
Abstract
Background
Because the Japanese native cattle Kuchinoshima-Ushi have been isolated in a small island and their lineage has been intensely protected, it has been assumed to date that numerous and valuable genomic variations are conserved in this cattle breed.
Results
In this study, we evaluated genetic features of this breed, including single nucleotide polymorphism (SNP) information, by whole-genome sequencing using a Genome Analyzer II. A total of 64.2 Gb of sequence was generated, of which 86% of the obtained reads were successfully mapped to the reference sequence (Btau 4.0) with BWA. On an average, 93% of the genome was covered by the reads and the number of mapped reads corresponded to 15.8-fold coverage across the covered region. From these data, we identified 6.3 million SNPs, of which more than 5.5 million (87%) were found to be new. Out of the SNPs annotated in the bovine sequence assembly, 20,432 were found in protein-coding regions containing 11,713 nonsynonymous SNPs in 4,643 genes. Furthermore, phylogenetic analysis using sequence data from 10 genes (more than 10 kbp) showed that Kuchinoshima-Ushi is clearly distinct from European domestic breeds of cattle.
Conclusions
These results provide a framework for further genetic studies in the Kuchinoshima-Ushi population and research on functions of SNP-containing genes, which would aid in understanding the molecular basis underlying phenotypic variation of economically important traits in cattle and in improving intrinsic defects in domestic cattle breeds.
Similar content being viewed by others

Background
The Japanese native cattle Kuchinoshima-Ushi has long been bred on Kuchinoshima Island in the Tokara Archipelago of Kagoshima Prefecture, Japan (Figure 1A). Kuchinoshima-Ushi, which have been used mainly as pack animals, are characterized genetically as being lean with a small body size (approximately 500-kg adult males and 300-kg females), wide breast, narrow waist, and horns. They have variable coat colours, including black, black with white spots, and brown. This phenotype is probably due to a population bottleneck caused by long-term isolation on this island. Similarities between ancient Japanese cattle described in historical records and Kuchinoshima-Ushi suggest that they have retained some features of ancient native cattle (Figure 1B, C). Currently, the four major Japanese domestic cattle breeds, namely, Japanese Black, Japanese Brown, Japanese Shorthorn, and Japanese Polled, are bred mainly for meat in Japan. These breeds were established by crossing Japanese native cattle with several European cattle breeds during the mid-19th century to improve the native stock. However, the specific characteristics inherited by modern Japanese domestic cattle are unknown.

Habitat and morphological characteristics of Kuchinoshima-Ushi. A: Kuchinoshima Island located in the Tokara Archipelago, Japan. B: Picture of Kuchinoshima-Ushi used in this study, which kept at Shitara Field, Nagoya University. C: Picture of ancient Japanese cattle pulling a traditional Japanese cart in the Heian Age. This picture scroll, entitled "Heiji-Monogatari-Emaki" and drawn in the 13th century, is a national treasure stored at the Tokyo National Museum. Image: TNM Image Archives http://TnmArchives.jp/.
Since the first whole-genome assembly of the human genome in 2001 [1], the sequencing and assembly of mammalian genomes have quickly progressed. The bovine genome was assembled by the international bovine whole-genome sequencing project through a combination of shotgun and bacterial artificial chromosome (BAC) sequencing of an inbred Hereford cow and her sire by using capillary sequencing [2]. Recently, Van Tassell et al. (2008) contributed more than 23,000 single nucleotide polymorphisms (SNPs) to the bovine SNP database (dbSNP) by next-generation sequencing using a dairy breed (Holstein) and seven major breeds of beef cattle [3], and more than 2 million SNPs have been submitted to the bovine dbSNP to date. Although the array approach with these SNPs is useful, the underlying SNP resource is far from complete for understanding genome structure [4, 5]. Eck et al. (2009) generated 24 gigabase (Gb) of sequence with an average 7.4-fold sequence depth from a single Fleckvieh bull by next-generation sequencing [6] and identified more than 2 million previously unknown SNPs and 115,000 small insertions and deletions (indels) in comparison with the reference sequence. Although the bovine genome and HapMap projects have progressed [2, 7], the sequences of Japanese cattle have not been utilized in the respective projects. Therefore, we conducted a whole-genome analysis to examine the genetic features of the Japanese native cattle Kuchinoshima-Ushi and to gain a better understanding of the genetic relationship between domestic cattle breeds and Kuchinoshima-Ushi. This study provides detailed genetic information of this breed of cattle based on 64.2 Gb of sequence data generated by next-generation sequencing.
Results and Discussion
Sequencing, mapping, and SNP/indel detection
Whole-genome sequencing was performed on a Genome Analyzer II (GAII) using the genomic DNA from a Kuchinoshima-Ushi male and generated 64.2 Gb of high quality sequence on 34 paired-end lanes (75-bp reads in 28 lanes and 36-bp reads in 6 lanes). Read mapping to the reference sequence (Btau 4.0) was performed using BWA [8], and 86% of the obtained reads were successfully mapped to a unique position on the bovine reference genome sequence (Btau4.0). Of the total mapped reads, 239,789,699 (26% of the total reads) were mapped to multiple chromosomal positions. We used uniquely mapped reads (551,136,389; 60% of the total reads) for further analysis (Figure 2). The chromosomal distribution of the mapped reads was unbiased except for chromosome 13 (Additional file 1). The number of reads mapped to chromosome 13 seemed higher than the average relative to the chromosome length. When we examined the read coverage of the chromosome 13 in detail, we found that most of the reads were mapped in a region known as the satellite repeat region. Although it is unknown why such a number of reads were mapped preferentially in the satellite region of chromosome 13, it is probably due to the feature of the mapping program BWA. We also performed mapping analysis using repeat-masking sequences ftp://hgdownload.cse.ucsc.edu/goldenPath/bosTau4/bigZips/, which showed that high number of reads mapped to BTA13 disappeared (Additional file 1). It shows that the region with high number of reads mapped is masked as repeat.

Mapped and unmapped reads to the bovine reference genome. Mapped reads were 790,926,088 (86%) of all the reads (916,449,194). Among the mapped reads, 239,789,699 reads (26% of the total reads) were mapped to multiple chromosomal positions and 551,136,389 reads (60% of the total reads) were uniquely mapped. The number of unmapped reads was 125,523,106 (14%).
On an average, 93% of the genome was covered by reads (Additional file 2), and the number of mapped reads corresponds to 15.8-fold coverage across the covered region. However, the previous study describing whole-genome sequencing of a Fleckvieh bull reported that 98% of the genome was covered by the reads at a relatively low read depth (7.4-fold) [6]. The relatively lower genome coverage in our study is probably due to the fact that compared to the Fleckvieh breed, the Kuchinoshima-Ushi breed is evolutionarily more distantly related to the breed of the reference genome (Hereford). The difference in coverage can also be due to the number of prepared libraries (three different libraries in the Fleckvieh breed, whereas only one library in the Kuchinoshima-Ushi breed).
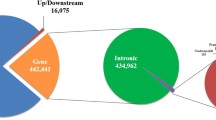
Using SAMtools [9], 6,303,790 SNPs were detected; 3,491,313 (55.4%) were heterozygous and 2,812,477 (44.6%) were homozygous. Of the identified SNPs, 5,031,648 (79.8%) were located in intergenic regions and 218,967 (3.5%) were within the 5-kb regions upstream or downstream from gene regions (Figure 3A). The remaining SNPs (1,053,175; 16.7%) were located in gene regions (Figure 3A). The number of SNPs in each chromosome decreased in accordance with the decrease of the length of chromosomes, and the SNP density in each 1-kb region of the total genome sequence showed that the chromosomal distribution of the SNPs was unbiased (Additional file 3, 4). The locations of the identified SNPs were also compared with those already published (dbSNP Build 129; latest version of dbSNP data ftp://ftp.ncbi.nlm.nih.gov/snp/organisms/cow_9913/chr_rpts/); 12.9% of the SNP sites were found in the database, and the remaining 87.1% were new. The percentage of newly identified SNPs in this study closely agreed with the results from a previous study [6]. We also investigated indel events and found 629,256 (284,007 insertions and 345,249 deletions). Of these indels, 498,101 (79.2%) were in intergenic regions, 23,686 (3.8%) were within 5-kb upstream or downstream of gene regions, and 107,469 (17.1%) were in gene regions (Figure 3B). The number of indels in each chromosome decreased in accordance with decrease of the length of the chromosomes (Additional file 3). The length of most indels was 1 bp (Additional file 5).

Identified SNPs and small indels. Identified SNPs (A) and small indels (B). We used the RefSeq and GenBank gene sets (16,635 genes) to annotate the detected variants. We found 1,003,695 intron SNPs, 17,684 untranslated regions (UTRs), 801 splice-site SNPs, 8,719 synonymous SNPs, and 11,713 nonsynonymous substitutions. Among the identified indels, 104,389 were found in intron regions, 2,942 in exon regions, and 138 in splice-sites.
Sequence data were deposited in the DDBJ Read Archive (DRA) [Accession #: DRA000172] and identified SNPs excluding ambiguous bases and those mapping to multiple locations unless providing more than 5 kbp flanking sequences were submitted to the single nucleotide polymorphism database (dbSNP) at NCBI under the handle TUANGRC. In addition to submitting data to the standard repositories, the positions of the SNPs for Kuchinoshima-Ushi can be viewed in a customized installation of the UCSC Genome Browser http://www.nodai-genome.org/btau/cgi-bin/hgGateway, along with supporting evidence (the number of reads for each allele and the density of the SNPs).
Functional annotation of nonsynonymous SNPs (nsSNPs) and nsSNP-containing genes
The SNPs in gene regions were annotated using the RefSeq and GenBank gene sets (16,635 genes). We found 1,004,496 SNPs in introns, 17,684 in untranslated regions (UTRs), 801 SNPs in splice-sites, and 20,432 coding SNPs leading to 11,713 nonsynonymous nucleotide substitutions (Figure 3A). The percentage of nonsynonymous changes in the coding region was 57.3%, which was higher than that of any other previous studies of whole-genome resequencing in humans and cattle [6, 10–15]. This finding would indicate on a possible occurrence of pseudogenes, the existence of proteins whose functions have been severely affected by extensive amino acid substitution, or significant differences in segmental duplications or copy number variants. Alternatively, the number of nsSNPs might have increased due to the false positives created by incomplete annotation information.
In our Kuchinoshima-Ushi data set, nsSNPs were detected in a total of 4,643 genes, which are listed in Additional file 6. Gene ontology (GO) terms associated with the 100 genes containing the highest number of nsSNPs were compared to those of all genes in the bovine whole genome by using the web-based tool agriGO [16, 17]. The analysis showed that the genes associated with molecular functions such as protein binding, catalytic activity, and metabolic pathways and their regulation were enriched among the top 100 nsSNP-containing genes in the Kuchinoshima-Ushi population (Figure 4A, Additional file 7). These results suggest the possibility that phenotypes associated with these genes may represent specific characteristics of the Kuchinoshima-Ushi breed. In contrast, genes involved in environmental adaptation such as sensory perception or immune function were not enriched in the list of the top 100 nsSNP containing genes.

Functional annotation of the genes containing nsSNPs. A. Gene ontology (GO) terms enriched in the 100 genes containing the highest number of nsSNPs. Blue columns show the percentage of genes among these nsSNP-containing genes, and red columns show the percentage of genes within the whole genome. In this chart, the secondary level terms are used as GO terms. B. Identified nsSNPs reportedly associated with phenotypes in the other breed of cattle. Six SNP sites exactly matched mutations reported in previous studies to be associated with economically important traits [25, 26, 29, 30, 35]. "-" implies the absence of record in the dbSNP database. EBV: estimated breeding values; SCS: somatic cell score.
We also examined whether nsSNP-containing genes were reportedly associated with economically important traits such as meat/milk production, growth rate, and domestication in other breeds of cattle. In order to extract information from studies reporting relationships between SNPs/genes and traits, we performed a PubMed search using economical traits such as growth rate or meat production as the query and limited the search to studies of Bos Taurus. From the result of the search, GenBank/EMBL/DDBJ nucleotide records reported in the obtained articles as well as sequences referred to in the articles as references (RefSeqs) were extracted and compared with the list of nsSNP-containing genes in our analysis of Kuchinoshima-Ushi.
Of the 4,643 nsSNP-containing genes, 708 matched to genes that have been reported to be potentially associated with economically important traits such as meat/milk production, growth rate, and domestication in other cattle breeds [18–22] (the list of genes containing nsSNPs that were reported as trait-associated in other cattle breeds is shown in Additional file 8). Some of the genes were also found to reside at positions of significant quantitative trait loci (QTL; data from Cattle QTLdb http://www.animalgenome.org/cgi-bin/QTLdb/BT/index); for example, body growth-associated genes such as genes for growth hormone (GH), growth hormone receptor (GHR), and leptin receptor (LEPR) [18–20]; lactation-related genes like prolactin receptor (PRLR) and caseins (CSN); and genes encoding immune-related proteins in milk, such as toll-like receptors (TLR s) [21, 22].
Recently, associations between SNPs and phenotypes have been reported in many studies, including cattle [23, 24]. Some SNP sites identified in Kuchinoshima-Ushi have already been reported to be associated with phenotypes in other cattle breeds (Figure 4B). For example, a mutation found in the fatty acid binding protein 4 (FABP4) was reported to be associated with palmitoleic acid (C16:1) content in intramuscular fat in Japanese Black cattle [25]. A mutation of the urotensin 2 receptor (UTS2R) was associated with skeletal muscle fat accumulation in Wagyu × Limousin population [26] and a mutation in calpain 1 (CAPN1) was associated with meat tenderness in Aberdeen Angus-cross cattle [27]. The nsSNPs found in the nucleotide-binding oligomerization domain containing 2 (NOD2/CARD15) gene were associated with the estimated breeding values for somatic cell score in Canadian Holstein cattle [28]. Identified SNP sites in the LEP and LEPR genes were associated with fat content in carcass meat of other cattle breeds including Nellore, Holstein-Friesian, Angus, Charolais, Hereford, and Simmental [19, 29, 30].
Nevertheless, population genetic studies in the Kuchinoshima-Ushi breed are required to validate the identified nsSNPs and to examine their association with relevant phenotypic traits in this population. Also, most nsSNPs identified in Kuchinoshima-Ushi have yet to be reported, nor have their functions or associations been investigated; new nsSNPs identified in this study will be valuable for future research.
Phylogeny of bovine-related species
To understand the genetic relationships between Kuchinoshima-Ushi and other breeds of cattle, we carried out a phylogenetic analysis. The resultant maximum likelihood tree of bovine-related species is shown in Figure 5. Bovinae subfamily relationships were largely consistent with those in an earlier study [31], with the exception of the phylogenetic position of bison and the sister-group relationship between East Asian river buffalo (Bubalus bubalis) and swamp buffalo (Bubalus carabasis). Two subtribes, namely, Bovina and Bubalina, formed a monophyletic group with high maximum likelihood (ML) bootstrap (BS) and Bayesian posterior probability (PP) support (BS > 99%, PP = 100%), respectively. In the Bovina clade, Hereford, Holstein, Tuli, and Kuchinoshima-Ushi comprised a monophyletic group with high statistical support (BS = 100%, PP = 100%), and Kuchinoshima-Ushi was the sister lineage of all other breeds of Bos taurus cattle with relatively high statistical support (BS = 74%, PP = 100%). Among species of wild cattle that have never been subjected to selective breeding, such as Banteng and Gaur, Kuchinoshima-Ushi was the nearest species of domesticated cattle. These data also suggest that Kuchinoshima-Ushi would be an appropriate breed for investigating adaptations associated with the domestication of wild cattle. When the sequence data of Japanese domestic breed of cattle such as Japanese Black will be available, the phylogenetic position of "Japanese native cattle" should be revealed more clearly.

Maximum likelihood tree of bovine-related species. Breeds of the species "Bos Taurus" (Hereford, Holstein, Tuli, and Kuchinoshima-Ushi) were boxed in pink. Numbers beside internal branches indicate bootstrap (BS) values (> 50%) from 1,000 replicates (left) and Bayesian posterior probabilities (right), respectively (shown as percentages). "-" indicates a node not recovered in the Bayesian analysis or <50% of BS values. "Mithan", also called "gayal", is a domesticated gaur.
Conclusions
In the present study, Japanese native cattle Kuchinoshima-Ushi, which have retained their phenotype over time without being affected genetically by European domestic breeds, were used for whole-genome resequencing with a next-generation sequencer. We identified 6.3 million SNPs, of which more than 5.5 million (87%) were found to be new. Among the identified SNPs, some nonsynonymous SNPs were found in genes that have been reported as candidate genes for QTL affecting economically important traits in other cattle breeds. The results of phylogenetic analysis showed the phylogenetic position of Kuchinoshima-Ushi. In the current status of domestic cattle, repeated selection for specific traits such as weight gain as well as milk and meat production has progressed, but problems have emerged with unconsidered traits such as reproduction and disease resistance. Our results provide a framework for future investigations aiming at the understanding of gene functions and identifying the molecular basis underlying phenotypic variation of economically important traits in domestic cattle breeds. It would contribute to the development of a new, more efficient breeding system preferentially for East Asian domestic cattle breeds including Japanese one. Furthermore, genome information for Kuchinoshima-Ushi is expected to be important not only in revealing the genetic structure of a geographically isolated breed originating from ancient, native Japanese cattle but also in identifying genetic traits that distinguish domestic breeds from original, undomesticated species.
Methods
Breeding conditions for Kuchinoshima-Ushi
For our experiments we used genomic DNA from a horned 119-month-old male Kuchinoshima-Ushi (#8915) (Figure 1B) that weighed 485 kg and was black with large white spots. The cattle in the wide field of Nagoya University are bred under good and relaxed conditions. In 1990, one male and three female Kuchinoshima-Ushi were captured on Kuchinoshima Island and transferred to the stock farm at Shitara Field, Nagoya University. In 1993, two males and three females were brought to the field from the Nagoya Animal Science Foundation. By 2004, the group had increased to 23 males and 22 females with inbred conditions in a closed colony.
DNA library construction and sequencing
Blood sampling was carried out according to the 'Regulations for Animal Experiments in Nagoya University' and the 'Guidelines for the Care and Use of Laboratory Animals', as specified by the Tokyo University of Agriculture.
Genomic DNA was extracted from blood using standard phenol/chloroform extraction methods [32]. A genomic DNA library was prepared using a paired-end DNA sample prep kit (Illumina Inc., San Diego, CA, USA) according to the manufacturer's instructions (Preparing Samples for Sequencing Genomic DNA, Part # 1003806 Rev.B, Illumina) with slight modification as follows: 2 μg of DNA were fragmented to a median fragment size of 200 bp using Adaptive Focused Acoustics (Covaris, Inc., Woburn, MA, USA). After size selection on a 2% agarose gel, 10 μl of the DNA fragments was enriched by 12 cycles of PCR. For quality control, an aliquot of the library was cloned into a sequencing vector (TOPO TA Cloning® Kit for Sequencing, Invitrogen Corp., Carlsbad, CA, USA) and 96 clones were sequenced by sanger sequencing. We found that all sequences were unique, and duplicates were not detected in the analyzed sample. BLAST search in the bovine genome sequence database using these sequences revealed high (90%) similarities to respective bovine sequences. DNA concentration was measured using UV-1800 UV-VIS Spectrophotometer (Shimadzu Corporation, Kyoto, Japan). The genomic DNA library was diluted to 10 nM, and a 2 μl aliquot was used to generate clusters on the Illumina Cluster Station using the Paired-End Cluster Generation Kit v2 (Illumina) and sequenced on the GAII with the SBS 36-cycle Sequencing Kit v3 following the manufacturer's instructions (Using the Paired-End Cluster Generation Kit v2 on the Cluster Station and Paired-End Module, Part #: 1005629 Rev.C; Using the SBS Sequencing Kit v3 on the Genome Analyzer, Part #: 1005637 Rev.A, Illumina) as 75-bp and 36-bp reads (28 and 6 paired-end lanes, respectively).
Software
For alignment and annotation of the sequence reads, we used the bovine genome assembly Btau 4.0 ftp://hgdownload.cse.ucsc.edu/goldenPath/bosTau4/chromosomes/ as a reference source. In this study, sequence scaffolds not yet assigned to specific chromosomes were excluded and no repeat masker was applied to the assembly.
Image analysis and ELAND alignment were performed with Illumina's Pipeline Analysis software ver. 1.4. Sequences passing through the standard Illumina GA pipeline filters (i.e., clusters with intensities greater than 0.6-times the average of the highest and the sum of the two highest intensities for the first 25 cycles) were retained for further analysis.
For short-read alignment and consensus assembly, we used a recently developed algorithm, BWA ver. 0.5.0 (the outline of the algorithm is available online http://bio-bwa.sourceforge.net/) [8]. The BWA default values for mapping were as follows: maximum edit distance (maxDiff) = 0.04, maximum number of gap opens (maxGapO) = 1, maximum number of gap extensions (maxGapE) = -1, disallow a long deletion within bp (nDelTail) = 16, disallow an indel within bp (nIndelEnd) = 5, take the first subsequence as seed (seedLen) = infinite, maximum edit distance in the seed (maxSeedDiff) = 2, number of threads (nThrds) = 1, mismatch penalty (misMsc) = 3, gap open penalty (gapOsc) = 11, gap extension penalty (gapEsc) = 4, and parameter for read trimming (trimQual) = 0. After read mapping, we discarded the reads mapped to multiple chromosomal positions and unmapped reads. Only reads mapped to a unique position on the reference genome sequence were used for SNP calling.
To call SNPs, we used SAMtools [9] and applied additional filters as follows: minimum read depth = 3, minimum read depth calling the SNP = 2, and a 30% cutoff of percent aligned reads calling the SNP per total mapped reads at the SNP sites. We also filtered these identified SNPs with more stringent parameters (i.e., minimum depth = 4, minimum SNP = 2, and 30% or higher aligned reads calling the SNP; and minimum depth = 5, minimum SNP = 3, and 30% or higher aligned reads calling the SNP), but the difference in the number of SNPs was small (Additional file 9). Heterozygous and homozygous SNPs were distinguished using an 80% cutoff of percent aligned reads calling the SNP. We also used BWA to estimate the sequence read depth, which influences the coverage and accuracy of SNP calling. After SNP calling, we annotated the SNPs using the GenBank and RefSeq gene sets (16,635 genes; the gene set is available from the UCSC download site ftp://hgdownload.cse.ucsc.edu/goldenPath/bosTau4/database).
Phylogenetic analysis
We used 15 species from the Bovinae subfamily (9 species from subtribe Bovina, 5 species from subtribe Bubalina, and one species from tribe Tragelaphini as an outgroup) for the phylogenetic analysis (Additional file 10). On the basis of a previous study [31], we used 10 nuclear genes (listed in Additional file 11). All of the sequence data except for that of Kuchinoshima-Ushi were previously determined and published (accession numbers can be seen in Additional file 11). Sequences of the genes from Kuchinoshima-Ushi were determined reflecting its SNP information in the reference sequence data. We concatenated newly-determined sequences of Kuchinoshima-Ushi and the published sequences and aligned them for the phylogenetic analysis. Phylogenetic trees were estimated using partitioned ML and partitioned Bayesian methods using RAxML ver. 7.0.3 [33] and MrBayes ver. 3.1.2 [34], respectively.
References
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, et al: The sequence of the human genome. Science. 2001, 291: 1304-1351. 10.1126/science.1058040.
Elsik CG, Tellam RL, Worley KC, Gibbs RA, Muzny DM, Weinstock GM, Adelson DL, Eichler EE, Elnitski L, Guigo R, Hamernik DL, Kappes SM, Lewin HA, Lynn DJ, Nicholas FW, Reymond A, Rijnkels M, Skow LC, Zdobnov EM, Schook L, Womack J, Alioto T, Antonarakis SE, Astashyn A, Chappie CE, Chen HC, Chrast J, Camara F, Ermolaeva O, Henrichsen CN, et al: The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science. 2009, 324: 522-528. 10.1126/science.1169588.
Van Tassell CP, Smith TP, Matukumalli LK, Taylor JF, Schnabel RD, Lawley CT, Haudenschild CD, Moore SS, Warren WC, Sonstegard TS: SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods. 2008, 5: 247-252. 10.1038/nmeth.1185.
Hayes BJ, Bowman PJ, Chamberlain AJ, Goddard ME: Invited review: Genomic selection in dairy cattle: progress and challenges. J Dairy Sci. 2009, 92: 433-443. 10.3168/jds.2008-1646.
VanRaden PM, Van Tassell CP, Wiggans GR, Sonstegard TS, Schnabel RD, Taylor JF, Schenkel FS: Invited review: reliability of genomic predictions for North American Holstein bulls. J Dairy Sci. 2009, 92: 16-24. 10.3168/jds.2008-1514.
Eck S, Benet-Pages A, Flisikowski K, Meitinger T, Fries R, Strom T: Whole genome sequencing of a single Bos taurus animal for single nucleotide polymorphism discovery. Genome Biology. 2009, 10: R82-10.1186/gb-2009-10-8-r82.
Gibbs RA, Taylor JF, Van Tassell CP, Barendse W, Eversoie KA, Gill CA, Green RD, Hamernik DL, Kappes SM, Lien S, Matukumalli LK, McEwan JC, Nazareth LV, Schnabel RD, Taylor JF, Weinstock GM, Wheeler DA, Ajmone-Marsan P, Barendse W, Boettcher PJ, Caetano AR, Garcia JF, Hanotte O, Mariani P, Skow LC, Williams JL, Caetano AR, Diallo B, Green RD, Hailemariam L, et al: Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009, 324: 528-532. 10.1126/science.1167936.
Li H, Durbin R: Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010, 26: 589-595. 10.1093/bioinformatics/btp698.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009, 25: 2078-2079. 10.1093/bioinformatics/btp352.
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR: Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008, 456: 53-59. 10.1038/nature07517.
Kim JI, Ju YS, Park H, Kim S, Lee S, Yi JH, Mudge J, Miller NA, Hong D, Bell CJ, Kim HS, Chung IS, Lee WC, Lee JS, Seo SH, Yun JY, Woo HN, Lee H, Suh D, Lee S, Kim HJ, Yavartanoo M, Kwak M, Zheng Y, Lee MK, Park H, Kim JY, Gokcumen O, Mills RE, Zaranek AW, et al: A highly annotated whole-genome sequence of a Korean individual. Nature. 2009, 460: 1011-1015.
Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, Pang AWC, Shago M, Stockwell TB, Tsiamouri A, Bafna V, Bansal V, Kravitz SA, Busam DA, Beeson KY, McIntosh TC, Remington KA, Abril JF, Gill J, Borman J, Rogers Y-H, Frazier ME, Scherer SW, Strausberg RL, et al: The diploid genome sequence of an individual human. PLoS Biol. 2007, 5: e254-10.1371/journal.pbio.0050254.
Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, Guo Y, Feng B, Li H, Lu Y, Fang X, Liang H, Du Z, Li D, Zhao Y, Hu Y, Yang Z, Zheng H, Hellmann I, Inouye M, Pool J, Yi X, Zhao J, Duan J, Zhou Y, Qin J, et al: The diploid genome sequence of an Asian individual. Nature. 2008, 456: 60-65. 10.1038/nature07484.
Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, Gomes X, Tartaro K, Niazi F, Turcotte CL, Irzyk GP, Lupski JR, Chinault C, Song XZ, Liu Y, Yuan Y, Nazareth L, Qin X, Muzny DM, Margulies M, Weinstock GM, Gibbs RA, Rothberg JM: The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008, 452: 872-876. 10.1038/nature06884.
Ahn SM, Kim TH, Lee S, Kim D, Ghang H, Kim DS, Kim BC, Kim SY, Kim WY, Kim C, Park D, Lee YS, Kim S, Reja R, Jho S, Kim CG, Cha JY, Kim KH, Lee B, Bhak J, Kim SJ: The first Korean genome sequence and analysis: Full genome sequencing for a socio-ethnic group. Genome Res. 2009, 19: 1622-1629. 10.1101/gr.092197.109.
Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, et al: The Gene Ontology (GO) database and informatics resource. Nucl Acids Res. 2004, 32: D258-261. 10.1093/nar/gkh036.
Zhou X, Su Z: EasyGO: Gene Ontology-based annotation and functional enrichment analysis tool for agronomical species. BMC Genomics. 2007, 8: 246-10.1186/1471-2164-8-246.
Thomas MG, Enns RM, Shirley KL, Garcia MD, Garrett AJ, Silver GA: Associations of DNA polymorphisms in growth hormone and its transcriptional regulators with growth and carcass traits in two populations of Brangus bulls. Gen Mol Res. 2007, 6: 222-237.
Ferraz JBS, Pinto LFB, Meirelles FV, Eler JP, De Rezende FM, Oliveira ECM, Almeida HB, Woodward B, Nkrumah D: Association of single nucleotide polymorphisms with carcass traits in Nellore cattle. Gen Mol Res. 2009, 8: 1360-1366. 10.4238/vol8-4gmr650.
Nkrumah JD, Li C, Yu J, Hansen C, Keisler DH, Moore SS: Polymorphisms in the bovine leptin promoter associated with serum leptin concentration, growth, feed intake, feeding behavior, and measures of carcass merit. J Anim Sci. 2005, 83: 20-28.
Bagnato A, Schiavini F, Rossoni A, Maltecca C, Dolezal M, Medugorac I, Sölkner J, Russo V, Fontanesi L, Friedmann A, Soller M, Lipkin E: Quantitative trait loci affecting milk yield and protein percentage in a three-country brown swiss population. J Dairy Sci. 2008, 91: 767-783. 10.3168/jds.2007-0507.
Hiendleder S, Thomsen H, Reinsch N, Bennewitz J, Leyhe-Horn B, Looft C, Xu N, Medjugorac I, Russ I, Kühn C, Brockmann GA, Blümel J, Brenig B, Reinhardt F, Reents R, Averdunk G, Schwerin M, Förster M, Kalm E, Erhardt G: Mapping of QTL for Body Conformation and Behavior in Cattle. J Hered. 2003, 94: 496-506. 10.1093/jhered/esg090.
Allan MF, Smith TPL: Present and future applications of DNA technologies to improve beef production. Meat Science. 2008, 80: 79-85. 10.1016/j.meatsci.2008.05.023.
Goddard ME, Hayes BJ: Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nature Reviews Genetics. 2009, 10: 381-391. 10.1038/nrg2575.
Hoashi S, Hinenoya T, Tanaka A, Ohsaki H, Sasazaki S, Taniguchi M, Oyama K, Mukai F, Mannen H: Association between fatty acid compositions and genotypes of FABP4 and LXR-alpha in Japanese Black cattle. BMC Genet. 2008, 9: 84-10.1186/1471-2156-9-84.
Jiang Z, Michal JJ, Tobey DJ, Wang Z, MacNeil MD, Magnuson NS: Comparative understanding of UTS2 and UTS2R genes for their involvement in type 2 diabetes mellitus. Int J Biol Sci. 2008, 4: 96-102.
Gill JL, Bishop SC, McCorquodale C, Williams JL, Wiener P: Association of selected SNP with carcass and taste panel assessed meat quality traits in a commercial population of Aberdeen Angus-sired beef cattle. Genet Sel Evol. 2009, 41: 36-10.1186/1297-9686-41-36.
Pant S, Schenkel F, Leyva-Baca I, Sharma B, Karrow N: Identification of single nucleotide polymorphisms in bovine CARD15 and their associations with health and production traits in Canadian Holsteins. BMC Genomics. 2007, 8: 421-10.1186/1471-2164-8-421.
Liefers SC, Veerkamp RF, Pas MFW, Delavaud C, Chilliard Y, Lende T: A missense mutation in the bovine leptin receptor gene is associated with leptin concentrations during late pregnancy. Anim Genet. 2004, 35: 138-141. 10.1111/j.1365-2052.2004.01115.x.
Buchanan FC, Fitzsimmons CJ, Kessel AGV, Thue TD, Winkelman-Sim DC, Schmutz SM: Association of a missense mutation in the bovine leptin gene with carcass fat content and leptin mRNA levels. Genet Sel Evol. 2002, 34: 105-116. 10.1186/1297-9686-34-1-105.
MacEachern S, McEwan J, Goddard M: Phylogenetic reconstruction and the identification of ancient polymorphism in the Bovini tribe (Bovidae, Bovinae). BMC Genomics. 2009, 10: 177-10.1186/1471-2164-10-177.
Maniatis T, Fritsch EF, Sambrook J: Molecular cloning: A Laboratory Manual. 1982, New York: Cold Spring Harbor Laboratory
Stamatakis A: RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006, 22: 2688-2690. 10.1093/bioinformatics/btl446.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574. 10.1093/bioinformatics/btg180.
Page BT, Casas E, Quaas RL, Thallman RM, Wheeler TL, Shackelford SD, Koohmaraie M, White SN, Bennett GL, Keele JW, Dikeman ME, Smith TPL: Association of markers in the bovine CAPN1 gene with meat tenderness in large crossbred populations that sample influential industry sires. J Anim Sci. 2004, 82: 3474-3481.
Acknowledgements
We thank Yutaka Yoshida, affiliated to the Tokyo University of Agriculture, for his help in preparation of the sample, Satoshi Sano and Misaki Imai for technical supports, and Yoshiaki Kikkawa, from the Tokyo University of Agriculture, Takahisa Yamada, from Niigata University, and Tetsuo Kunieda, from Okayama University, for their valuable discussions and suggestions. We also thank Hiroshi Ando, Nobue Tsuichihara, and Fumitaka Yoshimura for technical support of breeding and maintenance of this breed in the Field Science Center of Nagoya University. This study was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (S0801025).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SY, HY, and TK conceived of the study and participated in its design and coordination. SO and SE kept Kuchinoshima-Ushi and provided samples required for sequencing. RK, KT, YS, YK, TM, and YK performed the sample preparation and the sequencing experiments. RK, KT, and YS performed the data analysis. RK, KT, and TK drafted the manuscript. SY, HY, TK, SO, and SE supervised research and all authors contributed to and approved the final manuscript.
Ryouka Kawahara-Miki, Kaoru Tsuda contributed equally to this work.
Electronic supplementary material
12864_2010_9930_MOESM1_ESM.PDF
Additional file 1:Reads for all chromosomes of repeat masked and unmasked genome assembly. In each chromosome, left columns show the reads mapped to the assembly without repeat masking and right columns show those to the repeat-masked assembly. Among the reads that were mapped to the reference genome sequence, most were mapped in pairs (blue column in each chromosome). However, in some read pairs, only one was mapped (red column). Additionally, some read pairs were mapped, but the distances or directions were not adequate (green columns). Length of the chromosomes is shown in the yellow line. High number of reads mapped to BTA13 of the assembly without repeat masking (left column) was removed in the repeat-masked assembly (right column). (PDF 78 KB)
12864_2010_9930_MOESM2_ESM.PDF
Additional file 2:Length of the regions that are covered by reads for each chromosome. Length of the regions that are covered by reads for each chromosome. The length of chromosomes is shown in the blue columns, and that of the regions covered by reads is shown in the red columns. The percentage of the regions that are covered by reads in each chromosome is indicated by the green line. On an average, 93% of the genome is covered by reads. (PDF 72 KB)
12864_2010_9930_MOESM3_ESM.PDF
Additional file 3:The number of identified SNPs and indels for each chromosome. SNPs are shown in the blue columns, and indels are shown in the red columns. Length of chromosomes is indicated by the green line. (PDF 72 KB)
12864_2010_9930_MOESM4_ESM.PDF
Additional file 4:SNP distribution on each chromosome. SNP density (SNPs per 1 kbp) is plotted by physical position. Relative length of the chromosomes was correlated with the length of each chromosome without repeat regions. (PDF 149 KB)
12864_2010_9930_MOESM5_ESM.PDF
Additional file 5:Distribution of the size of indels. We identified 284,007 insertions (positive values) and 345,249 deletions (negative values). (PDF 29 KB)
12864_2010_9930_MOESM6_ESM.XLSX
Additional file 6:The list of genes containing nsSNPs. The list of genes containing nsSNPs along with the accession numbers (ss number). (XLSX 404 KB)
12864_2010_9930_MOESM8_ESM.XLSX
Additional file 8:The list of genes containing nsSNPs that were reported as trait-associated in other cattle breeds. (XLSX 54 KB)
12864_2010_9930_MOESM9_ESM.PDF
Additional file 9:The number of SNPs and indels with various filters. Detected SNPs and indels were filtered with additional filters and the number of homozygous and heterozygous SNPs and indels was compared. Parameters for the filters were (1) Depth: the number of reads mapped to the SNP sites, (2) SNPs: the number of reads calling SNP at the SNP site, and (3) Mutation: the cutoff value of percent aligned reads calling the SNP per total mapped reads at the SNP sites. Cut off value was 30% in all filters. In the table, "%" means the reduced percentage of the number of SNPs/indels compared with basic parameters (i.e., Depth≥ 3, SNPs≥ 2, and Mutation≥ 30). (PDF 63 KB)
12864_2010_9930_MOESM11_ESM.DOC
Additional file 11:Summary of the genes sequenced for the phylogenetic reconstruction. List of the genes and their accession numbers which were used for the phylogenetic analysis. (DOC 39 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article
Cite this article
Kawahara-Miki, R., Tsuda, K., Shiwa, Y. et al. Whole-genome resequencing shows numerous genes with nonsynonymous SNPs in the Japanese native cattle Kuchinoshima-Ushi. BMC Genomics 12, 103 (2011). https://doi.org/10.1186/1471-2164-12-103
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-12-103