figure S1). The genome-wide correlation coefficient between Fmax and δ was 0.0603 and that between Fmax and MA_δ was 0.1672. The correlations varied considerably across chromosomes (Table 1), with half of the chromosomes (14/29) showing negative correlations between Fmax and δ (and MA_δ). BTA5, 6 and 18 had positive correlations although the correlation coefficient was low for BTA6. For the chromosomes with a positive correlation between Fmax and δ, correlations were consistently higher between Fmax and MA_δ than between Fmax and δ (this was also true for FST except for BTA6 and BTA26, for which the correlations were very low). The highest chromosome-wide positive correlations between Fmax and δ were (in descending order) for BTA28, 18, 14, 5, 2 and 7 (for the correlations between Fmax and MA_δ, the order of BTA28 and BTA18 and that of BTA8 and BTA2 were switched but otherwise the same).

Patterns of genetic differentiation and linkage mapping results for chromosomes with large QTL . Patterns of the moving average of allele frequency differences between Holstein and Charolais cattle (MA_δ, represented by diamonds) and the maximum F-ratio of the linkage mapping study with Holstein and Charolais founders (Fmax, represented by the curve) across BTA5, 6 and 18. The horizontal lines show the average values of MA_δ for each chromosome. For BTA5, the black triangle on the x-axis indicates the position of the SILV gene. For BTA18, the black triangle indicates the position of the MC1R gene.
To test whether SNP ascertainment bias, and in particular the fact that the majority of SNPs were detected in Holsteins, may explain the lack of concordance between the interval mapping and SNP frequency, the effect of breed-of-origin on the value of δ was examined. This statistic was significantly higher (p < 0.001) for the SNPs detected in Holsteins (δ = 0.1565) than that detected in all other breeds (0.1372). The correlation coefficient between δ and Fmax values was recalculated as described above, for the 14,132 segregating SNPs that were detected in breeds other than Holsteins. For that subset of data, the overall correlation coefficient between Fmax and δ was 0.0427, lower than that calculated for all SNPs, thus the inclusion of the Holstein-derived SNPs did not reduce the overall correlation between Fmax and δ. However, for the non-Holstein SNP data, fewer chromosomes (13/29) had negative correlations between Fmax and δ (12/29 for MA_δ) than was found considering all SNPs (14/29).
The comparison of δ and MA_δ distributions above and below Fmax thresholds revealed little difference between regions with high and low Fmax values for the lowest Fmax threshold (F = 5), but there were differences for the higher thresholds (F = 7 and F = 10). Wilcoxon signed rank tests revealed significant differences in the mean δ values between highF and lowF positions for Fmax thresholds of both 7 and 10 (p < 0.05), but not for the threshold of 5. The same results were seen for comparisons of mean MA_δ values between highF and lowF positions (for FST and MA_ FST, only tests with threshold F = 10 were significant). These results are presented graphically in plots of the chromosome-wide mean MA_δ values for highF versus those for lowF positions (Figure 2). For F = 5, slightly more than half of the chromosomes (15/27) had greater mean MA_δ values for highF than lowF groups. For the cut-off of F = 7, nine out of 13 chromosomes had greater mean MA_δ values for highF than lowF groups. For the F = 10 cut-off, five out of the six chromosomes for which there were Fmax values greater than 10 had greater mean MA_δ values for highF than lowF groups. These five chromosomes included those with the highest Fmax values in this study (255.52, 63.67 and 39.86 for BTA5, 6 and 18, respectively), as well as BTA10 and 12 for which the highest Fmax values were between 10 and 12 (BTA22, for which the mean MA_δ value was greater for lowF than highF groups, also had its highest Fmax value in this range).

Genetic differentiation for positions with significant versus non-significant linkage mapping results for three significance thresholds. Mean moving average of allele frequency differences (MA_δ) values across chromosomal positions above and below linkage mapping maximum F-ratio (Fmax) thresholds of 5, 7, and 10. Each circle represents an individual chromosome; only chromosomes for which there were Fmax values exceeding the relevant threshold are included.
With regard to extreme values of the genetic differentiation measures across the genome: the top 1% of the δ distribution (values > 0.4981) covered 27 of the 29 autosomes. Of the 124 loci in the top 1% of the δ distribution, the greatest number of markers were found on BTA1 (9 positions), 5 (13 positions), 6 (10 positions) and 8 (9 positions). When positions were ranked by MA_δ values rather than by δ (Additional file 3, Table S2), the top 1% (> 0.2622) were present on 18 of the 29 autosomes, with the majority on BTA5 (20 positions), 6 (15 positions), 13 (15 positions) and 20 (13 positions). The most extreme values of MA_δ (top 0.1%) were found on BTA5 (5 positions, 61-64 Mbp), BTA6 (5 positions, 77-78 Mbp), and BTA18 (2 positions, 7.2-7.3 Mbp). The highest δ, MA_δ and MA_ FST values were found at 77.4 Mbp on BTA6. The highest FST value (and third highest δ value) was found at 63.6 Mbp on BTA5.
Discussion
The objective of the analysis reported here was to assess if there was consistency between the pattern of allele frequency differences between two cattle breeds and the pattern of statistical evidence for QTL identified in a cross-bred population of the same breeds. The motivation for this was to test whether the current trend to look for signatures of selection in data from high density genome scans could detect the genomic regions controlling phenotypic traits identified by traditional linkage mapping techniques. The results suggest that genomic differentiation patterns can pick up very large, qualitative phenotypic effects, but may have limitations as predictors of genomic regions associated with smaller, quantitative phenotypic differences.
In the comparison of genomic data from two cattle breeds carried out in the present study, allelic differentiation was amplified in regions where the largest genetic effects were detected by QTL mapping (BTA5 and 18), however, overall, there was low correlation between the test statistics from the two analyses. The BTA5 and 18 QTL were associated with Mendelian loci influencing coat colour: the Charolais-derived allele in a region of BTA5 (close to or at the SILV gene, 61.9 Mbp) causes complete dilution of coat pigmentation and gives this breed its characteristic white coat [3, 17] and the BTA18 QTL is associated with the MC1R locus (13.8 Mbp), which is the main regulator of the switch between red and black coat color pigments [18] and associated with black and red coat color in cattle, where the black allele is dominant [19, 20]. Charolais carry the red allele at MC1R, although it is not visible in the coat because of the dilution effect, whereas black and white Holsteins are predominantly homozygous for the black allele, with a low frequency of heterozygotes [3, 21]. The correlations between Fmax and MA_δ were 0.421 on BTA5 and 0.651 on BTA18, and as seen in Figure 1, the QTL peaks overlap with peaks in the MA_δ graphs. Furthermore, the positions of the known pigmentation genes are very close to the peak MA_δ values, particularly for the SILV gene on BTA5. However, there was only a weak positive correlation on BTA6 (0.075), where the third highest F-ratio in the linkage mapping study was found. The QTL corresponding to the high F-ratio was associated with birth weight and carcass-related traits [4] and was coincident with QTL for similar traits reported in other studies [22, 23]. As seen in Figure 1, there is concordance between this QTL peak and a region of high MA_δ although this was not in the top 1% of the distribution. The chromosome-wide correlation on BTA6 was reduced, however, because there are other regions further downstream with even higher values of MA_δ but where there is only weak evidence of QTLs, for bone weight and average feed intake. The regions of high MA_δ may be associated with traits that were not measured in the linkage mapping study or the linkage mapping study may not have been powerful enough to detect associations in this region. In the 2-Mbp region centered at 77.4 Mbp (the peak δ and MA_δ position), there are no annotated genes in the bovine genome, although there is homology to some expressed genes in the human genome (http://genome.ucsc.edu). The KIT gene, which is associated with the level of white spotting in Holstein cattle and other mammals [24], is located at 72.8 Mbp on this chromosome, nearly 5 Mbp downstream of the δ and MA_δ peak. While the level of spotting was not analyzed in the linkage mapping population, variation in this trait was observed in the F2/Backcross generation.
Interpretation of correlation coefficients calculated across the genome is made difficult because not all traits that differ between the breeds were measured in the interval mapping study. Indeed some loci under divergent selection in these two breeds may not have phenotypically obvious effects and will not have been analysed. However, this problem is avoided by the analysis where differentiation levels were compared at positions with low F-ratios versus high F-ratios. Where there is a positive association between the two analyses, there should be higher allele frequency differences for the high F-ratio group than the low F-ratio group, even if there are other QTL that were not detected. For the most extreme comparison of Fmax > 10 versus Fmax < 10, there was a clear distinction in mean δ values between the lowF and highF groups; 5 of the 6 chromosomes with Fmax values > 10 had greater mean δ values for positions with high F-values. The exception was BTA22, for which the mean δ (and MA_δ) value was higher for the lowF group. There were several QTL on this chromosome for carcass traits, but the MA_δ graph did not follow the QTL pattern. While the difference between the highF and lowF groups for the Fmax = 7 threshold was also statistically significant, the effect was not as pronounced as for the Fmax = 10 threshold; for 4 out of 13 chromosomes, MA_δ was greater in the lowF than the highF group.
Efficacy and power of differentiation-based "selection mapping"
While approaches based on allelic differentiation are currently being used for QTL mapping in some model species e.g. in Drosophila, Arabidopsis and maize [25–27], the general applicability of these methods is not yet clear. Theoretical predictions of allele frequency differences at markers linked to a locus under selection suggest that even for a locus under strong selection, the allele frequency differences between two selected lines will fall off rapidly with distance between the markers and the selected locus [28]. A similar effect should hold for multiple populations that are divergent for the same traits, as measured by FST or related statistics. This has been demonstrated in recent empirical studies: e.g. Sutter et al.[29] looked at the population structure for 22 large and small dog breeds using dense markers near the IGF 1 locus, a gene that is associated with body size, a trait which has been under strong selection in the development of dog breeds. In this region, they found a very narrow peak (< 0.2 Mbp) in extreme values of FST (their Figure two). In contrast, Akey et al.[30] identified a 1-Mbp region including several markers with high FST values between Shar-Pei and Dachshund dogs (their Figure three) in a region containing a strong candidate locus for skin wrinkling (a characteristic of the Shar-Pei breed). The extensive region of inflated FST in this case compared to the narrower region surrounding IGF 1 may reflect the more limited region of LD shared by several breeds compared to a broader LD region that may be shared by two breeds. The power of an approach based on allelic differentiation for identification of genes of large effect will depend, in part, on the levels of linkage disequilibrium in the genome. Further studies are required to quantify this relationship and to determine the marker density required to detect loci associated with selected traits in a given population.
Limitations of the current study
Factors in addition to LD levels may have influenced the results of the present study, for example, the interval mapping data was based on small numbers of individuals measured for some of the phenotypic traits (especially dairy-related traits). This may have increased the rates of both false positive and false negative linkage associations. Furthermore, the density of markers in the linkage study was relatively low such that QTL positions were not very precise and in many cases 95% confidence intervals covered most of the chromosome. However, marker density was enhanced in regions where QTL were detected in preliminary analysis [4], which increased the precision of positioning the QTL with largest effects and therefore should have increased the correlation between allele frequency difference and Fmax for these regions (e.g. BTA6). However, this was not observed in the analysis. In addition, performing interval mapping using genome sequence positions may also have influenced the scale of the Fmax values as there is not a one-to-one correspondence between the linkage map and the genome sequence due, in part, to variation in recombination rates across the genome. This should not, however, have had a substantial impact on the results pertaining to the moving average measures, which were spatially averaged. Finally, although the linkage mapping experiment was designed to detect QTL fixed for alternative alleles in the founder breeds, it may still have picked up large effects for which the QTL was segregating within breeds. In that case, allelic differentiation between breeds would be reduced compared to the case where alternative alleles were fixed in the two breeds.
In addition to the inherent limitations of the individual datasets, there may be problems in combining them: the QTL mapping study used British Charolais and Holstein animals as founders for the experimental crossbred population, while the HapMap SNP diversity study sampled North American Charolais and Holsteins. This should not have been a problem for the Holstein breed, which is very similar worldwide due to large-scale use of artificial insemination in the global dairy industry and the worldwide predominance of North American sire lines. However, there may be genetic differences between UK and North American Charolais due to genetic drift or different selection pressures in Europe and North America. Such differences are most likely to occur in regions associated with quantitative traits rather than highly-visible, categorical traits like coat colour which are key for breed identification.
High genetic differentiation signal
In addition to the high MA_δ values on BTA5, 6 and 18 discussed above, there were extreme values (> 0.3) on BTA13 (11.3 Mbp) and BTA20 (28.5-28.6 Mbp). The BTA13 region could not be associated with a gene known to be involved in any of the traits considered or with QTL for these traits from this study or others. On BTA20, a QTL for growth was detected slightly upstream of the Fmax peak (Additional file 2, Figure S1). Furthermore, the growth hormone receptor (GHR) gene, located at 33.9 Mbp on this chromosome, is associated with growth in cattle [31, 32] as well as milk yield and composition traits [33]. As in the case with KIT on BTA6, however, this gene is several Mbp downstream of the MA_δ peak.
Several of the regions with high levels of genetic differentiation were detected in previous studies to identify signatures of selection in cattle. Seven of the 13 regions of high FST reported by Flori et al.[34] in three dairy breeds (their Table two) were within 2 Mbp of the top 1% MA_δ positions reported in the current study. These included regions of BTA3, 4, 5, 6, 18 (near MC1R), 20 (near GHR) and 26. In a study of allele frequency differences between Holstein and Angus (another beef breed) cattle [35], of the 15 significant (p < 0.001) sliding window average differences (their Table two), three were within 2 Mbp of the top 1% MA_δ positions identified in the current study: on BTA14 (two positions) and BTA20, within 5 Mbp of GHR. There was less concordance between data reported here and results from Barendse et al.[36], which reported markers with high FST values for both the original HapMap dataset (19 beef and dairy breeds) and a set of 20 beef and dairy breeds from Australia (their Additional File two). Only 4 out of the 95 significant markers identified by Barendse et al.[36] were within 2 Mbp of peak regions reported in the current study. These were found on BTA1, 7, 12 and 13. None of the seven regions with high FST reported in the original HapMap study [12] (their Table one) overlapped with regions identified in the current study.However, in a separate analysis of the HapMap data [37], 11 out of 78 regions showing differences in allele frequencies between three or more dairy breeds and the complete dataset (see their Table S2) were located within 2 Mbp of peak regions in the current study. These covered seven chromosomes and included the region of BTA6 near the KIT gene. The correspondence between the current study and those of Flori et al. [34] and Stella et al. [37], which focused on dairy breeds, may provide some insight into the development of cattle breeds. It suggests that the major selective pressures have not been specifically for "meat" or "dairy" traits, but rather for more obvious characteristics like coat color or pattern and overall size. This is likely to be in part due to the relative length of time in which selection has been applied to these appearance-related traits as compared to less visible traits related to meat and dairy production, some of which may have been under strong selection only over the last few decades. In addition, color and appearance traits are generally under the control of one or relatively few genes (see the Online Mendelian Inheritance in Animals (OMIA) database, http://www.ncbi.nlm.nih.gov/sites/entrez?db=omia) whereas many meat and dairy traits are quantitative traits and thus under the control of multiple genes, which may fix more slowly than single genes [38]. It is likely that if the current trend of specialization in some cattle breeds (e.g. Holstein) continues, the level of differentiation at loci associated with production traits will also increase.
Conclusions
The study reported here describes a direct comparison of a linkage mapping study with a genetic differentiation scan for the identification of genomic regions associated with population/breed differences. Results from this study indicate that while the genome-wide analysis of genetic diversity does detect regions associated with large, qualitative phenotypic effects (such as coat color), this approach may not have sufficient power to detect smaller, quantitative effects, depending on the marker resolution used in the study and the levels of linkage disequilibrium in the populations under investigation.
References
Gutierrez-Gil B, Ball N, Burton D, Haskell M, Williams JL, Wiener P: Identification of quantitative trait loci affecting cattle temperament. J Hered. 2008, 99 (6): 629-638. 10.1093/jhered/esn060.
Gutierrez-Gil B, Wiener P, Nute GR, Burton D, Gill JL, Wood JD, Williams JL: Detection of quantitative trait loci for meat quality traits in cattle. Anim Genet. 2008, 39: 51-61. 10.1111/j.1365-2052.2007.01682.x.
Gutierrez-Gil B, Wiener P, Williams JL: Genetic effects on coat colour in cattle: dilution of eumelanin and phaeomelanin pigments in an F2-Backcross Charolais x Holstein population. BMC Genet. 2007, 8 (56):
Gutierrez-Gil B, Williams JL, Homer D, Burton D, Haley CS, Wiener P: Search for quantitative trait loci affecting growth and carcass traits in a cross population of beef and dairy cattle. J Anim Sci. 2009, 87 (1): 24-36. 10.2527/jas.2008-0922.
Young FJ, Woolliams JA, Williams JL, Glass EJ, O'Neill RG, Fitzpatrick JL: In vitro peripheral blood mononuclear cell proliferation in a crossbred cattle population. J Dairy Sci. 2005, 88 (7): 2643-2651. 10.3168/jds.S0022-0302(05)72941-6.
Green P, Falls K, Crooks S: Documentation for CRI-MAP, version 2.4. 1990, St. Louis, MO: Washington University School of Medicine
Ihara N, Takasuga A, Mizoshita K, Takeda H, Sugimoto M, Mizoguchi Y, Hirano T, Itoh T, Watanabe T, Reed KM, Snelling WM, Kappes SM, Beattie CW, Bennett GL, Sugimoto Y: A comprehensive genetic map of the cattle genome based on 3802 microsatellites. Genome Res. 2004, 14 (10A): 1987-1998. 10.1101/gr.2741704.
Liu Y, Qin X, Song XZH, Jiang HY, Shen YF, Durbin KJ, Lien S, Kent MP, Sodeland M, Ren YR, Zhang L, Sodergren E, Havlak P, Worley KC, Weinstock GM, Gibbs RA: Bos taurus genome assembly. BMC Genomics. 2009, 10 (180):
Zhang Z, Schwartz S, Wagner L, Miller W: A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000, 7 (1-2): 203-214. 10.1089/10665270050081478.
Seaton G, Haley CS, Knott SA, Kearsey M, Visscher PM: QTL Express: mapping quantitative trait loci in of simple and complex pedigrees. Bioinformatics. 2002, 18 (2): 339-340. 10.1093/bioinformatics/18.2.339.
Haley CS, Knott SA, Elsen JM: Mapping quantitative trait loci in crosses between outbred lines using least-squares. Genetics. 1994, 136 (3): 1195-1207.
Gibbs RA, Taylor JF, Van Tassell CP, Barendse W, Eversoie KA, Gill CA, Green RD, Hamernik DL, Kappes SM, Lien S, Matukumalli LK, McEwan JC, Nazareth LV, Schnabel RD, Weinstock GM, Wheeler DA, Ajmone-Marsan P, Boettcher PJ, Caetano AR, Garcia JF, Hanotte O, Mariani P, Skow LC, Williams JL, Diallo B, Hailemariam L, Martinez ML, Morris CA, Silva LOC, Spelman RJ, et al: Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009, 324 (5926): 528-532. 10.1126/science.1167936.
Smith MW, Lautenberger JA, Shin HD, Chretien JP, Shrestha S, Gilbert DA, O'Brien SJ: Markers for mapping by admixture linkage disequilibrium in African American and Hispanic populations. Am J Hum Genet. 2001, 69 (5): 1080-1094. 10.1086/323922.
Weir BS: Genetic Data Analysis II. 1996, Sunderland, Massachusetts: Sinauer Associations, Inc
Weir BS, Cockerham CC: Estimating F-statistics for the analysis of population structure. Evolution. 1984, 38 (6): 1358-1370. 10.2307/2408641.
Akey JM, Zhang G, Zhang K, Jin L, Shriver MD: Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002, 12 (12): 1805-1814. 10.1101/gr.631202.
Kuhn C, Weikard R: An investigation into the genetic background of coat colour dilution in a Charolais x German Holstein F-2 resource population. Anim Genet. 2007, 38 (2): 109-113. 10.1111/j.1365-2052.2007.01569.x.
Jackson IJ: Molecular genetics - Color-coded switches. Nature. 1993, 362 (6421): 587-588. 10.1038/362587a0.
Klungland H, Vage DI, Gomezraya L, Adalsteinsson S, Lien S: The role of Melanocyte-Stimulated Hormone (MSH) receptor in bovine coat color determination. Mamm Genome. 1995, 6 (9): 636-639. 10.1007/BF00352371.
Joerg H, Fries HR, Meijerink E, Stranzinger GF: Red coat color in Holstein cattle is associated with a deletion in the MSHR gene. Mamm Genome. 1996, 7 (4): 317-318. 10.1007/s003359900090.
Russo V, Fontanesi L, Scotti E, Tazzoli M, Dall'Olio S, Davoli R: Analysis of melanocortin 1 receptor (MC1R) gene polymorphisms in some cattle breeds: their usefulness and application for breed traceability and authentication of Parmigiano Reggiano cheese. Italian Journal of Animal Science. 2007, 6: 257-272.
Eberlein A, Takasuga A, Setoguchi K, Pfuhl R, Flisikowski K, Fries R, Klopp N, Furbass R, Weikard R, Kuhn C: Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the Non-SMC Condensin I Complex, Subunit G (NCAPG) gene. Genetics. 2009, 183 (3): 951-964. 10.1534/genetics.109.106476.
Setoguchi K, Furuta M, Hirano T, Nagao T, Watanabe T, Sugimoto Y, Takasuga A: Cross-breed comparisons identified a critical 591-kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-Met substitution in NCAPG as a positional candidate. BMC Genet. 2009, 10 (43):
Liu L, Harris B, Keehan M, Zhang Y: Genome scan for the degree of white spotting in dairy cattle. Anim Genet. 2009, 40 (6): 975-977. 10.1111/j.1365-2052.2009.01936.x.
Nuzhdin SV, Harshman LG, Zhou M, Harmon K: Genome-enabled hitchhiking mapping identifies QTLs for stress resistance in natural Drosophila. Heredity. 2007, 99 (3): 313-321. 10.1038/sj.hdy.6801003.
Turner TL, Von Wettberg EJ, Nuzhdin SV: Genomic analysis of differentiation between soil types reveals candidate genes for local adaptation in Arabidopsis lyrata. Plos One. 2008, 3 (9): e3183-10.1371/journal.pone.0003183.
Wisser RJ, Murray SC, Kolkman JM, Ceballos H, Nelson RJ: Selection mapping of loci for quantitative disease resistance in a diverse maize population. Genetics. 2008, 180 (1): 583-599. 10.1534/genetics.108.090118.
Keightley PD, Bulfield G: Detection of quantitative trait loci from frequency changes of marker alleles under selection. Genet Res. 1993, 62 (3): 195-203. 10.1017/S0016672300031906.
Sutter NB, Bustamante CD, Chase K, Gray MM, Zhao KY, Zhu L, Padhukasahasram B, Karlins E, Davis S, Jones PG, Quignon P, Johnson GS, Parker HG, Fretwell N, Mosher DS, Lawler DF, Satyaraj E, Nordborg M, Lark KG, Wayne RK, Ostrander EA: A single IGF1 allele is a major determinant of small size in dogs. Science. 2007, 316 (5821): 112-115. 10.1126/science.1137045.
Akey JM, Ruhe AL, Akey DT, Wong AK, Connelly CF, Madeoy J, Nicholas TJ, Neff MW: Tracking footprints of artificial selection in the dog genome. Proceedings of the National Academy of Sciences of the United States of America. 2010, 107 (3): 1160-1165. 10.1073/pnas.0909918107.
Curi RA, Palmieri DA, Suguisawa L, Ferraz ALJ, de Oliveira HN, Furlan LR, Silveira AC, Lopes CR: Effects of GHR gene polymorphisms on growth and carcass traits in Zebu and crossbred beef cattle. Livestock Science. 2006, 101 (1-3): 94-100. 10.1016/j.livprodsci.2005.09.015.
Sherman EL, Nkrumah JD, Murdoch BM, Li C, Wang Z, Fu A, Moore SS: Polymorphisms and haplotypes in the bovine neuropeptide Y, growth hormone receptor, ghrelin, insulin-like growth factor 2, and uncoupling proteins 2 and 3 genes and their associations with measures of growth, performance, feed efficiency, and carcass merit in beef cattle. J Anim Sci. 2008, 86 (1): 1-16. 10.2527/jas.2006-799.
Blott S, Kim JJ, Moisio S, Schmidt-Kuntzel A, Cornet A, Berzi P, Cambisano N, Ford C, Grisart B, Johnson D, Karim L, Simon P, Snell R, Spelman R, Wong J, Vilkki J, Georges M, Farnir F, Coppieters W: Molecular dissection of a quantitative trait locus: A phenylalanine-to-tyrosine substitution in the transmembrane domain of the bovine growth hormone receptor is associated with a major effect on milk yield and composition. Genetics. 2003, 163 (1): 253-266.
Flori L, Fritz S, Jaffrezic F, Boussaha M, Gut I, Heath S, Foulley JL, Gautier M: The genome response to artificial selection: A case study in dairy cattle. Plos One. 2009, 4 (8): e6595-10.1371/journal.pone.0006595.
Hayes BJ, Chamberlain AJ, Maceachern S, Savin K, McPartlan H, MacLeod I, Sethuraman L, Goddard ME: A genome map of divergent artificial selection between Bos taurus dairy cattle and Bos taurus beef cattle. Anim Genet. 2009, 40 (2): 176-184. 10.1111/j.1365-2052.2008.01815.x.
Barendse W, Harrison BE, Bunch RJ, Thomas MB, Turner LB: Genome wide signatures of positive selection: The comparison of independent samples and the identification of regions associated to traits. BMC Genomics. 2009, 10 (178):
Stella A, Ajmone-Marsan P, Lazzari B, Boettcher P: Identification of selection signatures in cattle breeds selected for dairy production. Genetics. 2010, 185 (4): 1451-1461. 10.1534/genetics.110.116111.
Kopp M, Hermisson J: The genetic basis of phenotypic adaptation I: Fixation of beneficial mutations in the moving optimum model. Genetics. 2009, 182 (1): 233-249. 10.1534/genetics.108.099820.
Acknowledgements
We thank the Bovine HapMap Consortium for access to the Bovine HapMap genotypes, Zen Lu for bioinformatics assistance and Ian Handel for assistance with figures. Defra LINK is acknowledged for funding the resource herd and associated data, which was used as the source of linkage mapping data used in this study. MAE would like to thank Isfahan University of Technology for providing funding during his sabbatical leave at The Roslin Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
PW planned the study, performed some of the data analyses and drafted the manuscript, MAE wrote and executed scripts to extract and manipulate the data, positioned the markers used in the linkage mapping study on the genome sequence, and performed some of the data analyses, JLW conceived and managed the study from which the linkage mapping results derived, was part of the Bovine HapMap Consortium and participated in writing the manuscript, DW advised on and assisted with the statistical analyses, AL wrote and executed scripts to extract and manipulate the data, JAW advised on the statistical analyses and was part of the Bovine HapMap Consortium, and BGG provided the linkage mapping data, assisted with positioning the markers on the genome sequence and helped prepare the manuscript. All authors approved the manuscript.
Electronic supplementary material
12864_2010_3919_MOESM2_ESM.PDF
Additional file 2:Figure S1: Patterns of the moving average of allele frequency differences between Holstein and Charolais cattle ( MA_δ , represented by diamonds) and the maximum F-ratio of the linkage mapping study with Holstein and Charolais founders (F max , represented by the curve) across the bovine autosomal genome (BTA1 - BTA29).(PDF 1 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wiener, P., Edriss, M.A., Williams, J.L. et al. Information content in genome-wide scans: concordance between patterns of genetic differentiation and linkage mapping associations. BMC Genomics 12, 65 (2011). https://doi.org/10.1186/1471-2164-12-65
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-12-65