
Abstract
Background
The N-acetyltransferase 2 (NAT2) gene plays a crucial role in the metabolism of many drugs and xenobiotics. As it represents a likely target of population-specific selection pressures, we fully sequenced the NAT2 coding region in 97 Mandenka individuals from Senegal, and compared these sequences to extant data on other African populations. The Mandenka data were further included in a worldwide dataset composed of 41 published population samples (6,727 individuals) from four continental regions that were adequately genotyped for all common NAT2 variants so as to provide further insights into the worldwide haplotype diversity and population structure at NAT2.
Results
The sequencing analysis of the NAT2 gene in the Mandenka sample revealed twelve polymorphic sites in the coding exon (two of which are newly identified mutations, C345T and C638T), defining 16 haplotypes. High diversity and no molecular signal of departure from neutrality were observed in this West African sample. On the basis of the worldwide genotyping survey dataset, we found a strong genetic structure differentiating East Asians from both Europeans and sub-Saharan Africans. This pattern could result from region- or population-specific selective pressures acting at this locus, as further suggested in the HapMap data by extremely high values of FST for a few SNPs positions in the NAT2 coding exon (T341C, C481T and A803G) in comparison to the empirical distribution of FST values accross the whole 400-kb region of the NAT gene family.
Conclusion
Patterns of sequence variation at NAT2 are consistent with selective neutrality in all sub-Saharan African populations investigated, whereas the high level of population differentiation between Europeans and East Asians inferred from SNPs could suggest population-specific selective pressures acting at this locus, probably caused by differences in diet or exposure to other environmental signals.
Similar content being viewed by others

Background
The N-acetyltransferase 2 (NAT2) gene plays a crucial role in the metabolism of xenobiotics, including many clinically useful drugs and exogenous chemicals present in the diet, cigarette smoke and the environment [1]. Extensive polymorphism in NAT2 gives rise to a wide interindividual variation in N-acetylation capacity. In particular, a clear bimodal distribution is observed that segregates the rapid acetylator phenotype, associated with a normal acetylation capacity, from the slow acetylator one, characterized by a reduced enzyme activity. These two main metabolic phenotypes occur with varying prevalence in populations of different ethnic origin [2].
The clinical consequences of the acetylation polymorphism can be severe if standard drug doses are applied, exposing patients to an increased risk of adverse drug reactions or a lack of therapeutic efficacy [3]. In addition, in the last decades, an increasing number of epidemiological studies have attempted to relate acetylation phenotype to a variety of complex human disorders, such as bladder cancer, atopic diseases, diabetes, Parkinson's disease and many others (see Butcher et al. [4] for a review). However, up to now, association studies in NAT2 have led to conflicting results among (and even within) human populations and most association findings have been difficult to replicate. One reason for these inconsistencies may relate to the fact that almost all studies focused on a limited number of candidate polymorphisms, which were not necessarily the same from one study to another [5, 6]. A shift toward a gene-based approach in which all common variation within a gene is considered jointly is advocated for future association studies [7]. By capturing all of the potential risk-conferring variations within NAT2, this approach should resolve much of the controversial issues of candidate-polymorphism studies.
There is now a large body of information on the distribution of NAT2 genetic variants all over the world [8]. However, most published reports used simplified protocols for NAT2 allele detection, omitting analysis of several polymorphic positions (such as G191A, C282T, T341C, and A803G within the coding region). They focused on a limited number of "indicator" mutations, thought to be tightly linked with other mutations and predictive of acetylator status, and have based allele designation on this. Such incomplete genotyping methods wrongly type different alleles as the same and may lead to misclassification of genotypes and deduced phenotypes [9]. Therefore, results of such investigations may be substantially biased and fail to provide an accurate picture of NAT2 allele distribution in worldwide populations.
In an attempt to better characterize the worldwide haplotype diversity and LD structure of NAT2, we performed an extensive survey of the literature to identify those samples that were adequately genotyped for all common variants in NAT2. In total, 41 population samples (including 6,727 individuals) from four continental regions (Africa, Europe, Asia, America) were selected and jointly analyzed. In addition, we performed full sequencing analysis of the NAT2 coding region in a large and ethnically well defined Mandenka sample from Eastern Senegal so as to further characterize African diversity at this locus and to detect novel variants not yet reported. Beyond a simple description of NAT2 gene diversity, the goal of the present study was to provide further insights into the evolutionary forces that most likely shaped NAT2 genetic variation in humans. In particular, three levels of diversity (intrapopulation, interpopulation, and interspecies variability) were used to investigate to what extent present NAT2 variation patterns solely reflect stochastic events of human evolution, or are distorted by natural selection.
Results
NAT2 sequence diversity in the Mandenka and variation in Sub-Saharan Africa
Results of the sequencing analysis of NAT2 in the Mandenka sample are reported in Table 1. A total of 12 polymorphic sites were identified, all located within the NAT2 coding exon. Two of them were singletons confirmed by resequencing. Apart from the seven SNPs that are commonly found in human populations, we found three additional variants that have been recently reported by Patin et al. [10] in several sub-Saharan African samples (C403G, G609T, G838A), and two novel nucleotide changes not yet described: C345T and C638T, which occurred at a frequency of 0.026 and 0.01, respectively. C638T leads to an amino acid change (P213L). Sixteen distinct haplotypes were inferred by the PHASE program, including two which were recently described in the Patin et al. data set [10] and four that are newly described here. Among these six haplotypes, four contain inactivating mutations and are thus predicted as 'slow alleles', whereas the other two (NAT2*12g and NAT2*12H) contain a nonsynonymous mutation (G609T or C403G) whose impact on phenotype is unknown. From diplotype configurations at NAT2 in the Mandenka, we inferred 48.5%, 39.2%, and 7.2% of slow, intermediate, and rapid acetylators, respectively. The remaining 5.1% individuals had an unknown acetylator status as they were carriers of either a NAT2*12g or a NAT2*12H haplotype.
Summary statistics of genetic variation at the NAT2 coding region (870 bp) in the Mandenka and the 12 African samples of Patin et al. [10] are reported in Table 2. Results for the entire surveyed region (1188 bp) in the Mandenka are also indicated. Patterns of diversity in this sample are entirely consistent with those displayed by the 12 other African samples. The mean values of the two nucleotide-variability measures, π and θ w , for the 13 samples are 0.268% and 0.221%, respectively. None of the tests of selective neutrality, performed on each sample both at the intrapopulation and at the interspecies levels, yielded significant results (not shown). This suggests that patterns of diversity at NAT2 are consistent with the hypothesis of selective neutrality and constant population size. The average sequence divergence between human and chimpanzee for NAT2 coding exon was of 1.6%, and the average substitution rate of 1.6 × 10-9 per nucleotide and year. All samples provided similar estimates both for Ne, the current effective population size, and TMRCA, the coalescence time back to the most recent common ancestor: average values were 14,218 individuals and 1.077 My, respectively, for the NAT2 coding exon. The age of mutations in the gene genealogy ranged from 96,200 (G857A) to 496,300 (A803G) years for the seven SNPs that commonly occur in human populations. The other polymorphisms, only reported to date in sub-Saharan Africans, had all estimated ages < 50,000 years.
NAT2 worldwide genotyping survey
A total of 6,727 individuals from 41 worldwide samples were analyzed for their genotype at the seven common SNPs of the NAT2 gene. All SNPs and populations were in Hardy-Weinberg equilibrium after Bonferroni correction for multiple testing. The seven SNPs defined 21 distinct haplotypes, whose composition in terms of SNP variants is given in Table 3.
Continental distribution of NAT2 SNP variants and haplotypes
Allele frequency variation of the seven common SNPs of NAT2 in the 41 sampled populations is shown in Figure 1, and the worldwide distribution of common NAT2 haplotypes is displayed in Figure 2A. Figure 1 clearly highlights the high correlation in frequency of the three variants 341C, 481T, and 803G in Europe, Asia and America. This reflects the high level of LD between these three SNPs which, in combination, form the NAT2*5B haplotype. This variant occurs at a high frequency in Europe but is rare in populations of Asian origin. In sub-Saharan Africa, a poorer correlation between 803G and the two other variants is observed. This can be explained by the higher frequency of NAT2*12A in Africa, in which the 803G variant occurs in isolation. The 191A variant (which defines the NAT2*14 cluster) is exclusively present in sub-Saharan Africa, and the 857A variant (which defines the NAT2*7 cluster) is mainly found in Asia and Central America. By contrast, the 590A variant occurs at roughly similar frequencies (~15–40%) in all worldwide samples, except in Amerindians where it is rare (0% and 3.7% in Ngawbe and Embera, respectively). The frequency of the 282T variant only slightly varies among human populations.

SNP frequencies in the 41 samples of the worldwide genotyping survey. Samples are ordered geographically (as in Additional file 1), thus including: SSAFR, sub-Saharan Africa (Tswana, Ateke Bantus, Bakola Pygmies, Baka Pygmies, Yoruba, Mandenka, Dogons, and Somali); NA, North Africa (Moroccans); EUR, Europe (Spanish, Sardinians, French, French-Canadians, UK Caucasians, both US Caucasian samples, Swedes, Saami, both German samples, Polish, Slovaks, Ashkenazi Jews, Romanians, Russians, and Turks); CSASIA, Central/South Asia (Gujarati, Turkmen, and Kyrgyz); EASIA, East Asia (the three Chinese, three Japanese, and two Korean samples, and Thai); AME, America (Embera, Ngawbe, and Nicaraguans).

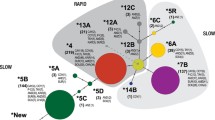
(A) Haplotype frequencies in the 28 samples genotyped for all seven common SNPs at NAT2 (i.e., without missing data), taken from the worldwide genotyping survey. Single populations are reported on the left side of the plot, with population codes in brackets; geographic areas are indicated on the right side, as follows: SSAFR, sub-Saharan Africa; NA, North Africa; EUR, Europe; CSASIA, Central/South Asia; EASIA, East Asia; AME, America. Only haplotypes with frequencies > 5% in at least one geographical region were represented individually; all other haplotypes were pooled into a single group (in white). Also, haplotypes NAT2*14A and NAT2*14B were pooled into the NAT2*14 cluster. (B) Median-joining networks of the inferred NAT2 haplotypes within three geographical regions: sub-Saharan Africa, Europe, and East Asia. Only haplotypes with frequencies > 0.005 within a geographic area were considered to construct the networks. Circle areas are proportional to the haplotypes' frequency, and branch lengths are proportional to the number of mutations separating haplotypes. Haplotypes' labels are shown in black; mutations are shown in red on corresponding network branches.
It is clearly apparent from Figures 2A and 2B that sub-Saharan African populations display greater haplotype diversity than either Europeans or Asians, and more complex relationships between NAT2 haplotypes are observed. A larger number of haplotypes of similar frequencies indeed occurs in these populations, generating a huge number of distinct genotypes. By contrast, in populations of Asian origin, only a few major haplotypes are found, namely NAT2*4, NAT2*6A and NAT2*7B. The mean haplotype diversity was estimated to be 0.80 ± 0.05 in Africans and 0.56 ± 0.09 in East Asians. Europeans displayed an intermediate value of 0.70 ± 0.04 [see Additional file 1].
In East Asia, the ancestral NAT2*4 haplotype associated with the rapid acetylator-phenotype accounts for more than 50% of the global variation (Thai excepted), while in Africa and Europe, the derived haplotypes associated with the slow-acetylator phenotype are predominant over NAT2*4. This observation explains the lower proportion of slow acetylators in East Asians compared to other human populations [see Additional file 2]. Derived haplotypes associated with the rapid acetylation phenotype (NAT2*12A, NAT2*13), are essentially found in Africa and are particularly frequent in Baka and Bakola Pygmies, which display a similar proportion of rapid acetylators as East Asians (83% and 90%, respectively).
Population structure
Several differentiated clusters of populations clearly appeared in the graphical representation of the MDS (Figure 3). The sub-Saharan African populations were found relatively dispersed while Europeans and East Asians (Thai excepted) formed tight clusters with small genetic distances between pairs of populations. The proportion of nonsignificant pairwise F ST 's at the 5% level within each continental area was 35% and 44% in Europe and East Asia, respectively, while it was only of 11% in sub-Saharan Africa. Among Africans, Somali were genetically the most similar to Europeans. Bakola Pygmies were found highly differentiated from all other populations (except from Baka Pygmies) which could easily be explained by strong genetic drift due to isolation and small effective size. Embera and Ngawbe Amerindians did not differentiate statistically from each other; while in close proximity to East Asians, they displayed significant F ST values with all other samples. In contrast, Nicaraguans were found very close to Europeans, in agreement with the Amerindian-European mixed origin of the sampled individuals. Turkmen and Kyrgyz displayed NAT2 allele frequencies intermediary to those observed in Europeans and East Asians, which points to the continuous nature of human genetic variation worldwide [11]. The MDS plot suggests that NAT2 genetic differentiation patterns are related to geography, which is confirmed by a high and significant correlation coefficient (r = 0.47, P < 10-5) observed between genetic and geographic distances.

Multidimensional scaling plot of Reynolds genetic distances among the 41 population samples of the worldwide genotyping survey. The stress value is 0.07, indicating a very good fit of the projection to the original data. Samples are numbered according to their population code [see Additional file 1 and Additional file 2]. The shaded areas highlight the distribution of samples from sub-Saharan Africa, Europe, and East Asia in the plot.
F ST gives a measure of the proportion of the genetic variance explained by differences among populations. The global F ST value estimated for the 41 worldwide samples was of 0.123 (P < 10-5). When grouping these 41 populations into five major geographic areas (sub-Saharan Africa, Europe/North Africa, Central/South Asia, East Asia, Central America), the vast majority of genetic variation was shown to occur within populations (83.5%), a high proportion (15%, P < 10-5) among geographic groups, and a mere 1.5% (P < 10-5) among populations within groups. When only three geographic groups were considered (35 samples grouped into sub-Saharan Africa, Europe/North Africa, and East Asia), the among groups component increased to 19%. The highest continental F ST value was found in sub-Saharan Africa (2.6%, P = 0.0001), whereas greater homogeneity was observed within East Asia (1.2%, P < 10-5), and even more so within Europe/North Africa (0.4%, P = 0.0002). In agreement with the MDS analysis, differentiation was very low between Europe (including Moroccans) and sub-Saharan Africa (F ST = 2.6%, P < 10-5), in contrast to the high F ST values observed between Asia and both Europe and Africa (22.2%, P < 10-5, and 23.2%, P < 10-5, respectively). Likewise, differences in the level of population differentiation were observed among the individual SNPs (Table 4). In particular, the C282T and G590A polymorphisms (defining the NAT2*6A haplotype) displayed strikingly low F ST values. By contrast, high F ST values were observed for the three SNPs T341C, C481T, and A803G which define the NAT2*5B haplotype. The highest F ST values at these three SNPs were observed between European and East Asian populations (around 40%).
LD analysis
We first tested whether the amount of LD (measured as the r2 coefficient between SNP pairs) differed between human populations. All population samples displayed similar levels of LD within each geographic area, except Somali who showed higher LD at NAT2 (average r2 value = 0.589) than other sub-Saharan African samples and were, in that respect, more similar to Europeans. The mean pairwise r2 value between the seven SNPs in the European samples (0.567 ± 0.075, including Moroccans) was significantly higher (Wilcoxon's test, P = 0.0002) than in both East Asians (0.276 ± 0.023) and Africans (0.243 ± 0.050, without Somali). No statistical differences in the level of LD were found between East Asian and African populations. However, the proportion of SNP pairs with r2 ≥ 0.5 was far smaller in sub-Saharan Africans (6.7%) than in both Europeans (40%) and East Asians (33.3%). Ashkenazi Jews exhibited the highest level of LD (average r2 value = 0.763); such an excess of LD is often observed in founder populations that recently grew from relatively small sizes [12]. We then tested whether the structure of LD was similar among populations by computing the correlation between r2 matrices of LD. Mantel's tests gave highly significant correlation values both between population pairs within geographic areas and for pairs of continental regions (P < 0.0001 with 10,000 permutations). Thus, although Europeans exhibited higher levels of LD at NAT2, the pattern of LD in this gene was found to be similar across human populations.
Discussion
This study provides a thorough description of NAT2 worldwide genetic diversity. By considering only the samples adequately characterized for the seven common SNPs of the NAT2 gene, we avoided biases arising from incomplete genotyping studies that may lead to both allele and phenotype misclassifications. These seven SNPs are the main polymorphisms occurring in human populations at NAT2 and their joint analysis has been shown to be highly predictive of the acetylation phenotype with a prediction rate close to 100% [13–17]. Such a high concordance between genotype and phenotype suggests that unknown NAT2 variants should be present at low frequencies and therefore may not substantially influence the phenotype prediction in population studies. Although these statements are tenable in populations that have been extensively studied at NAT2, such as Europeans or East Asians, it is not yet known if they hold in populations poorly or inadequately studied for NAT2 gene variation, such as sub-Saharan African populations. As shown by Patin et al. [10] and in this study, these populations usually display a greater allelic diversity and may contain novel variants not previously reported in populations of European or Asian origin. Although two new mutations were observed in our sequencing analysis of NAT2 in the Mandenka sample (C345T and C638T, see Table 1), we did not disclose yet any new major polymorphism apart from the seven acknowledged ones. Consequently, only a small proportion of subjects (5%) would have been classified differently regarding their acetylator status if they had been tested for only the seven common SNPs of NAT2 (these 5% individuals with an unknown acetylator status would have been classified as either intermediate or rapid acetylators). A similar observation has been made for several other African populations by Patin et al. [10] (Yoruba from Nigeria, Akele Bantus from Gabon, Mbuti Pygmies from the Democratic Republic of Congo, Chagga Bantu-speakers from Tanzania, Somali and !Kung San from Namibia). However, that same survey also detected novel variants occurring at non negligible frequencies in several Pygmy populations: up to one fourth of the individuals presented an unknown acetylator status due to the high prevalence of novel mutations with an unknown functional effect. Therefore, further sequencing studies that provide information about the entire frequency spectrum rather than pre-selected variants are required to provide an unbiased description of NAT2 sequence variation in not yet investigated human populations.
Genetic structure of human populations at NAT2
The genetic diversity patterns observed at NAT2 are largely consistent with those reported in many other studies of different gene regions in the human genome. The inferred levels of sequence diversity (Table 2) were found to be consistent with values reported at other highly variable human autosomal loci, such as LPL [18], GYPA [19], and CCR5 [20]. The average sequence divergence between human and chimpanzee (1.6%) was close to previous estimates of putatively neutral genomic regions [21], and the estimated Ne and TMRCA were also found to be in agreement with those of several other nuclear loci, which estimate the human Ne and TMRCA to be close to 10,000 and ~1 My, respectively [22, 23]. However, one should be aware that the stochastic nature of the coalescence process used to describe the genealogy, the assumptions that have to be made (for example, the absence of recombination and selection) and the removal of data (rare recombinant haplotypes) can all have important effects on inference and lead to imprecise estimations. Sub-Saharan Africans displayed the greatest haplotype diversity at NAT2 and also had the largest number of unique haplotypes [see Table 3]. Furthermore, haplotypes described outside Africa were essentially a subset of the collection of NAT2 haplotypes found within Africa. These features of molecular diversity are generally interpreted as strong evidence for the 'Out-of-Africa' model which hypothesizes that all modern populations emerged from a common ancestral population in Africa [22]. A linear diversity gradient away from Africa was indeed observed at NAT2 in this study, with African populations showing the highest heterozygosities, then successively decreasing in Europeans and East Asians [see Additional file 1]. This pattern is suggestive of a gradual loss of diversity in successive colonization bottlenecks as our species grew and spread all over the world [24]. Ngawbe and Embera Amerindians displayed comparable levels of haplotype diversity to East Asians (0.42 and 0.57, respectively). However, this last observation contrasts with the recent findings of Fuselli et al. [25] that demonstrated higher NAT2 intra-population genetic diversity in Native Americans than in East Asians, implying more complex processes in the evolution of populations at NAT2 than the simple linear model exposed here above.
Another line of evidence supporting a relatively recent African origin of modern humans came from our analysis of LD patterns at NAT2: the lowest levels of LD were found in African populations, a common finding in empirical studies of LD in human populations. This is consistent with a larger long-term effective size of African populations and/or a bottlenecked population history of non-African populations [22, 26]. Even within a small gene like NAT2, the SNP markers appeared to be poorly correlated in sub-Saharan Africans.
We observed a particular pattern of genetic diversity at NAT2 for the Thai sample compared to the other East Asian populations examined. Notably, the frequency of NAT2*4 was found to be significantly lower in Thai (0.30 versus around 0.50 in other Asian populations), resulting in a larger fraction of slow acetylators in this sample (50% versus 5–20%). Interestingly, among the East Asian populations investigated, the Thai sample is the only representative of the variation of NAT2 in Southern East Asia. Because the full NAT2 gene diversity has not yet been investigated in other Southeast Asian populations, it is not possible to conclude whether this population harbors a specific profile with respect to this genetic system, or if it resembles other Southeast Asians. Further data from these latter populations are needed to speculate on a possible genetic differentiation pattern between Northern and Southern East Asian populations at the NAT2 locus.
Possible selective pressures acting on NAT2
Because of its role in the detoxification of exogenous substances, the NAT2 gene has long been considered as a likely target of population-specific selective pressures. But many questions remain about the roles that population history and natural selection have played in shaping the diversity of NAT2. An intriguing point about this gene concerns the high frequency of poor-metabolizers and slow acetylator alleles in most human populations. This might represent the evolution of balanced polymorphisms, maintained by natural selection through heterozygote advantage or spatial-temporal selection of alternative alleles, as it has been shown for G6PD deficiencies or phenylketonuria [27, 28]. An alternative explanation is that NAT2 may evolve under no evolutionary constraint, this enzyme being not essential from an evolutionary perspective, maybe because it is dispensable or redundant with other enzymes. In terms of the detoxification of potentially harmful environmental aromatic amines, NAT1 seems indeed to be more active than NAT2, which preempts the latter's role as a key adaptation to increase the fitness of our species [16].
In this study, we investigated whether the patterns of sequence variation at NAT2 were consistent with a standard, neutral equilibrium model in 13 populations of sub-Saharan Africa. All neutrality tests found no evidence of a departure from selective neutrality. But making robust inferences on the action of natural selection at a particular locus requires a thorough characterization of population history, since the neutral null hypothesis is a composite hypothesis that also makes assumptions regarding the demography of the populations. It is typically assumed that the population is in equilibrium at constant size and with no population subdivision. But suppose that African populations did pass through a relatively narrow bottleneck in the late Pleistocene and then expand. In this case, the observed data might reflect the antagonistic effects of a bottleneck (which increases the frequency of rare alleles) and balancing selection (which decreases that frequency), and the apparent evidence in favour of a neutral model of evolution would be an artefact, produced by the confounded effects of these two opposing forces. However, the results of several studies have suggested that sub-Saharan African sequence diversity was compatible with an equilibrium model of long-term constant population size and random mating [29–32]. They showed that rapid growth from a small initial size was not compatible with the African sequence data and that, if a prehistoric growth occurred, it started from a relatively large Palaeolithic population. Therefore, under such an equilibrium model, NAT2 can be considered as a neutrally evolving gene, at least in the sub-Saharan African populations investigated. Further studies are needed to determine whether this finding can be generalized to all African populations. It would be also useful to increase both the number of individuals studied and the size of the genomic region surveyed (by investigating for instance the entire NAT2 gene sequence which spans around 10 kb) to increase the power of neutrality tests which remains weak when the sample sizes and the numbers of segregating sites are small.
The global level of genetic structure (F ST = 0.123, P < 10-5) was found to be remarkably consistent with the average F ST value for the human genome [33, 34], and the high correlation found between genetic and geographic distances (r = 0.47, P < 10-5) implies that patterns of human diversity at NAT2 can largely be accounted for by the simple interaction of drift and geographically-structured gene flow. However, while the overall degree of population structure at NAT2 was similar to previously reported values for neutral markers [35, 36], unusual patterns of differentiation were observed between geographic groups. In particular, a striking differentiation of East Asia both from Europe and from Africa was found, a result that could suggest the action of region- or of population-specific selective pressures. A large variance in F ST values was observed for individual SNPs (Table 4), with the highest F ST values being observed between European and East Asian populations for the three SNPs T341C, C481T, and A803G (around 40%). These values were compared to the empirical distribution of F ST across a 400-kb region encompassing the NAT gene family (composed of the two functional NAT1 and NAT2 genes, and the NATP pseudogene) [see Additional file 3]. F ST were computed for 550 individual markers spanning the 400-kb NATs region by using publicly available HapMap SNP data (the International HapMap Project [37] /Public Release #20). They all measured population differentiation between 60 Europeans and 90 East Asians. The F ST values displayed by the T341C, C481T, and A803G SNP variants (pointed by a black arrow in the Additional file 3) of the NAT2 coding region appeared to be exceptionally large since they differed by more than three standard deviations from the mean value computed for all 550 markers in the 400-kb region (solid horizontal line in Additional file 3). Although this criterion does not have any specific statistical significance, we may expect that such exceptional values are beyond those that might be accounted for by variation at neutral loci. High F ST values were also observed in the 25-kb region following the NAT2 gene. This may be explained by the strong LD linking sites within the NAT2 coding exon and sites in the noncoding region next to the NAT2 gene, observed in both European [see Additional file 4] and East Asian populations [see Additional file 5]. Indeed, if positive natural selection was acting on NAT2, such as through local adaptation, it would induce hitchhiking of nearby regions [38] and would increase interpopulation differentiation at linked neutral sites, as distinct haplotypes would be fixed in different populations. Besides, it is also interesting to note the high F ST values in the region surrounding the NAT1 gene and, surprisingly, in a noncoding segment preceding the NATP pseudogene.
By contrasting the F ST of individual SNPs to the empirical distribution of F ST across a genomic region, it is possible to identify those loci that exhibit unusual patterns of population differentiation as potential candidates for the action of natural selection [39, 40]. Several studies have used this strategy to detect the action of selection on specific genes or on a genome-wide scale [33, 34, 41]. However, genome-wide surveys of F ST have demonstrated substantial variation of F ST values across the genome, even among SNPs that are very close to each other [33, 34], thus stressing the difficulty to distinguish between non-random events such as local selection and random events such as extreme genetic drift as the agents responsible for the unusual patterns observed. Individual-marker F ST estimates are probably too variable to be reliable indicators of past selective events and other more powerful tests are required to provide unambiguous evidence of natural selection. The availability of genotype data for additional markers surrounding the NAT2 gene would enable the implementation of the long-range haplotype test [42] which has better power for identifying signatures of recent positive selection.
A possible explanation for the unusually large difference in allele frequencies observed between European and East Asian populations for the NAT2 variants could be the impact of population-specific selective pressures. The main molecular basis for the high discrepancy between Europeans and East Asians is that the most common allele at the NAT2 locus in Europeans (NAT2*5B) is very rare in East Asians and could represent a different selective advantage within the gene pools of these separate populations. Patin et al. [43] found evidence of a rapid increase in frequency of the NAT2*5B haplotype in Western and Central Eurasian populations in the last ~6,500 years in response to positive selection, suggesting that this slow allele probably conferred some selective advantage to its carriers in this part of the world. A thorough survey of NAT2 sequence variation in East Asians will be necessary to determine whether the predominance of the rapid-acetylator NAT2*4 allele over the slow ones is the result of local positive selection or whether it can be explained by stochastic processes such as genetic drift.
Conclusion
This study provides a thorough description of the worldwide haplotype diversity and LD structure of the NAT2 gene. We found that patterns of NAT2 sequence variation are consistent with selective neutrality in all sub-Saharan African populations investigated, whereas the high level of population differentiation between Europeans and East Asians inferred from SNPs may suggest population-specific selective pressures acting at this locus, probably caused by differences in diet or exposure to other environmental signals.
Methods
NAT2 sequencing of the Mandenka
Full sequence diversity of NAT2 exon 2, which contains the entire protein-coding region, was determined in 97 healthy unrelated individuals (62 men, 35 women) from the Niokholo Mandenka. This agriculturalist population from Eastern Senegal speaks a language belonging to Mande, a major primary branch of the Niger-Congo language family. We used DNA extracted from the lymphoblastoid cell lines (LCL) described in Excoffier et al. [44]. The sample size considered was sufficient to detect NAT2 variants present at a frequency ≥ 3%, with a probability of at least 99%.
A 1,211-bp fragment covering the entire coding region of NAT2 was amplified by PCR and subsequently sequenced on both DNA strands on an ABI 3730xl automated sequencer (Applied Biosystems) using three internal primer pairs, as described in Cascorbi et al. [45] (sequencing service provided by Macrogen, Seoul, Korea). The sequence variation of a 1188-bp fragment (from nt -59 to nt +1129) was fully surveyed, which includes the 870 bp of the NAT2 coding exon and 318 bp of non-coding flanking parts. All singletons were verified by PCR reamplification and resequencing the PCR products in both directions. The observed genotype frequencies at all polymorphic sites were in conformity with the assumptions of Hardy-Weinberg equilibrium when tested using Fisher's exact tests. Haplotype reconstruction was performed using the Bayesian method implemented in PHASE v.2.1 [46]. All 194 haplotype sequences were submitted to GenBank [GenBank:DQ904040-DQ904233].
For comparison purposes, we merged the NAT2 sequence data set with a collection of previously published sequences of the NAT2 coding exon (870 bp) [10]. This additional dataset consists in 285 individuals from 12 sub-Saharan African populations, including Yoruba from Nigeria (N = 31); Baka Pygmies (N = 31), Bakola Pygmies (N = 26), Bedzan Pygmies (N = 32) and Ngumba Bantus (N = 16) from Cameroon; Baka Pygmies (N = 16) and Akele Bantus (N = 26) from Gabon; Biaka Pygmies from Central African Republic (N = 24); Mbuti Pygmies from the Democratic Republic of Congo (N = 24); Chagga Bantu-speakers from Tanzania (N = 32); Somali (N = 20) and !Kung San from Namibia (N = 7).
NAT2 worldwide genotyping survey
We selected from published reports up to 2006 all the population samples that were genotyped for the seven most common SNPs at NAT2 and for which genotype data was available. Among these seven SNPs, all located in the coding exon, four result in an amino acid substitution that leads to a significant decrease in acetylation capacity (G191A, T341C, G590A, G857A). The other three are either silent mutations (C282T, C481T) or a non-synonymous substitution that does not alter the phenotype (A803G).
A complete list of the selected samples is provided in Additional file 1, along with a full description of each sample and references. The collected data consisted of 6,727 individuals (13,454 chromosomes) from 41 human populations representing major geographic regions: Europe (17 samples), North Africa (1), sub-Saharan Africa (8), Central/South Asia (3), East Asia (9), and Central America (3). The geographical distribution of the samples is shown in Additional file 2. Sample sizes range from 24 (Somali) to 1000 (Korean) individuals, with an average of 160 individuals per sample. For 13 samples, genotype data at one SNP (11 samples) or two SNPs (2 samples) out of the seven were missing. It involves either G191A (which has been shown to be extremely rare in Europeans and Asians) or the synonymous C282T polymorphism. These samples were excluded from LD analyses.
Statistical analyses
All sequence analyses were performed on both the Mandenka sample and on each of the 12 African samples of Patin et al. [10]. Homologous sequences from one chimpanzee (Pan troglodytes; Ensembl Chinpanzee genome) and one rhesus monkey (Macaca mulatta; GenBank XM_001098734) were used to infer SNPs' ancestral state. Each of these also served as an outgroup for evolutionary and population genetic tests.
DnaSP v.4.10 [47] was used to compute, in each sample, the nucleotide (π) and haplotype (H) diversity, Watterson's θ w [48], as well as to perform several neutrality tests to detect signals of natural selection: Tajima's D[49], Fu and Li's F* and D*[50], Fu's F s [51], and Fay and Wu's H[52]. The statistical significance of the tests was estimated from 10,000 coalescent simulations of an infinite site locus, conditional on sample size, both with and without recombination. The McDonald-Kreitman test [53] was applied to detect deviation from the neutral expectation of equal rates of nonsynonymous to synonymous polymorphism within humans and nonsynonymous to synonymous fixed substitutions between humans and chimpanzee.
A coalescence model for the ancestral history of a sample of genes was used to estimate the time scale of polymorphic variation in the NAT2 gene. The time to the most recent common ancestor (TMRCA) and mutation ages were estimated from the NAT2 gene tree, conditional on a maximum-likelihood estimate of θ(), the population mutation parameter. These estimates were computed with GeneTree v.9.0 [54] (running 106 replications), under a standard coalescence model assuming neutrality, the infinite-sites mutation model (haplotypes presumably affected by recurrent mutation or recombination were removed from the analysis), random mating, and constant population size. All estimates were inferred on individual population samples, to avoid biases due to population structure. Time, scaled in 2Ne units, was converted into years by use of a 25-year generation time and the value of the effective population size (Ne) obtained as divided by 4 μ. The neutral mutation rate per gene per generation (μ) was estimated based on human-chimpanzee sequence divergence, assuming a divergence time of 5 million years (My) [55].
For the worldwide genotyping survey, we inferred NAT2 haplotypes from the unphased multi-locus genotypes using PHASE v.2.1 software [46]. The individual acetylation phenotypes were then predicted from the haplotype combination at NAT2, in accordance with the acknowledged classification of NAT2* alleles based on their functional impact [see Table 3]: individuals with two low activity alleles were classified as slow acetylators, those with two functional alleles as rapid acetylators, and those with both a slow and a functional allele as intermediate acetylators.
Median-joining networks [56] describing the mutational relationships among the inferred NAT2 haplotypes were generated using Network 4.1.1 software [57].
Population structure in the worldwide genotyping survey set was investigated by an analysis of molecular variance (AMOVA) [58] that included the molecular distance matrix among NAT2 haplotypes. Population differentiation was tested by permutation tests (20,000 permutations) based on the F ST statistic. Coancestry coefficients, or linearized F ST values [59], were computed among populations and the resulting genetic distance matrix was used for multidimensional scaling analysis (MDS) [60] performed with the NTSYS v.2.1 software [61]. A Mantel test was applied to test the correlation of pairwise genetic distances with geographic distances, computed as great-circle distances between populations from their coordinates of latitude and longitude (US Caucasians and Ashkenazi Jews were excluded from the analysis since they could not be allocated precisely to a specific area). All calculations, including random-permutation procedures to assess statistical significance, were performed by use of the Arlequin v.3.0 package [62].
Pairwise LD between the seven genotyped SNPs was estimated by computing the r2 statistic [63] with DnaSP [47], after the exclusion, in each population of the worldwide genotyping survey, of SNPs with minor allele frequency (MAF) < 0.05. Statistical significance of LD between SNP pairs was assessed using Fisher's exact tests followed by Bonferroni corrections. Mantel tests to compare r2 matrices were performed using the program CADM [64]. Comparisons were made between populations within each continental group. Subsequently, r2 values were recalculated for populations pooled into geographical groups and Mantel tests were applied.
References
Hein DW: Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res. 2002, 506 (): 65-77.
Upton A, Johnson N, Sandy J, Sim E: Arylamine N-acetyltransferases – of mice, men and microorganisms. Trends Pharmacol Sci. 2001, 22: 140-146. 10.1016/S0165-6147(00)01639-4.
Meisel P: Arylamine N-acetyltransferases and drug response. Pharmacogenomics. 2002, 3: 349-366. 10.1517/14622416.3.3.349.
Butcher NJ, Boukouvala S, Sim E, Minchin RF: Pharmacogenomics J. 2002, 2: 30-42. 10.1038/sj.tpj.6500053.
Goldstein DB: Pharmacogenetics in the laboratory and the clinic. N Engl J Med. 2003, 348: 553-556. 10.1056/NEJMe020173.
Goldstein DB, Tate SK, Sisodiya SM: Pharmacogenetics goes genomic. Nat Rev Genet. 2003, 4: 937-947. 10.1038/nrg1229.
Neale BM, Sham PC: The future of association studies: gene-based analysis and replication. Am J Hum Genet. 2004, 75: 353-362. 10.1086/423901.
Hamdy SI, Hiratsuka M, Narahara K, Endo N, El-Enany M, Moursi N, Ahmed MS, Mizugaki M: Genotype and allele frequencies of TPMT, NAT2, GST, SULT1A1 and MDR-1 in the Egyptian population. Br J Clin Pharmacol. 2003, 55: 560-569. 10.1046/j.1365-2125.2003.01786.x.
Cascorbi I, Roots I: Pitfalls in N-acetyltransferase 2 genotyping. Pharmacogenetics. 1999, 9: 123-127. 10.1097/00008571-199902000-00016.
Patin E, Harmant C, Kidd KK, Kidd J, Froment A, Mehdi SQ, Sica L, Heyer E, Quintana-Murci L: Sub-Saharan African coding sequence variation and haplotype diversity at the NAT2 gene. Hum Mutat. 2006, 27: 720-10.1002/humu.9438.
Serre D, Paabo S: Evidence for gradients of human genetic diversity within and among continents. Genome Res. 2004, 14: 1679-1685. 10.1101/gr.2529604.
Shifman S, Darvasi A: The value of isolated populations. Nat Genet. 2001, 28: 309-310. 10.1038/91060.
Mrozikiewicz PM, Cascorbi I, Brockmoller J, Roots I: Determination and allelic allocation of seven nucleotide transitions within the arylamine N-acetyltransferase gene in the Polish population. Clin Pharmacol Ther. 1996, 59: 376-382. 10.1016/S0009-9236(96)90104-6.
Meisel P, Schroeder C, Wulff K, Siegmund W: Relationship between human genotype and phenotype of N-acetyltransferase (NAT2) as estimated by discriminant analysis and multiple linear regression:1. Genotype and N-acetylation in vivo. Pharmacogenetics. 1997, 7: 241-246. 10.1097/00008571-199706000-00009.
Gross M, Kruisselbrink T, Anderson K, Lang N, McGovern P, Delongchamp R, Kadlubar F: Distribution and concordance of N-acetyltransferase genotype and phenotype in an American population. Cancer Epidemiol Biomarkers Prev. 1999, 8: 683-692.
Jorge-Nebert LF, Eichelbaum M, Griese EU, Inaba T, Arias TD: Analysis of six SNPs of NAT2 in Ngawbe and Embera Amerindians of Panama and determination of the Embera acetylation phenotype using caffeine. Pharmacogenetics. 2002, 12: 39-48. 10.1097/00008571-200201000-00006.
Lee SY, Lee KA, Ki CS, Kwon OJ, Kim HJ, Chung MP, Suh GY, Kim JW: Complete sequencing of a genetic polymorphism in NAT2 in the Korean population. Clin Chem. 2002, 48: 775-777.
Clark AG, Weiss KM, Nickerson DA, Taylor SL, Buchanan A, Stengard J, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, Sing CF: Haplotype structure and population genetic inferences from nucleotide-sequence variation in human lipoprotein lipase. Am J Hum Genet. 1998, 63: 595-612. 10.1086/301977.
Baum J, Ward RH, Conway DJ: Natural selection on the erythrocyte surface. Mol Biol Evol. 2002, 19: 223-229.
Bamshad MJ, Mummidi S, Gonzalez E, Ahuja SS, Dunn DM, Watkins WS, Wooding S, Stone AC, Jorde LB, Weiss RB, Ahuja SK: A strong signature of balancing selection in the 5' cis-regulatory region of CCR5. Proc Natl Acad Sci USA. 2002, 99: 10539-10544. 10.1073/pnas.162046399.
Ebersberger I, Metzler D, Schwarz C, Paabo S: Genomewide comparison of DNA sequences between humans and chimpanzees. Am J Hum Genet. 2002, 70: 1490-1497. 10.1086/340787.
Excoffier L: Human demographic history: refining the recent African origin model. Curr Opin Genet Dev. 2002, 12: 675-682. 10.1016/S0959-437X(02)00350-7.
Tishkoff SA, Verrelli BC: Patterns of human genetic diversity: implications for human evolutionary history and disease. Annu Rev Genomics Hum Genet. 2003, 4: 293-340. 10.1146/annurev.genom.4.070802.110226.
Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL: Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Nat Acad Sci USA. 2005, 102: 15942-15947. 10.1073/pnas.0507611102.
Fuselli S, Gilman RH, Chanock SJ, Bonatto SL, De Stefano G, Evans CA, Labuda D, Luiselli D, Salzano FM, Soto G, Vallejo G, Sajantila A, Pettener D, Tarazona-Santos E: Analysis of nucleotide diversity of NAT2 coding region reveals homogeneity across Native American populations and high intra-population diversity. Pharmacogenomics J. 2007, 7: 144-152. 10.1038/sj.tpj.6500407.
De La Vega FM, Isaac H, Collins A, Scafe CR, Halldorsson BV, Su X, Lippert RA, Wang Y, Laig-Webster M, Koehler RT, Ziegle JS, Wogan LT, Stevens JF, Leinen KM, Olson SJ, Guegler KJ, You X, Xu LH, Hemken HG, Kalush F, Itakura M, Zheng Y, de The G, O'Brien SJ, Clark AG, Istrail S, Hunkapiller MW, Spier EG, Gilbert DA: The linkage disequilibrium maps of three human chromosomes across four populations reflect their demographic history and a common underlying recombination pattern. Genome Res. 2005, 15: 454-462. 10.1101/gr.3241705.
Verrelli BC, McDonald JH, Argyropoulos G, Destro-Bisol G, Froment A, Drousiotou A, Lefranc G, Helal AN, Loiselet J, Tishkoff SA: Evidence for balancing selection from nucleotide sequence analyses of human G6PD. Am J Hum Genet. 2002, 71: 1112-1128. 10.1086/344345.
Woolf LI: The heterozygote advantage in phenylketonuria. Am J Hum Genet. 1986, 38: 773-775.
Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A: Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am J Hum Genet. 2001, 69: 831-843. 10.1086/323612.
Pluzhnikov A, Di Rienzo A, Hudson RR: Inferences about human demography based on multilocus analyses of noncoding sequences. Genetics. 2002, 161: 1209-1218.
Stajich JE, Hahn MW: Disentangling the effects of demography and selection in human history. Mol Biol Evol. 2005, 22: 63-73. 10.1093/molbev/msh252.
Voight BF, Adams AM, Frisse LA, Qian Y, Hudson RR, Di Rienzo A: Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc Natl Acad Sci USA. 2005, 102: 18508-18513. 10.1073/pnas.0507325102.
Akey JM, Zhang G, Zhang K, Jin L, Shriver MD: Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002, 12: 1805-1814. 10.1101/gr.631202.
Weir BS, Cardon LR, Anderson AD, Nielsen DM, Hill WG: Measures of human population structure show heterogeneity among genomic regions. Genome Res. 2005, 15: 1468-1476. 10.1101/gr.4398405.
Excoffier L, Hamilton G: Comment on "Genetic structure of human populations". Science. 2003, 300: 1877-10.1126/science.1083411.
Barbujani G: Human races: Classifying people vs. understanding diversity. Current Genomics. 2005, 4: 215-226. 10.2174/1389202054395973.
The International HapMap Consortium: The International HapMap Project. Nature. 2003, 426: 789-796. 10.1038/nature02168.
Maynard Smith J, Haigh J: The hitch-hiking effect of a favourable gene. Genet Res. 1974, 23: 23-35.
Cavalli-Sforza LL: Population structure and human evolution. Proc R Soc Lond B Biol Sci. 1966, 164: 362-379.
Bowcock AM, Kidd JR, Mountain JL, Hebert JM, Carotenuto L, Kidd KK, Cavalli-Sforza LL: Drift, admixture, and selection in human evolution: a study with DNA polymorphisms. Proc Natl Acad Sci USA. 1991, 88: 839-843. 10.1073/pnas.88.3.839.
Fullerton SM, Bartoszewicz A, Ybazeta G, Horikawa Y, Bell GI, Kidd KK, Cox NJ, Hudson RR, Di Rienzo A: Geographic and haplotype structure of candidate type 2 diabetes susceptibility variants at the calpain-10 locus. Am J Hum Genet. 2002, 70: 1096-1106. 10.1086/339930.
Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, Ackerman HC, Campbell SJ, Altshuler D, Cooper R, Kwiatkowski D, Ward R, Lander ES: Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002, 419: 832-837. 10.1038/nature01140.
Patin E, Barreiro LB, Sabeti PC, Austerlitz F, Luca F, Sajantila A, Behar DM, Semino O, Sakuntabhai A, Guiso N, Gicquel B, McElreavey K, Harding RM, Heyer E, Quintana-Murci L: Deciphering the ancient and complex evolutionary history of human arylamine N-acetyltransferase genes. Am J Hum Genet. 2006, 78: 423-436. 10.1086/500614.
Excoffier L, Poloni ES, Santachiara-Benerecetti S, Semino O, Langaney A: The molecular diversity of the Niokholo Mandenkalu from Eastern Senegal: an insight into West Africa genetic history. Molecular Biology and Human Diversity. Edited by: Boyce AJ, Mascie-Taylor CGN. 1996, Cambridge: Cambridge University Press, 141-155.
Cascorbi I, Drakoulis N, Brockmoller J, Maurer A, Sperling K, Roots I: Arylamine N-acetyltransferase (NAT2) mutations and their allelic linkage in unrelated Caucasian individuals: correlation with phenotypic activity. Am J Hum Genet. 1995, 57: 581-592.
Stephens M, Donnelly P: A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003, 73: 1162-1169. 10.1086/379378.
Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Watterson GA: On the number of segregating sites in genetical models without recombination. Theor Pop Biol. 1975, 7 (2): 256-276. 10.1016/0040-5809(75)90020-9.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Fu YX, Li WH: Statistical tests of neutrality of mutations. Genetics. 1993, 133: 693-709.
Fu YX: Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997, 147: 915-925.
Fay JC, Wu CI: Hitchhiking under positive Darwinian selection. Genetics. 2000, 155: 1405-1413.
McDonald JH, Kreitman M: Adaptive evolution at the Adh locus in Drosophila. Nature. 1991, 351: 652-654. 10.1038/351652a0.
Griffiths RC, Tavaré S: Simulating probability distributions in the coalescent. Theor Popn Biol. 1994, 46: 131-159. 10.1006/tpbi.1994.1023.
Patterson N, Richter DJ, Gnerre S, Lander ES, Reich D: Genetic evidence for complex speciation of humans and chimpanzees. Nature. 2006, 441: 1103-1108. 10.1038/nature04789.
Bandelt HJ, Forster P, Rohl A: Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999, 16: 37-48.
Network 4.1.1 software. [http://www.fluxus-engineering.com]
Excoffier L, Smouse PE, Quattro JM: Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992, 131: 479-491.
Reynolds JJ, Weir BS, Cockerham CC: Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics. 1983, 105: 767-779.
Kruskal JB: Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika. 1964, 29: 1-27. 10.1007/BF02289565.
Rohlf FJ: 2000: NTSYS-PC, numerical taxonomy system for the PC. Exeter Software, Version 2.1. Applied Biostatistics Inc., Setauket, USA
Excoffier L, Laval G, Schneider S: Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005, 1: 47-50.
Hill WG, Robertson A: Linkage disequilibrium in finite populations. Theoret Appl Genet. 1968, 38: 226-231. 10.1007/BF01245622.
Legendre P, Lapointe F-J: Assessing the congruence among distance matrices: single malt Scotch whiskies revisited. Australian and New Zealand Journal of Statistics. 2004, 46: 615-629. 10.1111/j.1467-842X.2004.00357.x.
The consensus gene nomenclature of human NAT2 alleles. [http://www.louisville.edu/medschool/pharmacology/NAT2.html]
Acknowledgements
This work was planned and funded by the Laboratory of Genetics and Biometry, Department of Anthropology and Ecology, University of Geneva. We warmly thank Alicia Sanchez-Mazas for helpful advice on the project, Barbara Kervaire for unvaluable technical support and Yamama Naciri for profitable discussions of the results. We gratefully acknowledge Etienne Patin and Lluis Quintana-Murci for kindly sharing data. We also thank three anonymous reviewers for their constructive comments on earlier versions of this manuscript. A.S. was supported by the French Fondation pour la Recherche Médicale. Preliminary results were presented at the GALF meeting (Geneva 2007) supported by FNS 10CO12-117107 to A. Sanchez-Mazas.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AS conceived of the study, collected data, performed the majority of the statistical analyses and wrote the initial draft of the manuscript. AL and PD participated to study design, supervised analyses, and were involved in drafting the manuscript. AL and EP provided the Mandenka sample. NG and RK participated in the NAT2 gene sequencing in the Mandenka sample. EP provided the conceptual framework for the study, supervised the statistical analyses and finalized the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12863_2007_587_MOESM2_ESM.doc
Additional file 2: Distribution of NAT2 predicted acetylation phenotypes in the 41 samples of the worldwide genotyping survey. (DOC 74 KB)
12863_2007_587_MOESM3_ESM.doc
Additional file 3: Distribution of F ST values between Europeans and East Asians across a 400-kb segment encompassing the human NAT gene family on chromosome 8. (DOC 78 KB)
12863_2007_587_MOESM4_ESM.doc
Additional file 4: LD/block structure of the European HapMap sample across a 400-kb segment encompassing the human NAT gene family on chromosome 8. (DOC 187 KB)
12863_2007_587_MOESM5_ESM.doc
Additional file 5: LD/block structure of the East Asian HapMap sample across a 400-kb segment encompassing the human NAT gene family on chromosome 8. (DOC 142 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sabbagh, A., Langaney, A., Darlu, P. et al. Worldwide distribution of NAT2 diversity: Implications for NAT2 evolutionary history. BMC Genet 9, 21 (2008). https://doi.org/10.1186/1471-2156-9-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-9-21