
Abstract
Background
Evolution of the Brassica species has been recursively affected by polyploidy events, and comparison to their relative, Arabidopsis thaliana, provides means to explore their genomic complexity.
Results
A genome-wide physical map of a rapid-cycling strain of B. oleracea was constructed by integrating high-information-content fingerprinting (HICF) of Bacterial Artificial Chromosome (BAC) clones with hybridization to sequence-tagged probes. Using 2907 contigs of two or more BACs, we performed several lines of comparative genomic analysis. Interspecific DNA synteny is much better preserved in euchromatin than heterochromatin, showing the qualitative difference in evolution of these respective genomic domains. About 67% of contigs can be aligned to the Arabidopsis genome, with 96.5% corresponding to euchromatic regions, and 3.5% (shown to contain repetitive sequences) to pericentromeric regions. Overgo probe hybridization data showed that contigs aligned to Arabidopsis euchromatin contain ~80% of low-copy-number genes, while genes with high copy number are much more frequently associated with pericentromeric regions. We identified 39 interchromosomal breakpoints during the diversification of B. oleracea and Arabidopsis thaliana, a relatively high level of genomic change since their divergence. Comparison of the B. oleracea physical map with Arabidopsis and other available eudicot genomes showed appreciable 'shadowing' produced by more ancient polyploidies, resulting in a web of relatedness among contigs which increased genomic complexity.
Conclusions
A high-resolution genetically-anchored physical map sheds light on Brassica genome organization and advances positional cloning of specific genes, and may help to validate genome sequence assembly and alignment to chromosomes.
All the physical mapping data is freely shared at a WebFPC site (http://lulu.pgml.uga.edu/fpc/WebAGCoL/brassica/WebFPC/; Temporarily password-protected: account: pgml; password: 123qwe123.
Similar content being viewed by others


Background
Flowering plants have extensively and often recursively experienced polyploidization [1–4]. The resulting duplicated regions, especially those produced recently, offer the means to further study the contributions of segmental and/or whole-genome duplication/triplication to the evolution of a lineage, but add to genome complexity. The high abundance of repetitive DNA sequences in some flowering plants adds further to genome complexity. At present, many plant genomes have been or are being sequenced. Draft genome sequences can lack sufficient contiguity in many genomic regions to support cross-species comparison of genome organization and structure, which is crucial to understanding plant evolution and speciation. In concert with sequence assemblies, independent physical maps often facilitate the correct ordering of DNA segments on chromosomes and thus clarify the genome organization changes revealed by multiple species comparisons [5, 6].
Brassica is in the tribe Brassiceae, a well-defined clade in the family Brassicaceae that also includes Arabidopsis thaliana, the source of the first flowering plant genome to be sequenced. Brassica and Arabidopsis are thought to have shared common ancestry ~14-20 million years ago [7–10]. The genus Brassica has great scientific and economic importance [11]. Crops of the genus Brassica are widely used in the cuisine of many cultures and provide much of world-wide edible vegetable oil supplies. Six Brassica species are widely cultivated, including three diploids: B. rapa (AA, 2n = 20), B. nigra (BB, 2n = 16) and B. oleracea (CC, 2n = 18), and three amphidiploids (allotetraploids): B. juncea (AABB, 2n = 36), B. napus (AACC, 2n = 38) and B. carinata (BBCC, 2n = 34).
Study of B. oleracea offers particularly great promise of new insights into morphological evolution that complement and extend upon what is available in Arabidopsis [12–14]. In B. oleracea, morphological divergence has been unusually rapid relative to reproductive isolation, i.e., this single species has a stunning range of morphologies among genotypes that are readily intercrossed. While domestication of most crops resulted in enhancement of a single plant part for use by humans, such as the seeds/grains of cereal crops, the fruits of some trees, or the roots of some vegetable crops, the B. oleracea crops are a striking exception. They include forms that have been selected for enlarged vegetative meristems at the apex (cabbages, B. oleracea subspecies capitata) or in the leaf axils (Brussels sprouts, subsp. gemmifera), forms with proliferation of floral meristems (broccoli, subsp. italica) or even aborted floral meristems (cauliflower, subsp. botrytis), and forms with swollen bulbous stems (kohlrabi, subsp. gongylodes), or orate leaf patterns (kales, subsp. acephala). These morphologically divergent genotypes ('morphotypes') are freely intercrossing.
The plasticity of B. oleracea makes it a potential model for the study of plant morphological evolution in much the same manner that the dog (Canis spp.) is an attractive model for mammalian evolution. While a few genes like the homologs of Arabidopsis mutants such as "CAULIFLOWER" are thought to play roles in some Brassica morphologies [15–17], these morphologies are under complex genetic control [18–21]. Some Brassica QTLs map to locations that correspond to relevant Arabidopsis mutants, suggesting positional candidates -- but many do not, suggesting the opportunity to identify functions recalcitrant to mutation in Arabidopsis [22, 23] or that escaped detection due to small phenotypic effects [24].
Due to their close phylogenetic relationship, Brassica-Arabidopsis comparative genomics promises to identify genetic determinants of a much broader spectrum of variation than might be accessible using Arabidopsis alone [12–14]. The close relationship of Brassica to Arabidopsis motivated NSF-funded low-coverage (0.6×) sequencing of B. oleracea (BO) genotype TO 1000 [25]. However, while the physiology and developmental biology of Arabidopsis and Brassica are similar, the genomes of Brassica species are much more complex than that of A. thaliana[26–28]. The 'diploid' Brassica genomes are 3-5 times larger than that of Arabidopsis, ranging from 0.97 pg/2C (468 Mb/1C) for B. nigra to 1.37 pg/2C (662 Mb/1C) for B. oleracea, partially as a result of multiple rounds of polyploidy during their ancestry [29, 30]. One round of ancient whole-genome triplication (gamma) in an early eudicot ancestor and two whole-genome duplications (beta and alpha) occurred before the Arabidopsis-Brassica split [4, 31, 32]. Additional polyploidization(s) occurred in the Brassica lineage after its divergence from Arabidopsis, reflected by large duplicated segments in the genetic maps of each of three diploids [B. rapa (syn. rapa,), B. nigra and B. oleracea] [27, 33–36]. The corresponding duplicated structure of the B. rapa and B. oleracea maps indicates that species divergence was after polyploidization, resulting whole-genome triplication [29, 37–39]. It was estimated that the genome triplication event and the initial diversification of the Brassiceae must have occurred between 7.9 and 14.6 mya [29], which might be the hypothesized single and major evolutionary event that have gave rise to the early lineages [40]. According to the analysis of the FLOWERING Locus C region, it was further estimated that the Brassica triplication occurred 13 to 17 mya, very soon after the Arabidopsis and Brassica divergence at 17-18 mya [10].
Significant progress has been made in developing genomic resources to expedite Brassica research [41–44]. A detailed genetic linkage map of B. rapa has been constructed containing 545 sequence-tagged loci distributed on 10 linkage groups covering 1287 cM, with an average interval of 2.4 cM between markers [45]. Genetic linkage maps were constructed for four B. oleracea populations, with an average length of 863.6 cM and a total of 367 loci were detected in the constructed composite map with an average interval between loci of 2.35 cM [33], which revealed at least 19 chromosomal rearrangements differentiating B. oleracea and Arabidopsis. Linkage maps of immortal mapping populations of rapid cycling, self-compatible lines from B. rapa and B. oleracea were recently developed, which included 224 and 279 markers, respectively [46]. A genome-wide physical map of the B. rapa genome was constructed by high-information-content fingerprinting (HICF) [44], which facilitates improved physical map construction in both throughput and quality by exploiting the fluorescence-labeled finger-printing approach. The map provided 242 anchored contigs on 10 linkage groups to serve as seed points from which to continue bidirectional chromosome extension for genome sequencing. There are also efforts to refine genetic linkage maps. Genome sequencing projects involving "A" and "C" genomes are on-going or planned [47, 48]. The Multinational Brassica Genome Project (MBGP) and Brassica rapa Genome Sequencing Project (BrGSP) are aiming to completely sequence the genome of Brassica rapa inbred line 'Chiifu" (http://www.brassicagenome.org; http://www.brassica-rapa.org).
Here we report a physical map of a rapid-cycling strain of B. oleracea (accession TO1434), integrating high-information-content fingerprinting (HICF) of Bacterial Artificial Chromosome (BAC) clones with overgo hybridization data from 2882 probes, including about 600 that have been genetically mapped. By integrating the B. rapa physical map, we explored genome-wide microsynteny between Arabidopsis and Brassica, and found probable (peri)centromere-related contigs. Comparison of the B. oleracea map with Arabidopsis and other available eudicot genomes showed appreciable 'shadowing' produced by more ancient polyploidies, resulting in a web of relatedness among contigs which increased genomic complexity, and interchromosomal breakpoints during their diversification. This physical map is of immediate value for gene isolation, and will serve as a valuable genomic resource for Brassica "C" genome sequencing, assembly of BAC sequences and further comparative genomics between Brassica genomes.
Results
BAC fingerprinting and physical map assembly
We fingerprinted a total of 73728 clones from 192 384-well plates. Fingerprints containing less than 30 and more than 200 bands were excluded from FPC analysis, which used a dataset of 53048 clones.
FPC (Finger-Printed Contigs, v9.0) [49] was used to construct BAC contigs. To produce an FPC-accessible dataset (FPC does not accept color labels or fractional sizes), the size of each fragment was multiplied by 10, after which the decimal part was dropped. This resulted in fragments with sizes ranging from 500 to < 6000 units. Secondly, the color labels were converted to non-overlapping numeric ranges by adding offset values 6000, 12000, or 18000 to three of the four colors, which eventually resulted in fragments ranging from 0 to 25000 units.
We designed and used overgo hybridization probes to support contig construction. A total of 4226 probes were designed by using Arabidopsis and Brassica sequences, and they are often from conservative domains (see Methods for details). After removing the probes that hit > 50 BAC clones, a subset of 2882 probes were involved in the physical map assembly process.
Well-to-well contamination produces many problems during assembly of HICF data. Therefore, before running FPC to construct the contigs, we removed the likely contaminated BACs in the dataset by implementing a de-contamination function in FPC. After an initial round of contig construction (cutoff = 1e-50 and tolerance = 4 and best of 100 repetitive constructions), a FPC program named DQer was run to eliminate possible questionable clones (Q-clones) for contigs > 15% Q-clones. Multiple iterations of end-to-end and singleton-to-contig merges were then adopted with successively less and less stringent settings (Figure 1).

FPC analytical pipeline used to assemble the Brassica oleracea physical map.
During the optimization of our processes, and later to improve quality of some below-average batches of BACs, we repeated fingerprinting of some 96-well 'subplates', with 72 subplates (5184 BACs) duplicated, and 12 subplates (830 BACs) triplicated. For each BAC repeated, the fingerprint having numbers of bands nearest the global average (120) was used in assembly.
A total of 46,006 BACs were used in contig assembly, yielding 2907 contigs each containing 2 or more BACs, and 2323 singletons. An average contig contained 8.7 BACs and 3.2 overgo probes. Two contigs (ctg03293 and ctg02560) contain more than 1000 BACs, ~60% of whose end sequences could be linked to Brassica repetitive sequences determined by running BLAST. Five contigs contained more than 100 BACs. Sixteen contigs (ctg00857, ctg01639, ctg02159, ctg02194, ctg02197, ctg02490, ctg02560, ctg02626, ctg02695, ctg02754, ctg02830, ctg03202, ctg03304, ctg02470, ctg03571, ctg04056) are RNA-related, and may help to decipher the rRNA and tRNA genes in Brassica. Six contigs (ctg01958, ctg02241, ctg02829, ctg03476, ctg03627, ctg04065) are likely chloroplast-related, and five contigs (ctg01690, ctg01958, ctg02241, ctg02960, ctg04062) are likely mitochondrion-related, including two contigs that are both chloroplast- and mitochondrion-related. These contigs may be chimeric, involving both nuclear and organelle DNA, or just nuclear DNA produced by lateral gene transfer from organelle to nucleus as previously discussed in Arabidopsis [50], and sorghum [51]. DNA similarity between BESs and organelle DNA can provide some clue about the identity of potentially chimeric contigs: BESs from chimeric contigs may have high identity with organelle DNA, e.g., DNA similarity > 98% over a long stretch, while laterally transferred DNA may not. We infer that ctg04065 (265 BACs) may be a chimeric contig of chloroplast DNA (188 BACs) and nuclear DNA (77 BACs). The DNA similarity of most involved BESs against chloroplast DNA are often > 99% in up to 800 bp, but some BESs have DNA similarity < 95%, perhaps reflecting a mix of extant chloroplast DNA and laterally transferred ones. We also suggest that ctg02241 and ctg04062 are chimeric mitochondrion-nuclear contigs inferred based on similar criteria. The latter contains most mitochrondrial BACs (14 of 25 in the contig). BESs of other organelle-related contigs have low similarity with extant organelle DNA, suggesting their origins by lateral gene transfer.
Comparative genomic analysis
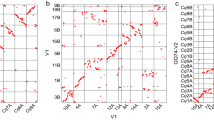
With the help of BAC end sequences and probe sequences, both B. oleracea and B. rapa contigs were mapped onto the Arabidopsis genome sequence (Figure 2). Neighboring hits < = 200 Kb from one another were used to infer DNA synteny between B. oleracea and Arabidopsis, and the longest syntenic region inferred is more than 870 Kb, with most regions less than 400 Kb (Figure 3A). For anchoring B. rapa contigs to Arabidopsis, the extension parameter was reduced to 100 Kb because a higher density of BESs made it easier to find cross-species synteny. A subset of 1990 B. oleracea and 1006 B. rapa contigs (68.5% and 70.4% of the total of respective datasets) hit one or more Arabidopsis regions. DNA sequence similarity revealed by the anchor sequences peaked at 92% (Figure 3B), which supports a 14.5-20.4 million year divergence time between Arabidopsis and Brassica [2, 3, 7–10]. Interestingly, the Blast E-value showed a bi-modal distribution (Figure 3C), which may imply at least two different sets of anchored regions in Arabidopsis, possibly reflecting the ancient duplication events.

An example of a Brassica FPC contig linked to different Arabidopsis regions. The contig was displayed with 2 or 3 rows, including assembled BAC clones, overgo probes, and merging information (if available) during contig assembly. Dashed lines between Brassica BAC clones, probes and Arabidopsis genomic regions show interspecific chromosomal synteny.

Characteristics of B. oleracea (Bo) contigs mapped onto the A. thaliana (At) genome. (A) Size of anchored regions based on length of Arabidopsis sequences covered; Sequence similarity (B) and BLAST E-values (C) between anchored Bo and At sequences.
We found clear evidence of ancient duplication events in the extant Brassica genomes. About 88% and 93% of Arabidopsis genome sequences have been covered by the anchored B. oleracea and B. rapa contigs, respectively (Figure 4). At least 70% of regions have been covered to a depth > = 2, surely a result of multiple homologous regions in Brassica (Figure 4). The peak is around 3, covering nearly 20-25% of Arabidopsis genome sequences. There is a sharp decrease from coverage 3 to 4, supporting previous propositions of triplication of at least portions of the Brassica ancestral genome after its divergence from Arabidopsis. The 13% of the Arabidopsis genome covered in depth 4, and total of 20% covered in depths > = 4, are shown below to be partly explained by the 'shadows' of more ancient genome duplications.

A map of Brassica oleracea and Brassica rapa contigs anchored to Arabidopsis chromosomes. Chromosomes are arranged in curved boxes, accompanied by gene densities (red), repetitive sequence densities (green), and distributions of overgo probes (blue ticks). The external light-blue and green blocks show the distribution of syntenic Brassica oleracea and Brassica rapa contigs along Arabidopsis chromosomes, respectively. Lines between chromosomes link syntenic genes in Arabidopsis, with colors distinguishing different duplicated blocks.
By checking Arabidopsis genomic regions known to correspond to one another due to ancient duplication, we revealed that 186 B. oleracea contigs (9.3% of all anchored ones) were anchored to both members of α-duplicated segment pairs and another 54 (2.7%) to β- or γ-duplicated regions. However, it is often possible to distinguish the orthologous regions from the outparalogous regions (produced by ancient duplications before the Arabidopsis-Brassica divergence). The inferred Arabidopsis-Brassica orthologous regions always share BLASTN E-values < 1e-30, while the outparalogous regions share E-values ~ 1e-10. Excluding the identified outparalogous regions from evaluation made the peak around coverage depth 2 and 3 even more prominent (Table 1), and the higher coverage-depth portion of Arabidopsis became smaller.
DNA breakages distinguishing Brassica and Arabidopsis
To locate DNA breakages distinguishing the two species, we divided Arabidopsis chromosomes into 'bins', which were further linked to Brassica BESs to find multiple associations of bins with different BESs. In total, we found 39 synteny discontinuities between the two lineages (Table 2), with 32 that imply interchromosomal rearrangements, and 7 that imply intra-chromosomal rearrangements. We identified tens of cases in which paired BAC ends fell in different duplicated regions. This added to the credibility of the analysis by showing that the approach finds actual associations in that the duplicated regions possibly share appreciable sequence similarity.
Heterochromatin vs. euchromatin
The chromosomal distribution of conserved Arabidopsis-Brassica synteny was striking, preserved almost universally in gene rich and repeat poor regions presumably representing the Arabidopsis euchromatin, and almost absent from the heterochromatin or pericentromeric regions (Figure 4). About 14% of Arabidopsis sequences were not covered by B. oleracea contigs, occurring mainly in the pericentromeric regions (Figure 4). Among 1990 anchored B. oleracea contigs (excluding the largest 5 contigs, suspected to be mosaics), 97% (1920) could be aligned to the 104 Mb euchromatic regions in Arabidopsis, involving 80% (2316) of anchored probes, which may correspond to low-copy-number genes in Brassica, and 91% (32415) of anchored BACs. In contrast, only 6.7% of contigs, 20% of anchored probes and 9% of anchored BACs aligned to the 15 Mb heterochromatic regions. About 3% of B. oleracea contigs can be anchored to both euchromatic and heterochromatic regions.
A total of 950 B. oleracea contigs that could not be aligned to Arabidopsis were hypothesized to be pericentromere-related, based on four lines of evidence. First, these contigs were gene-scarce, accounting for 33% of total contigs but less than 1% (25) of gene-derived probes. Second, these 33% of contigs account for only 16% of BACs, indicating that the underlying BACs are relatively recalcitrant to assembly, consistent with low DNA sequence complexity resulting from high repetitive DNA content. Third, 46% of the BACs were repeat-related based on their end-sequences (see above), the same as those aligned to the Arabidopsis heterochromatin and much higher than the 34% of BACs aligned to the euchromatin (P-value = 0). Fourth, we searched the BES against two Brassica-centromere-specific repeats (CentrBr1 and CentBr2, each 176 bp), and found that non-anchored contigs had a similar abundance of centromeric elements (18%) as known heterochromatin-aligned contigs (19%), and much more than euchromatin-aligned contigs (10%). Accordingly, many of the non-anchored contigs may be centromeric.
Ribosomal-RNA-related contigs correspond mainly to two pericentromeric regions on Arabidopsis chromosomes 2 and 3, showing possible expansion of their related orthologous copies in Brassica. The regions covered to the greatest depths are not RNA-related but possibly related to other repeats like transposons.
Evolution of centromeric repeats
Identified first in B. rapa[52], we found thousands of CentBr1 and CentBr2 repeat sequences in the BESs from both B. oleracea and B. rapa, which permitted us to perform a comparative analysis of their evolution. We hereafter refer to them as CentB1 and CentB2, since they are not confined to B. rapa. A subset of 791 and 563 B. oleracea BESs, or 2% of the total, are CentB1- and CentB2-related, respectively. Many B. rapa BESs (20%) were also related to these elements, and showed unbalanced relatedness to the two repeat classes, with 17156 and 1132 BESs related to two classes, respectively. About 50% of the BACs in both species were related to the same repeat class at both ends, while only a small fraction (~0.5%) were related to different elements at each end, suggesting a relatively separate distribution and expansion of the two element families in the Brassica genomes.
From the BESs we retrieved 2894 and 62222 sequences of the two centromeric repeat classes and randomly selected 100 B. oleracea sequences and 200 B. rapa sequences for phylogenetic analysis (Figure 5). As expected, the CentB2 repeats grouped together, forming a subtree in which repeats from each species form two subgroups, each clustered with repeats from the other species. This illustrates the separate divergence and expansion of family members in each species. The CentB1 repeats from the two species are much more interleaved with one another, though forming many clusters and showing separate expansion. This phylogenetic distribution suggests a clear origination and initial divergence of these repeat families in a rapa-oleracea common ancestor. Possible cross-species gene transfer cannot be ruled out due to the existence of many subgroups containing genes from both species.

Phylogeny of centromeric repeats in B. oleracea and B. rapa. B. oleracea repeat ids start with "To" and B. rapa repeat ids start with "Br". CentBr1 repeats end with "1". CentBr2 repeats (ending with "2") are denoted with red branches.
Discussion
Recursive polyploidizations and subsequent changes
Brassica provides an attractive system in which to study polyploidy and its consequences, having been affected by recursive polyploidizations including γ triplication in a common ancestor of most if not all rosids, β (< 70 mya) and α (< 32 mya) duplications in the Brassicales after divergence from papaya, triplication (< 20 mya) in the Brassica oleracea and B. rapa common ancestor, and very recent duplications to form B. juncea (AABB), B. napus (AACC), and B. carinata (BBCC). These provide good opportunities to study the relationship between speciation and genome doubling/tripling.
Genome macro-structural changes during lineage evolution can be enormous, but the types and rates of change differ widely among lineages. For example, the chromosome numbers of tetraploid Brassica species are the sum of the chromosome numbers of their parental diploids, showing no significant chromosomal changes after genome doubling. In contrast, there have been about 7 chromosomal fissions, fusions and merges in the A. thaliana lineage since its divergence from A. lyrata, the latter still showing near-perfect collinearity with a member of a different genus, Capsella rubella[53–57]. Genomic resources in preparation for an outgroup, Sisymbrium irio, may soon make it possible to deduce the levels and patterns of change in the diploid Brassicas since their divergence.
Gene losses after the Brassica triplication event have been very extensive. One chromosome segment from the rosid common ancestor would be represented in 36 copies through sequential episodes of two whole-genome triplications and two whole-genome duplications (3 × 2 × 2 × 3) in the B. oleracea (or B. rapa, or B. nigra) genomes if all doubled/tripled copies had been preserved, with such a genome containing more than 400000 genes. The angiosperm genomes sequenced to date are estimated to have about 25000 to 46000 protein-coding genes, with the largest set of predicted gene models from soybean (46430) [58]. All these genomes have been affected by 1 to 3 whole-genome duplications like Brassica. Therefore, the Brassica genomes must have preserved only a small fraction of duplicated genes, as reported previously [26]. The physical map reveals a clear impact of these recursive duplications on genome complexity, with a web of syntenic patterns among paleo-duplicated regions upon which the relatively recent triplication is superimposed, making the genome complicated to decipher.
Comparative analysis of B. napus and A. thaliana, has been proposed to define 24 genomic blocks in the ancestral Brassica karyotype (n = 8) [57]. These blocks were used to delineate the genome of B. rapa with each block in 1-3 copies, revealing ~44 major rearrangements during the evolution of B. rapa from the ancestral karyotype. Our present analysis likewise suggests 39 synteny discontinuities between B. oleracea and A. thaliana genome sequences. Since the genomic structure of Arabidopsis has been affected only by several major rearrangements [57], we predict that many of these synteny discontinuities occurred during the evolution of B. oleracea and its close ancestors, perhaps mostly during a period of genomic instability shortly after the lineage-specific whole-genome triplication. A similar analysis was performed by mapping B. rapa BAC clones onto the A. thaliana genome, inferring 19 inter-chromosomal rearrangements [59].
Synteny preservation and recombination
Most B. oleracea and B. rapa contigs and BACs, including the majority of low-copy DNA hybridization probes, could be anchored to the Arabidopsis euchromatin. Despite this synteny preserved between Brassica and Arabidopsis euchromatin after 15-20 mya of divergent evolution, pericentromeric regions tacitly assumed to be heterochromatic appear substantially rearranged, as few contigs can be anchored. Repetitive and centromeric probes are enriched in the few contigs anchored here as well as many non-anchored contigs, suggesting that the latter belong here too. Not only is cross-species synteny better preserved in euchromatin, but paralogous DNA synteny produced by whole-genome duplications also remains more evident (Figure 2). Indeed, the depth of coverage of the Arabidopsis genome by Brassica BAC contigs increases with distance from the Arabidopsis pericentromeric space. An attractive future study would be to compare on a nucleotide-for-nucleotide basis the entire centromeric regions of Arabidopsis and Brassica chromosomes, perhaps revealing small islands that are preserved by selection acting on key functions
The Arabidopsis-Brassica comparison provides further support for a model of genome evolution that has arisen from comparison of the monocots rice and sorghum [51] and is also supported by analysis of the soybean sequence [58]. Specifically, synteny preservation is high and repetitive DNA abundance is low in genomic regions where recombination is relatively frequent. In sorghum, very recent LTR retroelement insertions are approximately evenly distributed across the entire genome, while older insertions are largely in the heterochromatin [6]. Considering these data in view of Muller's ratchet [60], one would predict most rearrangements to be slightly deleterious, in that gene arrangement appears to be much more strongly preserved in recombinogenic than non-recombinogenic regions such as pericentromeric space [51].
The extensive duplicated regions in Brassica genomes provide much opportunity for illegitimate recombination, which could lead to reciprocal (crossing-over) or nonreciprocal (gene conversion) DNA information transfer, or homeologous nonreciprocal transposition [61]. Illegitimate recombination is often deleterious, incurring DNA mutations, deletions, and inversions. Gene conversion can be explained as a "copy and paste" process, which removes the information of one DNA segment but doubles the effect of its homologous segment, leading to changes in expression dosage. Illegitimate recombination has a much greater chance to occur between relatively young duplicated blocks [61], or to recur between ancient blocks that are kept very similar by its recurrence [6, 62, 63]. Different lines of cytological evidence show that exchanges can occur between homeologous chromosomes of both resynthesized and natural B. napus[64–66]. Though the Brassica triplication event may have occurred as much as 18 mya [10], evidence from rice-sorghum comparison supports illegitimate recombination between 70 million-year-old duplicated regions. Indeed, intragenomic study of rice shows that 70-my old duplicated regions have interacted as recently as the past 400,000 years [63]. Therefore, another important future study, when the required data are available, will be to investigate the impact of illegitimate recombination on the evolution of Brassica genes, genomes, and species.
Toward sequencing Brassica oleracea
Recursive polyploidizations may complicate assembly of Brassica genome sequences, especially if they are accompanied by frequent illegitimate recombination events that render 'islands' of paralogous DNA sequence (such as genes) homogeneous. Based on our findings herein and those in previous publications, there are many duplicated blocks, making Brassica genomes very complex to decipher. Though the frequency of homeologous recombination per generation is very low [61], its cumulative effect over many generations may be high. Gene conversion or homeologous DNA translocation could keep two homeologous DNA segments very similar, misleading efforts to reconstruct the evolutionary history of genes or genomic structures.
The physical map described herein, genetically anchored and rich in landmarks such as BAC end sequences and hybridization data to genetically-mapped markers, provides a valuable adjunct to efforts in progress to sequence the rapid-cycling genotype from which the BACs were made. Moreover, efforts are also in progress to investigate the genomic basis of the remarkable morphological diversity among cultivated forms that distinguishes B. oleracea from any other plant species we are aware of. The BACs provide an excellent bridge between the resolution that might be accomplished by QTL fine mapping [67], and the identification of determinant genes.
Based on the physical map of B. oleracea, we have done a very preliminary comparative genomics analysis with several eudicot plants. The future availability of whole-genome sequences from Brassica species will further expand scope for comparative analysis and shed light on both genome-level and single-gene-level changes that have contributed to the evolutionary trajectory of Brassica.
Conclusions
A genetically-anchored, sequence-rich physical map for B. oleracea sheds light on genome evolution of Brassicaceae species, and provides a valuable resource toward the assembly of genome sequences, especially using recent short-read technologies.
Methods
BAC library
BAC library BOTO1, constructed from the TO1434 line, was prepared from partial HindIII digest of Brassica oleracea genomic DNA. The library includes a total of 87168 clones, of which 73728 were gridded and finger-printed and used in overgo hybridization. The expected BAC size is ~100 Kb. Clones having < 30 or > 250 bands were removed from further analysis, which resulted in a total of 61871 clones.
Probe design and hybridization
A total of 4226 B. oleracea overgo probes were hybridized to the BOTO1 BAC clone library. Overgo probes, 40 bp each, were designed from Arabidopsis gene sequences, with 603 probes [BOVG0001-BOVG0602, and BOVG1153] designed from markers on genetic map, 490 probes [BOVG0603-BOVG1152] from Brassica genomic sequences matching α-singleton genes (defined in Bowers et al., 2003), 576 probes [BOVG1154-BOVG1729] from Brassica genomic sequences matching α-duplicated genes, and the remainder from an assortment of other Arabidopsis genes. For probe design, source sequences were searched with BLASTN (at most 4 mismatched sites are allowed and at least 31 bp in length of hit region) against all known plant sequences to find conserved domains, and compared to known plant repeats to screen out possible repetitive sequences. The selected sequences were then chopped into 40 bp segments and screened for GC content of between 40% and 60%.
Probes were labeled using P-32 and applied to macroarrays of 18,432 BACs per membrane following methods described previously [51]. Briefly, multiplex experiments were done by applying 576 probes at a time, in pools of 24 probes per bottle, by rows, columns and diagonals of a 24 × 24 array of probes. Films were manually scored, and scores digitized using text-recognition software (ABBYY FINEREADER). Data were deconvoluted and stored in our locally developed MS Access database system "BACMan". The hybridization data were involved to construct BAC contigs while running FPC.
BAC fingerprinting
The high-information-content fingerprinting (HICF) method was adopted, together with a commercially available SNAPshot labeling kit. Plasmids were digested with EcoRI, BamHI, XbaI, XhoI and HhaI. The ends of restriction fragments were differentially labeled using fluorochrome tagged ddNTPs after the first four enzyme cuts, and the last enzyme further reduced fragment size and produced a blunt end. The fingerprints were generated by an ABI sequencer and size files were generated by GeneMapper Software v4.0 after processing the chromatograms. Only the fragments from 50 to < 600 bp were preserved for further analysis, those beyond this range being considered unreliable.
Well-to-well contamination causes major problems in assembly. We screened possibly contaminated wells before assembly using a de-contamination function implemented in FPC v9.0. A clone was inferred to have been contaminated if it had a statistically significant number of overlapping bands (e.g. cutoff 1e-50) with any of its neighboring clones within a 7 × 7 square of wells. In total, 5477 clones were inferred at a cutoff 1e-50, and tolerance 4 to have been potentially contaminated, and were excluded from assembly. Well-to-well contamination also contributed much to forming an unexpectedly large contig.
BAC end sequencing and analysis
A subset of BACs were end-sequenced using methods described previously [68], yielding 85317 BAC end sequences (BESs) http://www.ncbi.nlm.nih.gov/. By searching against the TIGR Brassica Repeat Database and our extended Brassica repeats database, especially two Brassica-centromere-specific repetitive sequences [52], 'repeat-related' and 'centromere-repeat-related' BAC end sequences were identified.
Inferring RNA-, chloroplast- and mitochondrion-related contigs
The eudicot RNA gene sequences, Arabidopsis thaliana complete chloroplast genome sequence (AP000423.1), and Brassica napus complete mitochondrion genome sequence (AP006444.1), were downloaded from GenBank, against which B. oleracea BAC end sequences were searched at E-value < 1e-5. If more than 20% of BAC end sequences of a contig hit these specific sequences, it was inferred to be RNA-, chloroplast- and/or mitochondrion-related.
Comparative analysis of Brassica rapa and B. oleracea physical map
The previously published B. rapa contigs [44] were involved in the present analysis by anchoring them to the Arabidopsis genome sequence using 100,666 BAC end sequences http://www.ncbi.nlm.nih.gov/.
Mapping onto Arabidopsis genomes
Contigs were anchored to Arabidopsis [69] genome sequence by performing BLASTN search with BAC end sequences and probe sequences against the genome sequence (E-value < 1e-10 for Arabidopsis and E-value < 1e-5 for other eudicots). BAC end sequences and probe sequences having more than 50 hits were not used in synteny analysis. Syntenic regions were identified by linking neighboring hits < = 200 Kb on Arabidopsis genome sequences (Figure 6A). We checked whether a contig can be linked to Arabidopsis duplicated regions. A Brassica FPC contig may be linked to multiple duplicated regions for recursive whole genome replication events, including α, β, and γ [3]. If all replicated copies have been preserved, a contig may be linked to one α-orthologous region, one α-paralogous region, two β-paralogous regions and eight γ-paralogous regions (Figure 6B). However, wide-spread DNA losses following replication events often lead to a degenerate pattern of correspondence. One contig may be related to multiple Arabidopsis regions, and it is often possible distinguish orthology from paralogy if sequence similarity is considered (Figure 6C). To find possible chromosomal breakpoints distinguishing Brassica from Arabidopsis, we searched for paired B. oleracea BESs that hit different Arabidopsis regions (Figure 6D). The procedure is similar to the one used in B. rapa and Arabidopsis comparison [59]. To perform the search, Arabidopsis chromosomal sequences were divided into bins of selectable sizes of 500 Kb or 1 Mb. Each bin was linked to BESs by BLASTN at E_value < 1e-30 (a parameter used previously [59]), and was then systematically compared to every other bin to check for multiple associating (i.e. with 3 or more) pairs of BESs. Different bin sizes made little difference to the results, indicating the stability of the approach.

Comparative mapping of Brassica FPC contigs onto the Arabidopsis genome. In subfigures (cartoons, not based on real data) A, C and D, Brassica contigs are displayed with assembled BAC clones (depicted by overlapping lines), and interspecific chromosomal synteny is shown in dashed lines. A). Interspecific chromosomal synteny inference. B). A Brassica contig (shown with a hexagon shape) is expected to be linked to multiple homologous regions in Arabidopsis (shown with circles), at most one ortholog, one α-paralog, two β-paralogs, and eight γ-paralogs. DNA losses may have removed some of them (shown with dashed-lined circles). C). A Brassica contig is linked to Arabidopsis duplicated regions. Unbalanced synteny often permits one to distinguish between orthology and paralogy, or reveals differential gene losses among paralogous regions. D). Inference of synteny discontinuity is shown for a Brassica contig against two Arabidopsis regions, which may indicate a chromosomal breakpoint during the diversification of the two species.
References
Soltis DE, Buggs RJ, Barbazuk WB, Schnable PS, Soltis PS: On the Origins of Species: Does Evolution Repeat Itself in Polyploid Populations of Independent Origin?. Cold Spring Harb Symp Quant Biol. 2009
Blanc G, Hokamp K, Wolfe KH: A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 2003, 13 (2): 137-144. 10.1101/gr.751803.
Bowers JE, Chapman BA, Rong J, Paterson AH: Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003, 422 (6930): 433-438. 10.1038/nature01521.
Tang H, Bowers JE, Wang X, Ming R, Alam M, Paterson AH: Synteny and collinearity in plant genomes. Science. 2008, 320 (5875): 486-488. 10.1126/science.1153917.
Lewin HA, Larkin DM, Pontius J, O'Brien SJ: Every genome sequence needs a good map. Genome Res. 2009, 19 (11): 1925-1928. 10.1101/gr.094557.109.
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, et al: The Sorghum bicolor genome and the diversification of grasses. Nature. 2009, 457 (7229): 551-556. 10.1038/nature07723.
Yang YW, Lai KN, Tai PY, Li WH: Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. Journal of Molecular Evolution. 1999, 48 (5): 597-604. 10.1007/PL00006502.
Vision TJ, Brown DG, Tanksley SD: The origins of genomic duplications in Arabidopsis. Science. 2000, 290 (5499): 2114-2117. 10.1126/science.290.5499.2114.
Koch M, Al-Shebaz IA, Mummenhoff K: Molecular systematics, evolution, and population biology of the mustard family (Brassicaceae). Annals of the Missouri Botanical Garden. 2003, 90: 151-171. 10.2307/3298580.
Yang TJ, Kim JS, Kwon SJ, Lim KB, Choi BS, Kim JA, Jin M, Park JY, Lim MH, Kim HI, et al: Sequence-level analysis of the diploidization process in the triplicated FLOWERING LOCUS C region of Brassica rapa. The Plant cell. 2006, 18: 1339-1347. 10.1105/tpc.105.040535.
Paterson AH, Lan TH, Amasino R, Osborn TC, Quiros C: Brassica genomics: a complement to, and early beneficiary of, the Arabidopsis sequence. Genome Biol. 2001, 2 (3): 1339-1347.
Paterson AH, Lan TH, Amasino R, Osborn TC, Quiros CF: Brassica genomics - a complement to, and early beneficiary of, the Arabidopsis sequence. Genome Biol. 2001, 2: 1339-1347.
Schranz ME, Song BH, Windsor AJ, Mitchell-Olds T: Comparative genomics in the Brassicaceae: a family-wide perspective. Curr Opin Plant Biol. 2007, 10 (2): 168-175. 10.1016/j.pbi.2007.01.014.
Bowman JL: Molecules and morphology: comparative developmental genetics of the Brassicaceae. Plant Systematics and Evolution. 2006, 259 (2-4): 199-215. 10.1007/s00606-006-0419-8.
Kempin SA, Savidge B, Yanofsky MF: Molecular-Basis of the Cauliflower Phenotype in Arabidopsis. Science. 1995, 267 (5197): 522-525. 10.1126/science.7824951.
Smith LB, King GJ: The distribution of BoCAL-a alleles in Brassica oleracea is consistent with a genetic model for curd development and domestication of the cauliflower. Molecular Breeding. 2000, 6 (6): 603-613. 10.1023/A:1011370525688.
Purugganan MD, Boyles AL, Suddith JI: Variation and selection at the CAULIFLOWER floral homeotic gene accompanying the evolution of domesticated Brassica oleracea. Genetics. 2000, 155 (2): 855-862.
Pease MS: Genetic studies in Brassica oleracea. Journal of Genetics. 1926, 16: 363-10.1007/BF02983007.
Currence T: Results from hybridizing cabbage with brussels sprouts. Proc Am Soc Hort Sci. 1934, 32: 485-487.
Detjen LR, McCue CA: Cabbage characters and their heredity. Delaware AES Technical Bulletin. 1933, 180: 1-127.
Yeager AE: The characteristics of crosses between botanical varieties of cabbage (Brassica oleracea). Proc Am Soc Hort Sci. 1943, 43: 199-200.
Lan TH, Paterson AH: Comparative mapping of quantitative trait loci sculpting the curd of Brassica oleracea. Genetics. 2000, 155 (4): 1927-1954.
Lan TH, Paterson AH: Comparative mapping of QTLs determining the plant size of Brassica oleracea. Theoretical and Applied Genetics. 2001, 103 (2-3): 383-397. 10.1007/s001220100615.
Meinke DW, Meinke LK, Showalter TC, Schissel AM, Mueller LA, Tzafrir I: A sequence-based map of Arabidopsis genes with mutant phenotypes. Plant Physiol. 2003, 131 (2): 409-418. 10.1104/pp.014134.
Wortman JR, Haas BJ, Hannick LI, Smith RK, Maiti R, Ronning CM, Chan AP, Yu CH, Ayele M, Whitelaw CA, et al: Annotation of the Arabidopsis genome. Plant Physiol. 2003, 132 (2): 461-468. 10.1104/pp.103.022251.
Town CD, Cheung F, Maiti R, Crabtree J, Haas BJ, Wortman J, Hine EE, Althoff R, Arbogast TS, Tallon LJ, et al: Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell. 2006, 18: 1348-1359. 10.1105/tpc.106.041665.
Parkin IA, Sharpe AG, Lydiate DJ: Patterns of genome duplication within the Brassica napus genome. Genome. 2003, 46 (2): 291-303. 10.1139/g03-006.
Mun JH, Kwon SJ, Yang TJ, Seol YJ, Jin M, Kim JA, Lim MH, Kim JS, Baek S, Choi BS, et al: Genome-wide comparative analysis of the Brassica rapa gene space reveals genome shrinkage and differential loss of duplicated genes after whole genome triplication. Genome biology. 2009, 10 (10): R111-10.1186/gb-2009-10-10-r111.
Lysak MA, Koch MA, Pecinka A, Schubert I: Chromosome triplication found across the tribe Brassiceae. Genome Res. 2005, 15 (4): 516-525. 10.1101/gr.3531105.
Schranz ME, Lysak MA, Mitchell-Olds T: The ABC's of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends in Plant Science. 2006, 11 (11): 535-542. 10.1016/j.tplants.2006.09.002.
Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KL, et al: The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature. 2008, 452 (7190): 991-996. 10.1038/nature06856.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, et al: The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007, 449 (7161): 463-467. 10.1038/nature06148.
Lan TH, DelMonte TA, Reischmann KP, Hyman J, Kowalski SP, McFerson J, Kresovich S, Paterson AH: An EST-enriched comparative map of Brassica oleracea and Arabidopsis thaliana. Genome Res. 2000, 10 (6): 776-788. 10.1101/gr.10.6.776.
Schmidt R, Acarkan A, Boivin K: Comparative structural genomics in the Brassicaceae family. Plant Physiology and Biochemistry. 2001, 39 (3-4): 253-262. 10.1016/S0981-9428(01)01239-6.
Babula D, Kaczmarek M, Barakat A, Delseny M, Quiros CF, Sadowski J: Chromosomal mapping of Brassica oleracea based on ESTs from Arabidopsis thaliana: complexity of the comparative map. Molecular Genetics and Genomics. 2003, 268 (5): 656-665.
Lukens L, Zou F, Lydiate D, Parkin I, Osborn T: Comparison of a Brassica oleracea genetic map with the genome of Arabidopsis thaliana. Genetics. 2003, 164 (1): 359-372.
Schranz ME, Quijada P, Sung SB, Lukens L, Amasino R, Osborn TC: Characterization and effects of the replicated flowering time gene FLC in Brassica rapa. Genetics. 2002, 162 (3): 1457-1468.
O'Neill CM, Bancroft I: Comparative physical mapping of segments of the genome of Brassica oleracea var. alboglabra that are homoeologous to sequenced regions of chromosomes 4 and 5 of Arabidopsis thaliana. Plant Journal. 2000, 23 (2): 233-243. 10.1046/j.1365-313x.2000.00781.x.
Parkin IAP, Gulden SM, Sharpe AG, Lukens L, Trick M, Osborn TC, Lydiate DJ: Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics. 2005, 171 (2): 765-781. 10.1534/genetics.105.042093.
Warwick SI, Black LD: Molecular Systematics of Brassica and Allied Genera (Subtribe Brassicinae, Brassiceae) - Chloroplast Genome and Cytodeme Congruence. Theoretical and Applied Genetics. 1991, 82 (1): 81-92.
Mun JH, Kwon SJ, Seol YJ, Kim JA, Jin M, Kim JS, Lim MH, Lee SI, Hong JK, Park TH, et al: Sequence and structure of Brassica rapa chromosome A3. Genome Biol. 2010, 11 (9): R94-10.1186/gb-2010-11-9-r94.
Choi SR, Teakle GR, Plaha P, Kim JH, Allender CJ, Beynon E, Piao ZY, Soengas P, Han TH, King GJ, et al: The reference genetic linkage map for the multinational Brassica rapa genome sequencing project. Theor Appl Genet. 2007, 115 (6): 777-792. 10.1007/s00122-007-0608-z.
Kaczmarek M, Koczyk G, Ziolkowski PA, Babula-Skowronska D, Sadowski J: Comparative analysis of the Brassica oleracea genetic map and the Arabidopsis thaliana genome. Genome. 2009, 52 (7): 620-633. 10.1139/G09-035.
Mun JH, Kwon SJ, Yang TJ, Kim HS, Choi BS, Baek S, Kim JS, Jin M, Kim JA, Lim MH, et al: The first generation of a BAC-based physical map of Brassica rapa. BMC Genomics. 2008, 9: 280-10.1186/1471-2164-9-280.
Kim JS, Chung TY, King GJ, Jin M, Yang TJ, Jin YM, Kim HI, Park BS: A sequence-tagged linkage map of Brassica rapa. Genetics. 2006, 174 (1): 29-39. 10.1534/genetics.106.060152.
Iniguez-Luy FL, Lukens L, Farnham MW, Amasino RM, Osborn TC: Development of public immortal mapping populations, molecular markers and linkage maps for rapid cycling Brassica rapa and B. oleracea. Theor Appl Genet. 2009, 31-43.
Ayele M, Haas BJ, Kumar N, Wu H, Xiao Y, Van Aken S, Utterback TR, Wortman JR, White OR, Town CD: Whole genome shotgun sequencing of Brassica oleracea and its application to gene discovery and annotation in Arabidopsis. Genome Res. 2005, 15 (4): 487-495. 10.1101/gr.3176505.
Yang TJ, Kim JS, Lim KB, Kwon SJ, Kim JA, Jin M, Park JY, Lim MH, Kim HI, Kim SH, et al: The Korea Brassica Genome Project: A glimpse of the Brassica genome based on comparative genome analysis with Arabidopsis. Comparative and Functional Genomics. 2005, 6 (3): 138-146. 10.1002/cfg.465.
Soderlund C, Humphray S, Dunham A, French L: Contigs built with fingerprints, markers, and FPC V4.7. Genome Res. 2000, 10 (11): 1772-1787. 10.1101/gr.GR-1375R.
Martin W: Gene transfer from organelles to the nucleus: frequent and in big chunks. Proc Natl Acad Sci USA. 2003, 100 (15): 8612-8614. 10.1073/pnas.1633606100.
Bowers JE, Arias MA, Asher R, Avise JA, Ball RT, Brewer GA, Buss RW, Chen AH, Edwards TM, Estill JC, et al: Comparative physical mapping links conservation of microsynteny to chromosome structure and recombination in grasses. Proc Natl Acad Sci USA. 2005, 102 (37): 13206-13211. 10.1073/pnas.0502365102.
Lim KB, Yang TJ, Hwang YJ, Kim JS, Park JY, Kwon SJ, Kim J, Choi BS, Lim MH, Jin M, et al: Characterization of the centromere and peri-centromere retrotransposons in Brassica rapa and their distribution in related Brassica species. Plant J. 2007, 49 (2): 173-183.
Kawabe A, Hansson B, Hagenblad J, Forrest A, Charlesworth D: Centromere locations and associated chromosome rearrangements in Arabidopsis lyrata and A-thaliana. Genetics. 2006, 173 (3): 1613-1619. 10.1534/genetics.106.057182.
Hansson B, Kawabe A, Preuss S, Kuittinen H, Charlesworth D: Comparative gene mapping in Arabidopsis lyrata chromosomes 1 and 2 and the corresponding A. thaliana chromosome 1: recombination rates, rearrangements and centromere location. Genetical Research. 2006, 87 (2): 75-85. 10.1017/S0016672306008020.
Koch MA, Kiefer M: Genome evolution among cruciferous plants: A lecture from the comparison of the genetic maps of three diploid species - Capsella rubella, Arabidopsis lyrata subsp Petraea, and A. thaliana. American Journal of Botany. 2005, 92 (4): 761-767. 10.3732/ajb.92.4.761.
Kuittinen H, de Haan AA, Vogl C, Oikarinen S, Leppala J, Koch M, Mitchell-Olds T, Langley CH, Savolainen O: Comparing the linkage maps of the close relatives Arabidopsis lyrata and A-thaliana. Genetics. 2004, 168 (3): 1575-1584. 10.1534/genetics.103.022343.
Lysak MA, Berr A, Pecinka A, Schmidt R, McBreen K, Schubert I: Mechanisms of chromosome number reduction in Arabidopsis thaliana and related Brassicaceae species. Proc Natl Acad Sci USA. 2006, 103 (13): 5224-5229. 10.1073/pnas.0510791103.
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, et al: Genome sequence of the palaeopolyploid soybean. Nature. 2010, 463 (7278): 178-183. 10.1038/nature08670.
Trick M, Kwon SJ, Choi SR, Fraser F, Soumpourou E, Drou N, Wang Z, Lee SY, Yang TJ, Mun JH, et al: Complexity of genome evolution by segmental rearrangement in Brassica rapa revealed by sequence-level analysis. BMC Genomics. 2009, 10: 539-10.1186/1471-2164-10-539.
Muller HJ: The Relation of Recombination to Mutational Advance. Mutation Research. 1964, 1 (1): 2-9. 10.1016/0027-5107(64)90047-8.
Udall JA, Quijada PA, Osborn TC: Detection of chromosomal rearrangements derived from homeologous recombination in four mapping populations of Brassica napus L. Genetics. 2005, 169 (2): 967-979. 10.1534/genetics.104.033209.
Wang X, Tang H, Bowers JE, Paterson AH: Comparative inference of illegitimate recombination between rice and sorghum duplicated genes produced by polyploidization. Genome Res. 2009, 19 (6): 1026-1032. 10.1101/gr.087288.108.
Wang X, Tang H, Bowers JE, Feltus FA, Paterson AH: Extensive concerted evolution of rice paralogs and the road to regaining independence. Genetics. 2007, 177 (3): 1753-1763. 10.1534/genetics.107.073197.
Attia T, R G: Meiotic pairing in haploids and amphihaploids of spontaneous versus synthetic origin in rape, Brassica napus L. Can J Genet Cytol. 1986, 28: 5-
Attia T, R G: Cytogenetic relationship within cultivated Brassica analyzed in amphihaploids from three diploid ancestors. Can J Genet Cytol. 1986, 323-329. 28
Newell CA, R ML, Bidney DL: Cytogenetic analysis of plants regenerated from tissue explants and mesophyll protoplasts of winter rape, Brassica napus L. Canadian Journal of Genetics and Cytology. 1984, 26: 10-
Paterson AH, Deverna JW, Lanini B, Tanksley SD: Fine Mapping of Quantitative Trait Loci Using Selected Overlapping Recombinant Chromosomes, in an Interspecies Cross of Tomato. Genetics. 1990, 124 (3): 735-742.
Thon MR, Martin SL, Goff S, Wing RA, Dean RA: BAC end sequences and a physical map reveal transposable element content and clustering patterns in the genome of Magnaporthe grisea. Fungal Genet Biol. 2004, 41 (7): 657-666. 10.1016/j.fgb.2004.02.003.
The Arabidopsis Genome Initiative: Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000, 408 (6814): 796-815. 10.1038/35048692.
Acknowledgements
We acknowledge financial support from the US Department of Agriculture Initiative for Future Agricultural and Food Systems (00-52100-9685), and the National Science Foundation Plant Genome Comparative Sequencing Program (IOS 0638418), Genes and Genome Systems Program (MCB 1021718), and Advances in Biological Informatics Program (DBI 0849896).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AHP and XW conceived the research. GP, CL, LN, BY, JEB, LL, EE, HS, CR, SK, JI, EG, CFQ, and RMA performed the experiments. XW, MT, JEB, JM, YS, BP, and YX performed data analysis. BM constructed the online service. AHP, TCO, JCP, and CT led the research. XW and AHP drafted the manuscript. All authors read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, X., Torres, M.J., Pierce, G. et al. A physical map of Brassica oleracea shows complexity of chromosomal changes following recursive paleopolyploidizations. BMC Genomics 12, 470 (2011). https://doi.org/10.1186/1471-2164-12-470
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-12-470