
Abstract
Background
In the last few years, efforts have been made to identify large effect QTL for grain yield under drought in rice. However, identification of most precise and consistent QTL across the environments and genetics backgrounds is essential for their successful use in Marker-assisted Selection. In this study, an attempt was made to locate consistent QTL regions associated with yield increase under drought by applying a genome-wide QTL meta-analysis approach.
Results
The integration of 15 maps resulted in a consensus map with 531 markers and a total map length of 1821 cM. Fifty-three yield QTL reported in 15 studies were projected on a consensus map and meta-analysis was performed. Fourteen meta-QTL were obtained on seven chromosomes. MQTL1.2, MQTL1.3, MQTL1.4, and MQTL12.1 were around 700 kb and corresponded to a reasonably small genetic distance of 1.8 to 5 cM and they are suitable for use in marker-assisted selection (MAS). The meta-QTL for grain yield under drought coincided with at least one of the meta-QTL identified for root and leaf morphology traits under drought in earlier reports. Validation of major-effect QTL on a panel of random drought-tolerant lines revealed the presence of at least one major QTL in each line. DTY 12.1 was present in 85% of the lines, followed by DTY 4.1 in 79% and DTY 1.1 in 64% of the lines. Comparative genomics of meta-QTL with other cereals revealed that the homologous regions of MQTL1.4 and MQTL3.2 had QTL for grain yield under drought in maize, wheat, and barley respectively. The genes in the meta-QTL regions were analyzed by a comparative genomics approach and candidate genes were deduced for grain yield under drought. Three groups of genes such as stress-inducible genes, growth and development-related genes, and sugar transport-related genes were found in clusters in most of the meta-QTL.
Conclusions
Meta-QTL with small genetic and physical intervals could be useful in Marker-assisted selection individually and in combinations. Validation and comparative genomics of the major-effect QTL confirmed their consistency within and across the species. The shortlisted candidate genes can be cloned to unravel the molecular mechanism regulating grain yield under drought.
Similar content being viewed by others


Background
Drought is a severe abiotic stress that affects the production and productivity of rice. Drought stress at the reproductive stage is the most devastating [1, 2]. Because of the ongoing process of climate change, the rainfall pattern has become more irregular in the cropping season, causing widespread drought in rice-growing areas, which results in severe yield losses [3, 4]. The development of drought-tolerant varieties that maintain good yield under drought is a priority area of rice research for sustainable rice production.
Marker-assisted mapping and introgression of major-effect QTL for grain yield under drought could be an efficient and fast-track approach for breeding drought-tolerant rice varieties [5]. However, the successful use of QTL in marker-assisted selection depends on their effect and consistency across genetic backgrounds and environments. Most of the QTL for grain yield under drought have been mapped against a single genetic background in early-segregating generations (F3, BC2, and BC2F2) evaluated in a limited number of environments. Such QTL may not provide a consistent effect because of variation in the genetic background and environment. Additionally, the QTL may not be transferrable to other backgrounds because of unfavorable epistatic interactions resulting in reduced or even no effects in a new genetic background [6, 7]. Considering all these facts, it is difficult to predict the usefulness of QTL for MAS based only on their performance in an individual genetic background in any particular study.
A more efficient way to select QTL for MAS is to compare the identified QTL with earlier reported studies for their consistency of location and effect across genetic backgrounds and environments. Consistently identified QTL at the same chromosomal location, explaining high phenotypic variance and having a major effect on a trait, can be effectively used in MAS [8–10].
QTL meta-analysis is an approach to identify consensus QTL across studies, to validate QTL effects across environments/genetic backgrounds, and also to refine QTL positions on the consensus map [11]. QTL meta-analysis requires independent QTL for the same trait obtained from different populations, different locations, or different environmental conditions [11]. The consistent QTL identified by meta-analysis for a set of QTL at a confidence interval (CI) of 95% are called meta-QTL (MQTL). The meta-QTL with the smallest CI and having a consistent and large effect on a trait are useful in MAS. In plants, the concept of meta-analysis has been applied to the analysis of QTL/genes for blast resistance [12], root traits and drought tolerance in rice [9, 10], lint fiber length in cotton [13], cyst nematode resistance in soybean [14], fusarium head blight resistance in wheat [15], flowering time [16], drought tolerance in maize [17], and disease resistance in cocoa [18].
QTL validation is another approach to confirm the effect of QTL across different genetic backgrounds. QTL regions harbor many genes; among them, a few key genes could be more important in the regulation of a complex trait. Meta-QTL regions with refined positions are more accurate for short-listing of candidate genes. The common candidate genes short-listed across the meta-QTL are more likely candidates that regulate yield [9].
In this study, QTL meta-analysis was carried out for yield QTL under drought to develop a consensus map and to identify consensus yield QTL under drought with the objective to provide markers of MQTL with high effects and small confidence intervals for possible use in MAS or for fine-mapping QTL for gene discovery. Also, markers linked to 12 major QTL for grain yield were validated on a set of random drought-tolerant lines, including landraces and improved drought breeding lines developed at IRRI, to know the frequency of their universal presence. Further, a comparative genomics approach was used to identify the homologous regions of MQTL in other cereal crops such as maize, sorghum, wheat, and barley (http://www.gramene.org/,http://www.maizegdb.org/, http://www.graingenes.org).
Materials and methods
Meta-QTL analysis
Three steps were employed for the identification of a consensus QTL for grain yield under drought. First, in a bibliographic review, reliable data on QTL for yield per plant were compiled. Second, a consensus map was created and on this map the QTL of individual studies was projected. In the third step, a meta-analysis was performed on QTL clusters to identify the consensus MQTL.
Bibliographic review and synthesis of yield QTL data
QTL information was collected from published reports involving mapping of QTL for grain yield under drought. There were 15 reports of a QTL mapping for grain yield under drought. The details of the parents used in developing the mapping population, size of the mapping population, markers used, and yield QTL identified are given in Table 1. In all, 53 QTL were reported for yield.
Development of a consensus map
A consensus genetic map was constructed and meta-analysis was performed using Biomercator v2.0 (http://www.genoplante.com/). The rice genetic linkage map of Temnykh et al. [19] was used as a reference map, on which the markers of 15 studies were projected to develop a consensus map. Chromosomes connected with fewer than two common markers to the reference map were excluded before the creation of the consensus map. Inversions of marker sequences were filtered out by discarding inconsistent loci with the exception of very closely linked markers. After the integration of all maps, the consensus map contained 531 markers, including SSR, RFLP, AFLP markers, and genes. The consensus map covered a total length of 1821 cM, with an average distance of 3.5 cM between markers.
QTL projections
For all studies, the 95% confidence intervals of initial QTL on their original maps were estimated using the approach described by Darvasi and Soller [20]:

Where N is the population size and R2 the proportion of the phenotypic variance explained by the QTL. The CI was re-estimated to control the heterogeneity of CI calculation methods across studies. Projection of QTL positions was performed by using a simple scaling rule between the original QTL flanking marker interval and the corresponding interval on the consensus chromosome. For a given QTL position, the new CI on the consensus linkage group was approximated with a Gaussian distribution around the most likely QTL position. All projections of QTL onto the consensus map were performed using the Biomercator (2.0) (http://www.genoplante.com/).
Meta-analysis
Meta-analysis was performed on the QTL clusters on each chromosome using Biomercator (2.0) (http://www.genoplante.com). The Akaike Information Criterion (AIC) was used to select the QTL model on each chromosome [21]. According to this, the QTL model with the lowest AIC value is considered a significant model indicating the number of meta-QTL. QTL meta-analysis requires independent QTL for the same trait obtained from different plant populations, different locations, or different environmental conditions [11].
QTL validation
Genotyping
All molecular marker work was conducted in the Gene Array and Molecular Marker Analysis (GAMMA) Laboratory, Plant Breeding, Genetics and Biotechnology (PBGB) division, IRRI. For DNA extraction, freeze-dried samples were used. Freeze-dried leaf samples were cut in eppendorf tubes and ground through a GENO grinder. Extraction was carried out by the modified CTAB method. DNA samples were stored in 2-mL deep-well plates (Axygen Scientific, California, USA). DNA samples were quantified on 0.8% agarose gel and concentration adjusted to approximately 25 ng μL-1. PCR amplification was done with a 15-μL reaction mixture having 40 ng DNA, 1 × PCR buffer, 100 μM dNTPs, 250 μM primers, and 1 unit Taq polymerase enzyme. The PCR profiles started with an initial denaturation of DNA at 94°C for 5 minutes, followed by 35 amplification cycles of denaturation at 94°C for 1 minute, annealing temperatures varied from 55°C to 58°C for 45 seconds based on the primer, extension at 72°C for 1 minute and final extension at 72°C for 7 minutes. The PCR products were resolved on 8% non-denaturing polyacrilamide gels (PAGE). The gels were scored taking respective QTL donor alleles as reference band and scores were used for QTL validation. The details of the peak markers of the 12 major effect QTL are given in Additional File 1.
Twelve major effect drought grain yield QTL were validated on a panel of 92 drought tolerant lines consisting of traditional drought tolerant donors, drought tolerant breeding lines developed through conventional breeding approaches and random high yielding lines under drought from QTL mapping populations. The peak marker of all the twelve major effect QTL were amplified on the drought panel lines. The lines were scored taking QTL donor allele as a base. The list of lines is given in the Additional File 2.
Gene content analysis
The 14 meta-QTL were analyzed for gene content to know the presence of genes and gene clusters responsible for drought. A comparative genomics approach was followed to analyze the genes present in meta-QTL. Gene content was noted based on annotated data of homologous regions in Nipponbare using RAP, Build5 (http://rapdb.dna.affrc.go.jp/download/index.html). It is assumed that the genes identified in Nipponbare regions are homologous and collinear to those underlying the yield QTL under drought mapped in different studies involving different donors and recipients.
Comparative genomics to identify homologous regions in cereals
A comparative genomics approach was followed to identify homologous regions between rice and maize using the genomic databases (http://www.gramene.org). Homologous regions identified were checked for the presence of drought grain yield QTL of maize (http://www.maizegdb.org). In sorghum, wheat, and barley, grain yield QTL reported were collected from a literature survey and these were compared with the meta-QTL using the comparative maps available in the Gramene database (http://www.gramene.org).
Results and discussion
Overview of QTL and development of a consensus map
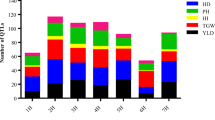
In the 15 populations of rice screened for drought tolerance to map QTL, population size ranged from 150 [22] to 436 lines [5]. The number of markers used ranged from 13 to 315 [1, 23]. The number of locations for phenotyping varied from 1 to 3. From the 15 studies, 53 yield QTL were reported, which were distributed on all the chromosomes except chromosome 11 (Table 1). The number of QTL per population ranged from 1 to 7. The proportion of QTL per chromosome ranged from one QTL each on chromosomes 5 and 7 to 18 yield QTL on chromosome 1. The distribution of yield QTL on different chromosomes showed that chromosomes 1, 2, and 10 have the highest number, 18, 7, and 7 QTL, respectively (Figure 1). The phenotypic variance of the initial QTL varied from 3.2% to 40% and the confidence interval of the markers varied from 2 to 30 cM. The rice genetic map of Temnykh et al. [19] was used as a reference map to develop a consensus map as this is a widely used genetic map of rice and it contained most of the markers used in the different studies. The consensus map consisted of 531 markers with a total map length of 1821 cM. The average distance between the markers was 3.5 cM, thus enabling the identification of a precise location of QTL. There were very few marker inversions in the consensus map, which were discarded from the final map and further analysis.

Distribution of grain yield QTL on rice chromosomes. The bar diagram depicts the frequency of drought grain yield QTLs on rice chromosomes
Meta-analysis and QTL validation
It is widely believed that QTL are accurate and can be positioned onto chromosomal locations by molecular mapping [24, 25]. However, their complex nature and context dependency in different genetic backgrounds and environments are constraints in identifying their precise location. The identification of the most accurate and precise major-effect QTL across genetic backgrounds and environments is a prerequisite for the successful use of QTL in MAS. Meta-analysis of QTL identified in different studies helps to identify the most precise and concise QTL, which can be further pursued for MAS or the identification of candidate genes. In our study, we attempted to identify the meta-QTL for grain yield under drought by genome-wide meta-analysis. From a bibliographic survey, a total of 53 QTL were short-listed for grain yield under drought from 15 studies. All 53 QTL were projected on a consensus map. The chromosomal regions with only one QTL were not considered for meta-analysis since meta-analysis by definition involves more than one QTL. Thus, 38 QTL were used for meta-analysis and meta-QTL were short-listed based on the Akaike Information Criterion (AIC). Accordingly, the QTL model with the lowest AIC value was considered a significant model indicating the number of meta-QTL. The number of meta-QTL along with their AIC values and confidence intervals are given in Table 2. In total, 14 independent meta-QTL were identified at a confidence interval of 95% on seven chromosomes, and meta-analysis successfully reduced the total QTL by 63% (Figures. 2, 3, 4, 5). The meta-QTL identified on each chromosome varied from 1 to 4. There were four meta-QTL on chromosome 1; two on chromosomes 2, 3, 8 and 10; and one each on chromosomes 4 and 12. The phenotypic variance of the meta-QTL varied from 4% to 28%. At 10 of the 14 meta-QTL, the mean phenotypic variance was more than 10%. In general, the confidence intervals at most of the meta-QTL were narrower than their respective original QTL. At nine loci on chromosomes 1, 2, 3, 4, 10, and 12, meta-QTL were narrower than the mean of their initial QTL. However, at five loci, the meta-QTL were broader than the mean of the initial QTL. The confidence intervals of the meta-QTL varied from 2.4 cM between the marker intervals RG109 and RM431 on chromosome 1 to 40.8 cM between the marker intervals RM337 and RM902 on chromosome 8. At two regions, meta-QTL1.4 (MQTL1.4) and MQTL12.1, the CI declined to around 2 cM. The physical intervals of the meta-QTL varied from 0.16 Mb to 5.3 Mb. Three meta-QTL were less than 500 kb. The meta-QTL regions with small genetic and physical intervals are useful in MAS. It is significant to see that seven QTL that had less than 1.3 Mb intervals also had a genetic interval of around 6 cM with a phenotypic variance of more than 10% (Figure 6). Three of these meta-QTL were on chromosome 1 and one each on chromosomes 2, 3 and 12. The physical intervals of MQTL1.2, MQTL1.3, and MQTL1.4 were less than 400 kb, that of MQTL12.1 was 700 kb, and those of MQTL3.2, MQTL4.1, and MQTL2.1 were 1Mb, 1.3 Mb, and 1.2 Mb, respectively. MQTL1.2, MQTL3.2, and MQTL12.1 had phenotypic variance of more than 20%. The seven MQTL regions with small genetic and physical intervals are important regions for MAS, fine mapping, candidate gene identification, and functional analysis. These QTL can be introgressed in popular rice mega-varieties to develop drought-tolerant and high-yielding lines. In addition to meta-analysis of QTL, the markers linked to the 12 major-effect QTL for grain yield were also validated on a panel of drought-tolerant lines to confirm their presence in larger set of lines. It is notable that major-effect QTL DTY 12.1 was present in 85% of the lines. DTY 3.2 , DTY 4.2 DTY 1.1 , DTY 8.1 , and DTY 1.2 were present in more than 50% of the lines (Figure 7). The amplification of the RM523 and RM11943 peak markers of DTY 3.2 and DTY 1.1 in a set of 92 drought tolerant panel lines is presented in Additional File 3. The result indicates the presence of at least one of the major-effect grain yield QTL in the drought panel lines. In general, the major-effect QTL identified for grain yield under drought have a genetic gain of 10% to 30%, with a yield advantage of around 150 to 500 kg/ha over recipient parents. However, considering practical benefit to farmers, the development of drought-tolerant rice varieties with a yield advantage of at least 1 ton/ha could be the desired target for rice breeders. The marker-aided QTL pyramiding of the major-effect MQTL identified in this study can be considered as an option for achieving this target.

Meta-QTLs identified on chromosomes 1 and 2 by Meta- analysis of reported yield QTLs. The picture shows the Meta-QTLs on chromosomes 1 and 2. Vertical lines on the left of chromosomes indicate the confidence interval, horizontal lines indicate the variance, MQTL are in red. Markers and genetic distance (cM) are shown on the right of chromosomes.

Meta-QTLs identified on chromosomes 3 and 4 by Meta- analysis of reported yield QTLs. The picture shows the Meta-QTLs on chromosomes 3 and 4. Vertical lines on the left of chromosomes indicate the confidence interval, horizontal lines indicate the variance, MQTL are in red. Markers and genetic distance (cM) are shown on the right of chromosomes.

Meta-QTLs identified on chromosomes 8 and 10 by Meta- analysis of reported yield QTLs. The picture shows the Meta-QTLs on chromosomes 8 and 10. Vertical lines on the left of chromosomes indicate the confidence interval, horizontal lines indicate the variance, MQTL are in red. Markers and genetic distance (cM) are shown on the right of chromosomes.

Meta-QTLs identified on chromosome 12 by Meta- analysis of reported yield QTLs. The picture shows the Meta-QTLs on chromosome 12. Vertical lines on the left of chromosomes indicate the confidence interval, horizontal lines indicate the variance, MQTL are in red. Markers and genetic distance (cM) are shown on the right of chromosomes.

Genetic and physical intervals of MQTL. The diagram depicts the genetic and physical intervals of the MQTLs. Solid bars indicates genetic interval (cM) and hollow bars indicates the physical interval (Mb) of the Meta-QTL.

Frequency of drought grain yield QTL in drought panel lines. The diagram depicts frequency of major effect drought grain yield QTLs in a drought panel consisting of 92 lines.
A comparison was made between the meta-QTL identified in this study with the meta-QTL identified for root traits in two earlier studies [9, 10]. It is very interesting to note that MQTL1.2, MQTL2.2, MQTL3.1, MQTL4.1, and MQTL8.2 coincided with QTL clusters for root and leaf morphology traits associated with drought tolerance/avoidance in rice [9]. All the 14 independent meta-QTL coincided with at least one meta-QTL identified for root traits under drought [10]. Earlier studies on meta-analysis of QTL for root traits [9, 10] and blast resistance in rice [12], fusarium head blight resistance in wheat [15], flowering time in maize [16], nematode resistance in soybean [14], and lint fiber length in cotton [13] identified precise and concise meta-QTL. Meta-QTL were also used to deduce candidate genes and were recommended for MAS in some of these studies.
Comparative genomics of MQTL
The existence of an evolutionary relationship among the grass families is a well-known fact. The syntenic relationship can be used to identify the homologous regions among these species, which in turn is useful in defining their role in plant growth, development, and adaptation across species. We compared meta-QTL regions for synteny in other cereal crops. The major-effect MQTL1.4 was also found in maize on chromosome 3 near marker msu2, in wheat on chromosome 4B near marker Rht-b1, and in barley on chromosome 6H near marker Bmac0316, while major-effect MQTL3.2 was also found in maize on chromosome 1 near marker Umc107a (Figure 8). All these markers were linked to grain yield under drought in their respective crops. The largest parts of chromosomes 1 and 3 of rice have a syntenic relation with chromosomes 3 and 1 of maize, so their respective homologous QTL were also found on the corresponding chromosomes. An interesting observation is that, near the sd1 locus on chromosome 1 of rice, QTL for grain yield under drought were identified most frequently. Sd1 is a major locus responsible for semidwarf plant stature in rice and its corresponding locus in wheat is Rht-b1 on chromosome 4B. MQTL1.4 is near the sd1 locus and also on its corresponding locus Rht-b1 in wheat, major QTL for grain yield under drought were detected.

Comparative map of MQTL 1.4 in rice with its corresponding grain yield QTL near Rht-b1 in wheat. The picture shows the comparative location of major effect Meta-QTL for grain yield under drought in rice MQTL1.4 on a wheat genetic map.
Gene content analysis and identification of candidate genes
Meta QTL with precise and narrow confidence intervals are useful in short listing the candidate genes. Using the annotated gene information available in the rice database, the genes present in the 14 meta-QTL regions were analyzed by comparative genomics approach and candidate genes were shortlisted. The short-listed candidate genes can be further confirmed by transgenic approaches by loss or gain of function studies. Most of the genes present in the MQTL were genes for hypothetical and expressed proteins, pseudo genes, genes for signal transduction, and transposable elements. However, there were many annotated genes/gene families that were common across the MQTL regions; these are probable candidate genes for yield under drought. It was found that three kinds of genes frequently occurred together in these regions. The genes/gene families were stress-inducible genes, growth and development-related genes, and sugar transport-related genes. Table 3 lists the important genes underlying MQTL for grain yield under drought. In six MQTL with less than a 1 Mb region, LRR kinase, leucine zipper, cell division-controlling proteins, sugar transport protein-like genes, no apical meristem (NAM), pentatricopeptide repeat proteins, cytokinin oxidase, F-box proteins, AP2-domain containing proteins, and zinc-finger transcription factors were present. The candidacy of these genes for yield and yield traits has already been proved in rice and other crops. Cytochrome P450 has a role in bassinosteroid homeostasis and had an influence on leaf angle leading to increased yield in rice [26, 27]. Pentatricopeptide repeats are present in the promoter region of Rf genes, which restore fertility and also play a role in embryogenesis in Arabidopsis[28, 29]. Zinc-finger (AN1-like)-like proteins are known to be involved in stress tolerance. Zinc-finger protein in rice are induced after different types of stresses, namely, cold, desiccation, salt, submergence, heavy metals, and mechanical injury. Over expression of the zinc-finger gene in transgenic tobacco conferred tolerance of cold, dehydration, and salt stress at the seed germination/seedling stage [30, 31]. F-box proteins play an important role in floral development and stress tolerance. In addition, F-box proteins appear to serve as the key components of the machinery involved in regulating plant growth and development throughout the plant's life cycle and their expression is influenced by light and abiotic stresses [32]. Leucine zippers are a class of transcription factor involved in ABA-independent stress tolerance. Over expression of OsbZIP23 in rice triggered clusters of genes regulating stress adaptations [33]. The no apical meristem gene (NAM) plays an important role in the growth and development of meristematic tissue. The root-specific expression of this gene resulted in enhanced root growth and improved drought tolerance in rice [34]. The other important genes that harbored the meta-QTL were the ERECTA and DREB genes. ERECTA is a leucine-rich repeat receptor-like kinase gene known for its influence on inflorescence development, stomatal density, epidermal cell expansion, and mesophyll cell proliferation. This gene is mainly involved in transpiration efficiency and enhanced drought response [35]. DREB is a well-known transcription factor that is induced by drought and it activates many down stream stress-responsive genes to ultimately improve the drought and chilling tolerance of rice [36]. Some of these short-listed genes can be considered as positional candidate genes that determine grain yield under drought. However, it is also well known that yield and adaptability to stress are complex in nature and highly negatively correlated. The QTL/genes for these two are often co-located. Even though individual genes have been proved to regulate yield under controlled drought experiments, a well-coordinated response of many genes is essential for drought tolerance under field conditions. This is evident from the presence of three different groups of gene clusters in most of the meta-QTL regions.
Conclusions
Meta-analysis of grain yield QTL is an effective approach in identifying concise and precise consensus QTL. The seven meta-QTL identified with small genetic and physical intervals could be useful in MAS/pyramiding. Validation of the major-effect QTL confirmed the consistency of the major-effect grain yield QTL under drought in different drought-tolerant panel lines. The comparative genomics approach to identify the consistency of drought grain yield QTL across species revealed the conservation of some of the loci, indicating their evolutionary significance. The presence of gene clusters in the meta-QTL indicates that a well-coordinated response of many genes is essential to achieve drought tolerance under field conditions.
References
Lanceras CJ, Pantuwan G, Jongdee B, Toojinda T: Quantitative trait loci associated with drought tolerance at reproductive stage in rice. Plant Physiology. 2004, 135: 384-399. 10.1104/pp.103.035527.
Venuprasad R, Dalid CO, Del Valle M, Zhao D, Espiritu M, Sta Cruz MT, Amante M, Kumar A, Atlin GN: Identification and characterization of large-effect quantitative trait loci for grain yield under lowland drought stress in rice using bulk-segregant analysis. Theoretical and Applied Genetics. 2009, 120: 177-190. 10.1007/s00122-009-1168-1.
Wassmann R, Jagadish SVK, Sumfleth K, Pathak H, Howell G, Ismail A, Serraj R, Redoña E, Singh RK, Heuer S: Regional vulnerability of climate change impacts on Asian rice production and scope for adaptation. Advances in Agronomy. 2009, 102: 91-133.
Bates BC, Kundzewicz ZW, Wu S, Palutikof JP: Climate change and water. In technical paper of the Intergovernmental Panel on Climate Change. 2008, IPCC Secretariat, Geneva, 210-[http://www.ipcc.ch/]
Bernier J, Kumar A, Ramaiah V, Spaner D, Atlin GN: A large-effect QTL for grain yield under reproductive-stage drought stress in upland Rice. Crop Science. 2007, 47: 507-518. 10.2135/cropsci2006.07.0495.
Podlich DW, Winkler CR, Cooper M: Mapping as you go: an effective approach for marker-assisted selection of complex traits. Crop Science. 2004, 44: 1560-1571. 10.2135/cropsci2004.1560.
Collins NC, Tardieu F, Tuberosa R: Quantitative trait loci and crop performance under abiotic stress: Where do we stand?. Plant Physiology. 2008, 147: 469-486. 10.1104/pp.108.118117.
Swamy BPM, Sarla N: Yield enhancing QTLs from wild species. Biotechnology Advances. 2008, 26: 106-111. 10.1016/j.biotechadv.2007.09.005.
Khowaja FS, Norton GJ, Courtois B, Price AH: Improved resolution in the position of drought-related QTLs in a single mapping population of rice by meta-analysis. BMC Genomics. 2009, 10: 276-10.1186/1471-2164-10-276.
Courtois B, Ahmadi N, Khowaja F, Price A, Rami JF, Frouin J, Hamelin C, Ruiz M: Rice root genetic architecture: meta-analysis from a QTL database improves resolution to a few candidate genes. Rice. 2009, 2: 115-128. 10.1007/s12284-009-9028-9.
Goffinet B, Gerber S: Quantitative trait loci: a meta-analysis. Genetics. 2000, 155: 463-473.
Ballini E, Morel JB, Droc G, Price AH, Courtois B, Nottehem JL, Tharreau DA: Genome wide meta analysis of rice blast resistance genes and quantitative trait loci provides new insights into partial and complete resistance. Molecular and Microbe Interaction. 2008, 21: 859-868. 10.1094/MPMI-21-7-0859.
Rong J, Feltus FA, Waghmare VN, Pierce GJ, Chee PW, Draye X, Saranga Y, Wright RJ, Wilkins TA, May OL, Smith CW, Gannaway JR, Wendel JF, Paterson AH: Meta-analysis of polyploid cotton QTL shows unequal contributions of sub genomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics. 2007, 176: 2577-2588. 10.1534/genetics.107.074518.
Guo B, Sleper DA, Lu P, Shannon JG, Nguyen HT, Arelli PR: QTLs associated with resistance to soybean cyst nematode in soybean meta-analysis of QTL locations. Crop Science. 2006, 46: 202-208. 10.2135/cropsci2005.04-0036.
Loffler M, Schon CC, Miedaner T: Revealing the genetic architecture of FHB resistance in hexaploid wheat (Triticum aestivum L.) by QTL meta-analysis. Molecular Breeding. 2009, 23: 473-488. 10.1007/s11032-008-9250-y.
Chardon F, Virlon B, Moreau L, Falque M, Joets J, Decousset L, Murigneux A, Charcosset A: Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with rice genome. Genetics. 2004, 168: 2169-2185. 10.1534/genetics.104.032375.
Hao Z, Li X, Liu X, Xie C, Li M, Zhang D, Zhang S: Meta-analysis of constitutive and adaptive QTL for drought tolerance in maize. Euphytica. 2010, 174: 165-177. 10.1007/s10681-009-0091-5.
Lanaud C, Fouet O, Clement D, Boccara M, Risterucci AM, Maharaj SS, Legavre T, Argout X: A meta-QTL analysis of disease resistance traits of Theobroma cacao L. Molecular Breeding. 2009, 24: 361-374. 10.1007/s11032-009-9297-4.
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch SR: Computational and experimental analysis of microsatellite in rice (Oryza sativa L.): frequency, length variation, transposon associations and genetic marker potential. Genome Research. 2001, 1: 1441-1452.
Darvasi A, Soller M: A simple method to calculate resolving power and confidence interval of QTL map location. Behavioural Genetics. 1997, 27: 125-132. 10.1023/A:1025685324830.
Hirotugu A: A new look at the statistical model identification. IEEE Transactions on Automatic Control. 1974, 19: 716-723. 10.1109/TAC.1974.1100705.
Beena : Studies on physio-morphological traits and genetic markers associated with drought response in rice (O. sativa). 2005, Tamil Nadu Agricultural University, Coimbatore, India, PhD thesis.
Prashant V: Development and morpho-molecular analysis of Swarna (Oryza sativa L.) introgression lines for drought tolerance. 2010, Veer Bahadur Singh Purvanchal University, Jaunpur, Uttar Pradesh, India
Price AH: Believe it or not, QTLs are accurate. Trends in Plant Science. 2006, 11: 213-216. 10.1016/j.tplants.2006.03.006.
Ashikari M, Matsuoka M: Identification, isolation and pyramiding of quantitative trait loci for rice breeding. Trends in Plant Science. 2006, 11: 344-350. 10.1016/j.tplants.2006.05.008.
Ashikari M, Sakakibara H, Lin S, Ymamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M: Cytokinin oxidase regulates rice grain production. Science. 2005, 309: 741-745. 10.1126/science.1113373.
Sakamoto T, Morinaka Y, Ohnishi T, Sunohara H, Fujioka S, Tanaka MU, Mizutani M, Sakata K, Takatsuto S, Yoshida S, Tanaka H, Kitano H, Matsuoka M: Erect leaves caused by brassinosteroid deficiency increase biomass production and grain yield in rice. Nature Biotechnology. 2005, 24: 105-109.
Bentolila S, Alfonso AA, Hanson MR: A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male-sterile plants. Proceedings of the National Academy of Sciences, USA. 2002, 99: 10887-10892. 10.1073/pnas.102301599.
Daniel A, Cushing E, Nancy R, Daniel R, Daniel M: Arabidopsis emb175 and other ppr knockout mutants reveal essential roles for pentatricopeptide repeat (PPR) proteins in plant embryogenesis. Planta. 2005, 221: 424-436. 10.1007/s00425-004-1452-x.
Mukhopadhyay A, Vij S, Tyagi AK: Overexpression of a zinc-finger protein gene from rice confers tolerance to cold, dehydration, and salt stress in transgenic tobacco. Proceedings of the National Academy of Sciences, USA. 2004, 101: 6309-6314. 10.1073/pnas.0401572101.
Wang L, Xu Y, Zhang C, Ma Q, Joo SH, Kim SK, Xu Z, Chong K: OsLIC, a novel CCCH-type zinc finger protein with transcription activation, mediates rice architecture via brassinosteroids signaling. PLOS One. 2008, 3: e3521-10.1371/journal.pone.0003521.
Jain M, Nijhawan A, Arora R, Agarwal P, Ray S, Sharma P, Kapoor S, Tyagi AK, Khurana JP: F-box proteins in rice: genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiology. 2007, 143: 1467-1483. 10.1104/pp.106.091900.
Xiang Y, Tang N, Du H, Ye H, Xiong L: Characterization of OsbZIP23 as a key player of the basic leucine zipper transcription factor family for conferring abscisic acid sensitivity and salinity and drought tolerance in rice. Plant Physiology. 2008, 48: 1938-1952.
Jeong JS, Kim YS, Baek KH, Jung H, Ha SH, Choi YD, Kim M, Reuzeau C, Kim JK: Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiology. 2010, 153: 185-197. 10.1104/pp.110.154773.
Masle J, Gilmore SR, Farquhar GD: The ERECTA gene regulates plant transpiration efficiency in Arabidopsis. Nature. 2005, 436: 866-870. 10.1038/nature03835.
Ito Y, Katsura K, Maruyama K, Taji T, Kobayashi M, Seki M, Shinozaki K, Shinozaki KY: Functional analysis of rice DREB1/CBF-type transcription factors involved in cold-responsive gene expression in transgenic rice. Plant and Cell Physiology. 2006, 47: 141-153.
Chandra Babu R, Nguyen BD, Chamarerk V, Shanmugasundaram P, Chezhian P, Jeyaprakash P, Ganesh SK, Palchamy A, Sadasivam S, Sarkarung S, Wade LJ, Nguyen HT: Genetic analysis of drought resistance in rice by molecular markers: association between secondary traits and field performance. Crop Science. 2003, 43: 1457-1469. 10.2135/cropsci2003.1457.
Yue B, Xiong L, Xue W, Xing Y, Luo L, Xu C: Genetic analysis for drought resistance of rice at reproductive stage in field with different types of soil. Theoretical and Applied Genetics. 2005, 111: 1127-1136. 10.1007/s00122-005-0040-1.
Zou GH, Mei HW, Liu HY, Liu GL, Hu SP, Yu XQ, Li MS, Wu JH, Luo LJ: Grain yield responses to moisture regimes in a rice population: association among traits and genetic markers. Theoretical and Applied Genetics. 2005, 112: 106-113. 10.1007/s00122-005-0111-3.
Yue B, Xue W, Xiong L, Yu X, Luo L, Cui K, Jin D, Xing Y, Zhang Q: Genetic basis of drought resistance at reproductive stage in rice: separation of drought tolerance from drought avoidance. Genetics. 2006, 172: 1213-1228.
Gomez MS, Satheesh Kumar S, Jeyaprakash P, Suresh R, Biji KR, Boopathi NM, Price AH, Chandra Babu R: Mapping QTLs linked to physio-morphological and plant production traits under drought stress in rice (Oryza sativa L.) in the target environment. American Journal of Biochemistry and Biotechnology. 2006, 2: 161-169.
Kumar R, Venuprasad R, Atlin GN: Genetic analysis of rainfed lowland rice drought tolerance under naturally-occurring stress in eastern India: heritability and QTL effects. Field Crops Research. 2007, 103: 42-52. 10.1016/j.fcr.2007.04.013.
Acknowledgements
Financial support to this study was provided by the Bill & Melinda Gates Foundation, USA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AK conceived the idea of Meta- analysis, QTL validation and comparative genomics of grain yield QTL under drought. BPMS compiled and analyzed the data, carried out the QTL validation and comparative genomics. SD helped in data compilation and analysis. BPMS, PV, SD, HUA and AK were responsible for drafting and editing the manuscript. All authors have read and approved the final manuscript.
Electronic supplementary material
12864_2010_3515_MOESM1_ESM.DOC
Additional File 1:Details of the markers used for QTL validation. This file contains the list of major effect QTLs for grain yield under drought and peak markers of the QTLs. Primer sequence, product size of the markers and annealing temperatures (Tm) used for amplifying the markers. (DOC 36 KB)
12864_2010_3515_MOESM2_ESM.XLS
Additional File 2:Drought panel lines for QTL validation. This table shows the list of drought panel lines and type of the breeding material. These lines were used for validating the major effect QTLs for grain yield under drought. (XLS 37 KB)
12864_2010_3515_MOESM3_ESM.XLS
Additional File 3:Amplification of RM523 and RM11943 peak markers of QTL 3.2 and QTL 1.1 in a set of 92 drought tolerant panel lines. The gel picture shows the amplication of RM523 and RM11943 peak markers of QTL 3.2 and QTL 1.1 in a set of 92 drought tolerant panel lines. (XLS 563 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Swamy, B.M., Vikram, P., Dixit, S. et al. Meta-analysis of grain yield QTL identified during agricultural drought in grasses showed consensus. BMC Genomics 12, 319 (2011). https://doi.org/10.1186/1471-2164-12-319
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-12-319