
Abstract
Background
Bovine whole genome linkage disequilibrium maps were constructed for eight breeds of cattle. These data provide fundamental information concerning bovine genome organization which will allow the design of studies to associate genetic variation with economically important traits and also provides background information concerning the extent of long range linkage disequilibrium in cattle.
Results
Linkage disequilibrium was assessed using r2 among all pairs of syntenic markers within eight breeds of cattle from the Bos taurus and Bos indicus subspecies. Bos taurus breeds included Angus, Charolais, Dutch Black and White Dairy, Holstein, Japanese Black and Limousin while Bos indicus breeds included Brahman and Nelore. Approximately 2670 markers spanning the entire bovine autosomal genome were used to estimate pairwise r2 values. We found that the extent of linkage disequilibrium is no more than 0.5 Mb in these eight breeds of cattle.
Conclusion
Linkage disequilibrium in cattle has previously been reported to extend several tens of centimorgans. Our results, based on a much larger sample of marker loci and across eight breeds of cattle indicate that in cattle linkage disequilibrium persists over much more limited distances. Our findings suggest that 30,000–50,000 loci will be needed to conduct whole genome association studies in cattle.
Similar content being viewed by others
Background
Linkage disequilibrium (LD) maps are fundamental tools for exploring the genetic basis of economically important traits in cattle. Likewise, comparative LD maps enable us to explore the degree of diversity between breeds of cattle and to detect genomic regions that have been subject to selective sweeps within the different dairy and beef breeds which represent different biological attributes (e.g. Continental European vs. British). The currently available information regarding LD in cattle is primarily based on microsatellite studies performed in dairy cattle. The most extensive of these studies used 284 genome-wide microsatellites in a population of Dutch Black and White Dairy cattle [1] to show that syntenic LD extended up to several tens of centimorgans (cM). Haplotypes for 581 maternally inherited gametes were used to estimate LD using Lewontin's normalized D'. The results indicated high levels of LD not only between closely linked markers but for markers located as much as 40 cM (~40 Mb) apart. Two subsequent studies examined the extent of LD in cattle although both used fewer animals and microsatellites [2, 3]. Vallejo [2] selected distantly related animals to quantify the level of genetic diversity in United States Holstein cattle. While only 23 Holstein bulls were genotyped with 54 microsatellite loci that spanned most of the autosomal genome, extensive LD was detected in the United States Holstein population in agreement with the findings of Farnir et al. [1]. Tenesa et al. [3] genotyped 50 Holstein bulls for 13 microsatellites spanning BTA2 and BTA6, to determine the extent of LD in the United Kingdom Holstein population. The average D' value was 44% with significant LD reported only for distances less than 10.3 cM. Linkage disequilibrium among non syntenic loci was not significant.
More recently Khatkar et al. [4] scored 220 BTA6 single nucleotide polymorphisms (SNPs) in a sample of 433 Australian dairy bulls and estimated LD between marker pairs using D'. While they found that LD decayed with increasing distance between markers, D' did not reach background until an average distance of 20 Mb separated the markers. They also found that there was extensive variability in the magnitude of D' at any one distance. The rate of decay of LD estimated using SNPs [4] was much greater than that estimated using microsatellites [1], which is consistent with the findings of Varilo et al. [5] that more informative marker systems are able to detect LD over greater physical distances.
Only recently has the extent of LD been examined in beef cattle populations. A sample of 162 half-sib progeny from a Japanese black sire and 406 half-sib Japanese brown cattle were genotyped with 246 and 156 autosomal microsatellite markers, respectively [6]. For syntenic markers, the mean D' was 16.3% for Japanese Brown and 25.1% for Japanese Black. Characteristic of D' as a measure of LD, significant LD was observed for marker pairs separated by as much as 40 cM in both breeds.
Quantifying the extent of LD in the bovine genome is a necessary first step for determining the number of markers that will be sufficient for QTL mapping by linkage disequilibrium. The previous studies which used microsatellite markers were either too narrowly focused on particular chromosomes, or were of insufficient resolution to precisely estimate genome-wide LD and almost certainly were unable to precisely estimate short-range LD. The high density and low inherent rates of mutation of SNPs relative to microsatellites within mammalian genomes allows for the identification of ancestral haplotype blocks and the estimation of identity by descent probabilities which are crucial for haplotype-based association studies [7]. In this study, we estimated LD in 8 breeds of cattle utilizing 2670 single nucleotide polymorphism (SNP) markers that were derived from the bovine genome sequence and were aligned to the Btau_3.1 genome sequence assembly.
Results and Discussion
Haplotype Estimation
The program GENOPROB 2.0 [8, 9] which utilizes multi-generation pedigrees including both genotyped and non-genotyped animals was used for the estimation of phased haplotypes. For all breeds, greater than 97% of the scored genotypes were determined by GENOPROB 2.0 to have a probability of at least 95% of being correct conditional on the pedigree and marker map [10] (Figure 1a, Additional file 1). While the overall level of genotype accuracy was high, the level of genotype certainty was clearly dependent on pedigree structure. The Holstein, Limousin and Angus samples were obtained from the most complex pedigrees and produced the most accurately estimated genotypes and phased chromosomes (Figure 1b, Additional file 1). The depth of the pedigree as well as the location of the genotyped individuals within the pedigree (generation) had the largest influence on the estimation of phase probabilities (oGmx). Figure 1b clearly demonstrates that the breeds with the greatest pedigree complexity produced the highest probabilities of correctly phased genotypes. The cumulative proportion of heterozygous genotypes which could be phased by GENOPROB 2.0 with order probabilities >0.99 was 87.6%, 76.9% and 69.2% for Holstein, Limousin and Angus, respectively. The Brahman sample comprises several independent three generation pedigrees consisting of grandparent – parent and multiple offspring in which only one parent and grandparent was genotyped and there were no additional close pedigree relationships between individuals within or between families. This is in contrast to the Nelore sample which represents a two generation pedigree and which, in most cases, both parents of each animal were genotyped. For these pedigree structures, the three generation Brahman pedigree produced 5.3% (BR 63.4%, NEL 58.1%) more heterozygous genotypes with a phase order probability of >0.99. However, using a three generation pedigree structure with larger numbers of individuals per generation and including complete pedigree relationships among ungenotyped and genotyped animals such as in the Holstein, Limousin and Angus samples, produced a significant increase in the proportion of heterozygous genotypes which could be accurately phased.

a and b. Summary of genotype (pGmx) and phase (oGmx) probabilities for each breed based on GENOPROB 2.0 results. Only genotyped progeny with at least one genotyped parent were used.
General LD Findings
Comparative linkage disequilibrium maps were generated for eight breeds of cattle for the 29 bovine autosomes. The majority of the SNPs used in this study were chosen because they had previously been identified as putatively being variable within Bos taurus. This ascertainment bias resulted in the SNP minor allele frequencies being substantially lower in the two Bos indicus breeds than in the Bos taurus breeds (Figure 2). It also resulted in a set of SNPs in which common SNPs within the Bos taurus genome were over-represented. However, even though these loci were identified from Bos taurus derived sequences, more than 50% of the loci were polymorphic in Bos indicus and had a minor allele frequency >0.05 (Figure 2). This indicates that a substantial fraction of the loci identified by the Bovine Genome Sequencing Project will have utility for QTL mapping within Bos indicus breeds.

Minor allele frequencies (MAF) for all breeds.
The current estimate of the size of the bovine genome is 2.87 Gb [11] and with equal spacing, the 2,670 SNP loci used in this study would have an inter-marker distance of approximately 1 Mb. However, the loci were selected according to genomic location, likely assay conversion rate and minor allele frequency in Bos taurus and consequently were not uniformly distributed (Figure 3), 30% of the loci have inter-marker distances less than 0.5 Mb, while 13% are separated by more than 3 Mb. The non-uniform distribution of marker locations allows the estimation of LD across several orders of magnitude of differences in physical distance.

Percentage of loci within each bin. Proportion of loci for each bin are shown based on inter-marker distances measured in Mb. The horizontal axis depicts the maximum inter-marker distance for each bin. Here, a majority of the markers fall within the 0.50 Mb bin, This bin includes all markers with an inter-marker distance greater than 0.10 Mb but less than or equal to 0.50 Mb.
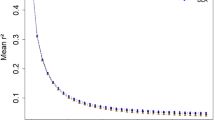
The r2 values for pairs of loci were binned according to the physical distance separating the loci and were averaged within each breed (Figure 4). As has previously been observed there is an inverse relationship between LD and physical or genetic distance [12] and r2 is essentially at long-range background levels in all eight breeds by a locus separation of approximately 500 kb. A similar study performed in pigs found that average r2 values had fallen to 0.1 for SNPs with an inter-marker distance of 3 cM [13]; similarly, linkage disequilibrium within dog breeds extends across several Mb [14]. However LD in humans extends for only tens of kb [15] which is consistent with the large effective size and rapid recent expansion of human populations.

Overall average r2 values. Average r2 values are shown for each breed. The maximum value for each bin is shown on the horizontal axis.
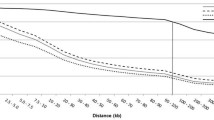
Our findings indicate a substantially shorter range of LD than has previously been reported in cattle [1, 6]. We attribute the differences between previous reports and our findings to the differences in measures used to report LD, namely D' versus r2. The Dutch Black and White Dairy cattle used in this experiment are a subset of the animals used in Farnir et al. [1] and the Holstein cattle are a subset of the animals previously used to fine map milk production traits on BTA6 [16]. To provide a direct comparison between the approaches, we estimated genome-wide average measures of LD using both r2 and D' in these two breeds (Figure 5). Both estimates of LD show an inverse relationship between LD and distance, however, in general, D' overestimates the extent of LD [17, 18]. The use of D' suggests that LD extends for several tens of centimorgans (or Mb), consistent with the earlier reports (Figure 5). However, the use of r2 indicates that LD is at background levels by approximately 0.5 Mb. Similar differences between measures of LD have recently been reported in cattle [19, 20].

Estimates of D' and r2 for Holstein and Dutch Black and White Dairy cattle. Average LD is shown for each breed in each bin. The maximum inter-marker value for each bin is shown on the horizontal axis.
It has been suggested that when large differences exist between marker allele frequencies, due to the presence of a rare allele, these two measures of LD are divergent [20]. D' estimates historical recombination through allelic association whereas r2 measures the squared correlation coefficient between locus allele frequencies and is strongly influenced by the order in which the mutations arose (genealogy) and not necessarily the physical distance between loci [21]. In the context of QTL mapping, r2 is the preferred measure of LD, because it quantifies the amount of information that can be inferred about one (perhaps nonobservable quantitative trait or disease) locus from another [22, 23], and can therefore be used to estimate the number of loci needed for association studies [23, 24]. For this reason we have used r2 as the primary measure of LD in this study.
Variation in average r2 values between breeds is evident in Figures 4 and 6. Considering the similarity between the Holstein and Dutch Black and White Dairy breeds, we expected comparable average r2 values between these breeds (Figure 4). In fact the extent of LD is quite similar within all of the Bos taurus and within the Bos indicus breeds, however, the Bos indicus appear to have substantially lower levels of LD at short inter-marker distances than do the Bos taurus. This could be the result of ascertainment bias as the SNPs used in this study were detected because they were common SNPs within Bos taurus and their average minor allele frequency was much lower in Bos indicus [25]. An alternative hypothesis is that the lower levels of LD at short inter-marker distances could also reflect historically larger effective population sizes [26], which seems particularly appropriate for the Nelore. On the other hand, long range LD in Brahman appears to be greater than for the Nelore and other Bos taurus breeds which suggests a smaller current effective population size which is consistent with the relatively recent formation of the breed as an admixture between extant Bos taurus and several imported Bos indicus breeds imported into the U.S. between 1854 and 1926 [27].
r2 by Chromosome
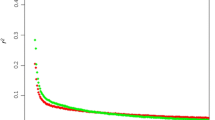
Variation in LD between chromosomes and breeds was examined using the 18.7% of all possible syntenic locus pairs (Additional file 2 and Figure 2) that were separated by less than 1 Kb. The average r2 values by chromosome and for each breed are shown in Figure 6 (Additional file 3). In Figure 6, breeds are grouped according to subspecies and the primary agricultural purpose of each breed. The first six Bos taurus breeds include Angus, Charolais, Limousin, and Japanese Black representing meat breeds, followed by Dutch Black and White Dairy and Holstein which are dairy breeds. While the Brahman is used primarily for meat in the U.S. and Australia, the Nelore is a Bos indicus breed used for both milk and meat production in South America. With the exception of BTA7, 12 and 21, the average r2 across the Bos taurus breeds was 0.5603 with minimum and maximum r2 values obtained on BTA29 in Limousin (0.12) and BTA14 in Holstein (0.91), respectively. The average r2 values across the Bos indicus breeds was 0.37 with minimum and maximum r2 values on BTA20 in Nelore (0.06) and BTA22 in Nelore (0.69). The relatively low level of LD at short inter-marker distances contrasts with previously published reports in cattle [1, 6]. We found comparable results for pairs of syntenic loci separated by approximately 100 kb and 500 kb (Additional files 4 and 5), respectively, which further supports our contention that useful LD [24] does not extend beyond 0.5 Mb and that average r2 values drop below 0.1 by 1 Mb (Figure 4). These findings have a profound impact on the number of loci and the number of individuals that will need to be tested in association-based QTL scans.

Average r2 values for inter-marker distances <1 kb for each breed and chromosome. Data are not presented for breed by chromosome combinations for which there were less than 5 informative locus pairs (BTA19).
BTA 7, 12 and 21
Figure 6 indicates that the average r2 values are low for all breeds on BTA 7, 12 and 21 when compared to all other autosomes. The average r2 values on these chromosomes appears not to be a sampling artifact, since the number of loci used to calculate the average r2 values was 19, 21 and 7 locus pairs on BTA 7, 12 and 21, respectively, which is not significantly different than for the other autosomes (Additional file 3). Additionally, each of the loci on these chromosomes had similar allele frequencies to those loci on the other autosomes (data not shown). This suggests that the loci on BTA 7, 12 and 21 may have been clustered around one or more recombination hotspots on each of these chromosomes. To determine if the loci were clustered, we plotted the location of the SNP pairs along each chromosome (Figure 7). Figure 7 demonstrates that the SNP pairs used to examine the extent of short range LD are distributed along the length of these three chromosomes and we therefore conclude that BTA 7, 12 and 21 have intrinsically lower levels of LD than do the other autosomes.

Distribution of SNPs along BTA6, 7, 12 and 21 respectively. Measures of LD, r2, are shown for Angus and Holstein. BTA6 was included for comparison purposes.
We have two theories as to why lower than average LD may exist on BTA 7, 12 and 21. First, because cattle have been selected for production traits for at least 50 generations, there is the possibility that selection on QTL distributed throughout the genome has generated different patterns of LD on individual chromosomes. However, compared to chromosomes of similar size, there does not appear to be fewer QTLs on BTA7, 12 and 21 [28, 29] and selection should have resulted in similar patterns of LD on these chromosomes as on all others. Second, it is possible that chromosomes 7, 12 and 21 have higher than average rates of recombination than do the other autosomes. A comparison between the physical [30] and genetic [31] maps of BTA7, 12 and 21 as well as chromosomes of similar physical size, indicates that the physical to genetic size relationship is similar for BTA7, 12 and 21 and other chromosomes of similar physical size. However, regions of increased recombination have been detected on human chromosomes 14 and 15 [32], which are partially orthologous to BTA21. A complete exploration of these chromosomes in order to study aspects of genome organization that potentially affect recombination rate will require additional markers and animals.
Conclusion
While we included loci in this analysis for which order was consistent between the Btau_3.1 assembly and our radiation hybrid map [10], the genome coordinates for each locus were obtained from the Btau_3.1 assembly. This assembly spans only 2.43 Gb and while an additional 319 Mb of sequence exists as contigs which are unassigned to chromosomes, we expect the final sequence to be much closer to 2.8 Gb. The unassembled contigs are likely to be biased towards centromeric and telomeric sequences and duplications which are difficult to assemble, but some are no doubt interstitial to chromosomes. The fact that these are unassembled would likely cause a systematic bias towards the underestimation of the physical distance between loci. There also appear to be a significant number or problems with the ordering and orientation of scaffolds within the assembly and these errors are likely to produce random effects on the estimation of distance between syntenic loci. Thus overall, we suspect that the incomplete nature of the assembly results in about a 10% underestimate of the distance between loci. This has only a minor affect on our conclusions and the extent of LD available for association analysis still does not significantly exceed 500 kb. At a physical distance of 100 kb separating flanking SNP loci, the average r2 is 0.15–0.2 and the average r2 between these markers and a QTL located at mid-interval is about 0.3 (Figure. 4). This would appear to be the lowest desirable resolution for whole genome association mapping in bovine and assuming a 2.87 Gb genome, it would require 28,700 fully informative SNPs to saturate the genome at an average resolution of 100 kb. Since the number of validated bovine SNPs is currently insufficient to achieve an even spacing and because many SNPs are likely to have low minor allele frequencies leading to their being uninformative in many populations, we believe that 50,000 SNPs will be the minimum required for whole genome association studies in cattle. Furthermore, the extent of LD on BTA 7, 12 and 21 appears to be much lower than for the autosomal genome as a whole and suggests that SNP density may need to be enhanced on these chromosomes. The construction of a high resolution LD map of the bovine genome will provide further insight into the effects of selection and evolutionary forces upon the genomes of breeds which have been selected for different agricultural purposes.
Methods
DNA Collection
DNA was collected from 70 Angus (USA), 20 Canadian Angus, 40 Charolais (Canada), 40 Brahman (USA), 97 Dutch Black and White Dairy cattle (Belgium), 48 Holstein (USA), 65 Japanese Black (Japan), 43 Limousin (USA) and 97 Nelore (Brazil) cattle. In order to phase the chromosomes using linkage information, we selected small families where members within the families were closely related but the families themselves were not closely related. Family structure and the number of individuals per family varied between the breeds but the general family structure consisted of a male grandparent, male parent and three or more progeny (Additional file 6). This three generation family structure allowed for the efficient estimation of marker phase relationships in the progeny and also produced the most likely phase relationships in each of the parents/grandparents.
Marker Selection and Genotyping
Sequence information for SNPs was obtained from public databases [33, 34]. Loci included in this study met the following criteria; minor allele frequency (MAF) ≥ 0.05 in Angus based on previous screens (data not shown) and concordant order determined by radiation hybrid (RH) mapping [10] and genomic sequence location. Oligonucleotides were designed, synthesized and assembled into oligo pooled assays (OPA) by Illumina Inc. (San Diego, CA). Genotyping was performed using the manufacturer's protocol for the Illumina® BeadStation 500G [35, 36]).
Locus locations within the bovine genome sequence assembly
Chromosomal coordinates for each SNP were obtained by aligning approximately 250 bp flanking each SNP by BLAST to the latest release of the bovine genome sequence assembly, Btau_3.1. These physical coordinates were compared to the linkage and RH maps of McKay et al[10]. Thirty four markers were excluded from the analysis because their assignment in the sequence assembly was to a chromosome that differed to their linkage or RH map assignment or because they had no chromosomal assignment in Btau_3.1. Marker information can be found in Additional file 7.
Haplotypes and LD Analysis
GENOPROB V2.0 [8, 9] was used to assess genotype score quality and produce whole chromosome phased haplotypes based on the pedigree and physical map locations of the loci. Briefly, GENOPROB uses an allelic peeling algorithm to estimate both the probability that a genotype is correct, denoted as pGmx, and the probability that the order (phase) of the alleles are correct, denoted as oGmx. Only genotypes with a pGmx ≥ 0.95 were used for LD analysis but no restriction was placed on order probability, oGmx. This produced a set of whole chromosome haplotypes comprised of accurately scored genotypes that were in the most likely phase configuration. LD was assessed by generating r2 values using GOLD [37] independently for the maternally- and paternally-inherited haplotypes. LD data presented here is based only on the maternally inherited haplotypes which avoids the overrepresentation of paternally inherited haplotypes within the primarily male pedigrees.
References
Farnir F, Coppieters W, Arranz JJ, Berzi P, Cambisano N, Grisart B, Karim L, Marcq F, Moreau L, Mni M, Nezer C, Simon P, Vanmanshoven P, Wagenaar D, Georges M: Extensive genome-wide linkage disequilibrium in cattle. Genome Res. 2000, 10 (2): 220-227. 10.1101/gr.10.2.220.
Vallejo RL, Li YL, Rogers GW, Ashwell MS: Genetic diversity and background linkage disequilibrium in the North American Holstein cattle population. J Dairy Sci. 2003, 86 (12): 4137-4147.
Tenesa A, Knott SA, Ward D, Smith D, Williams JL, Visscher PM: Estimation of linkage disequilibrium in a sample of the United Kingdom dairy cattle population using unphased genotypes. J Anim Sci. 2003, 81 (3): 617-623.
Khatkar MS, Collins A, Cavanagh JA, Hawken RJ, Hobbs M, Zenger KR, Barris W, McClintock AE, Thomson PC, Nicholas FW, Raadsma HW: A first-generation metric linkage disequilibrium map of bovine chromosome 6. Genetics. 2006, 174 (1): 79-85. 10.1534/genetics.106.060418.
Varilo T, Paunio T, Parker A, Perola M, Meyer J, Terwilliger JD, Peltonen L: The interval of linkage disequilibrium (LD) detected with microsatellite and SNP markers in chromosomes of Finnish populations with different histories. Hum Mol Genet. 2003, 12 (1): 51-59. 10.1093/hmg/ddg005.
Odani M, Narita A, Watanabe T, Yokouchi K, Sugimoto Y, Fujita T, Oguni T, Matsumoto M, Sasaki Y: Genome-wide linkage disequilibrium in two Japanese beef cattle breeds. Anim Genet. 2006, 37 (2): 139-144. 10.1111/j.1365-2052.2005.01400.x.
Vignal A, Milan D, SanCristobal M, Eggen A: A review on SNP and other types of molecular markers and their use in animal genetics. Genet Sel Evol. 2002, 34 (3): 275-305. 10.1051/gse:2002009.
Thallman RM, Bennett GL, Keele JW, Kappes SM: Efficient computation of genotype probabilities for loci with many alleles: I. Allelic peeling. J Anim Sci. 2001, 79 (1): 26-33.
Thallman RM, Bennett GL, Keele JW, Kappes SM: Efficient computation of genotype probabilities for loci with many alleles: II. Iterative method for large, complex pedigrees. J Anim Sci. 2001, 79 (1): 34-44.
McKay SD, Schnabel RD, Murdoch BM, Aerts J, Gill CA, Gao C, Li C, Matukumalli LK, Stothard P, Wang Z, Van Tassell CP, Williams JL, Taylor JF, Moore SS: Construction of bovine whole-genome radiation hybrid and linkage maps using high-throughput genotyping. Anim Genet. 2007, 38 (2): 120-125. 10.1111/j.1365-2052.2006.01564.x.
Baylor College of Medicine- Bovine Genome Project Website: http://www.hgsc.bcm.tmc.edu/projects/bovine/.
Schaeffer SW, Miller EL: Estimates of linkage disequilibrium and the recombination parameter determined from segregating nucleotide sites in the alcohol dehydrogenase region of Drosophila pseudoobscura. Genetics. 1993, 135 (2): 541-552.
Du FX, Clutter AC, Lohuis MM: Characterizing linkage disequilibrium in pig populations. Int J Biol Sci. 2007, 3 (3): 166-178.
Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, Mauceli E, Xie X, Breen M, Wayne RK, Ostrander EA, Ponting CP, Galibert F, Smith DR, DeJong PJ, Kirkness E, Alvarez P, Biagi T, Brockman W, Butler J, Chin CW, Cook A, Cuff J, Daly MJ, DeCaprio D, Gnerre S, Grabherr M, Kellis M, Kleber M, Bardeleben C, Goodstadt L, Heger A, Hitte C, Kim L, Koepfli KP, Parker HG, Pollinger JP, Searle SM, Sutter NB, Thomas R, Webber C, Baldwin J, Abebe A, Abouelleil A, Aftuck L, Ait-Zahra M, Aldredge T, Allen N, An P, Anderson S, Antoine C, Arachchi H, Aslam A, Ayotte L, Bachantsang P, Barry A, Bayul T, Benamara M, Berlin A, Bessette D, Blitshteyn B, Bloom T, Blye J, Boguslavskiy L, Bonnet C, Boukhgalter B, Brown A, Cahill P, Calixte N, Camarata J, Cheshatsang Y, Chu J, Citroen M, Collymore A, Cooke P, Dawoe T, Daza R, Decktor K, DeGray S, Dhargay N, Dooley K, Dooley K, Dorje P, Dorjee K, Dorris L, Duffey N, Dupes A, Egbiremolen O, Elong R, Falk J, Farina A, Faro S, Ferguson D, Ferreira P, Fisher S, FitzGerald M, Foley K, Foley C, Franke A, Friedrich D, Gage D, Garber M, Gearin G, Giannoukos G, Goode T, Goyette A, Graham J, Grandbois E, Gyaltsen K, Hafez N, Hagopian D, Hagos B, Hall J, Healy C, Hegarty R, Honan T, Horn A, Houde N, Hughes L, Hunnicutt L, Husby M, Jester B, Jones C, Kamat A, Kanga B, Kells C, Khazanovich D, Kieu AC, Kisner P, Kumar M, Lance K, Landers T, Lara M, Lee W, Leger JP, Lennon N, Leuper L, LeVine S, Liu J, Liu X, Lokyitsang Y, Lokyitsang T, Lui A, Macdonald J, Major J, Marabella R, Maru K, Matthews C, McDonough S, Mehta T, Meldrim J, Melnikov A, Meneus L, Mihalev A, Mihova T, Miller K, Mittelman R, Mlenga V, Mulrain L, Munson G, Navidi A, Naylor J, Nguyen T, Nguyen N, Nguyen C, Nguyen T, Nicol R, Norbu N, Norbu C, Novod N, Nyima T, Olandt P, O'Neill B, O'Neill K, Osman S, Oyono L, Patti C, Perrin D, Phunkhang P, Pierre F, Priest M, Rachupka A, Raghuraman S, Rameau R, Ray V, Raymond C, Rege F, Rise C, Rogers J, Rogov P, Sahalie J, Settipalli S, Sharpe T, Shea T, Sheehan M, Sherpa N, Shi J, Shih D, Sloan J, Smith C, Sparrow T, Stalker J, Stange-Thomann N, Stavropoulos S, Stone C, Stone S, Sykes S, Tchuinga P, Tenzing P, Tesfaye S, Thoulutsang D, Thoulutsang Y, Topham K, Topping I, Tsamla T, Vassiliev H, Venkataraman V, Vo A, Wangchuk T, Wangdi T, Weiand M, Wilkinson J, Wilson A, Yadav S, Yang S, Yang X, Young G, Yu Q, Zainoun J, Zembek L, Zimmer A, Lander ES: Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005, 438 (7069): 803-819. 10.1038/nature04338.
Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, Frazer KA, Cox DR: Whole-genome patterns of common DNA variation in three human populations. Science. 2005, 307 (5712): 1072-1079. 10.1126/science.1105436.
Schnabel RD, Kim JJ, Ashwell MS, Sonstegard TS, Van Tassell CP, Connor EE, Taylor JF: Fine-mapping milk production quantitative trait loci on BTA6: analysis of the bovine osteopontin gene. Proc Natl Acad Sci U S A. 2005, 102 (19): 6896-6901. 10.1073/pnas.0502398102.
Ardlie KG, Kruglyak L, Seielstad M: Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002, 3 (4): 299-309. 10.1038/nrg777.
Ke X, Hunt S, Tapper W, Lawrence R, Stavrides G, Ghori J, Whittaker P, Collins A, Morris AP, Bentley D, Cardon LR, Deloukas P: The impact of SNP density on fine-scale patterns of linkage disequilibrium. Hum Mol Genet. 2004, 13 (6): 577-588. 10.1093/hmg/ddh060.
Khatkar MS, Zenger KR, Hobbs M, Hawken RJ, Cavanagh JA, Barris W, McClintock AE, McClintock S, Thomson PC, Tier B, Nicholas FW, Raadsma HW: A primary assembly of a bovine haplotype block map based on a 15,036-single-nucleotide polymorphism panel genotyped in holstein-friesian cattle. Genetics. 2007, 176 (2): 763-772. 10.1534/genetics.106.069369.
Boyles AL, Scott WK, Martin ER, Schmidt S, Li YJ, Ashley-Koch A, Bass MP, Schmidt M, Pericak-Vance MA, Speer MC, Hauser ER: Linkage disequilibrium inflates type I error rates in multipoint linkage analysis when parental genotypes are missing. Hum Hered. 2005, 59 (4): 220-227. 10.1159/000087122.
Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES: High-resolution haplotype structure in the human genome. Nat Genet. 2001, 29 (2): 229-232. 10.1038/ng1001-229.
Zhao H, Nettleton D, Soller M, Dekkers JC: Evaluation of linkage disequilibrium measures between multi-allelic markers as predictors of linkage disequilibrium between markers and QTL. Genet Res. 2005, 86 (1): 77-87. 10.1017/S001667230500769X.
Pritchard JK, Przeworski M: Linkage disequilibrium in humans: models and data. Am J Hum Genet. 2001, 69 (1): 1-14. 10.1086/321275.
Kruglyak L: Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet. 1999, 22 (2): 139-144. 10.1038/9642.
Weiss KM, Clark AG: Linkage disequilibrium and the mapping of complex human traits. Trends Genet. 2002, 18 (1): 19-24. 10.1016/S0168-9525(01)02550-1.
Tenesa A, Navarro P, Hayes BJ, Duffy DL, Clarke GM, Goddard ME, Visscher PM: Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007, 17 (4): 520-526. 10.1101/gr.6023607.
Sanders JO: History and development of Zebu cattle in the United States. J Anim Sci. 1980, 50: 1188-1200.
Hu ZL, Reecy JM: Animal QTLdb: beyond a repository : A public platform for QTL comparisons and integration with diverse types of structural genomic information. Mamm Genome. 2007, 18 (1): 1-4. 10.1007/s00335-006-0105-8.
Polineni P, Aragonda P, Xavier SR, Furuta R, Adelson DL: The bovine QTL viewer: a web accessible database of bovine Quantitative Trait Loci. BMC Bioinformatics. 2006, 7: 283-10.1186/1471-2105-7-283.
Website, NCBI, Mapview: . [http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi?taxid=9913]
Website, USDA, MARC: . [http://www.marc.usda.gov/genome/genome.html]
Yu A, Zhao C, Fan Y, Jang W, Mungall AJ, Deloukas P, Olsen A, Doggett NA, Ghebranious N, Broman KW, Weber JL: Comparison of human genetic and sequence-based physical maps. Nature. 2001, 409 (6822): 951-953. 10.1038/35057185.
Website, NCBI, dbSNP: . [http://www.ncbi.nlm.nih.gov/projects/SNP/]
Website, Medicine BC, FTP: . [ftp://ftp.hgsc.bcm.tmc.edu/pub/data/Btaurus/snp]
Oliphant A, Barker DL, Stuelpnagel JR, Chee MS: BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques. 2002, Suppl: 56-8, 60-1..
Website, Illumina: . [http://www.illumina.com]
Abecasis GR, Cookson WO: GOLD--graphical overview of linkage disequilibrium. Bioinformatics. 2000, 16 (2): 182-183. 10.1093/bioinformatics/16.2.182.
Acknowledgements
The authors thank Michel Georges for intellectual contributions. This work was supported through Grant Number 2003A245R awarded to S.S. Moore by the Alberta Agriculture Research Institute. Taylor and Schnabel were supported by the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service (CSREES) grants #2005-35205-15448; #2006-35616-16697 and #2006-35205-16701. J. Aerts was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) grant BBS/B13454 "Bovine genome annotation and analysis."
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declares that there are no competing interests.
Authors' contributions
SDM conceived the study and participated in its design, data collection, analysis and manuscript preparation. RDS participated in study design, data analysis and manuscript preparation. BM participated in study design, genotyping and data analysis. LKM provided bioinformatics support. JA provided bioinformatics support. WC organized collection of Dutch Black and White samples, DC provided Charolais samples, EDN organized collection of Nelore samples, CAG provided bioinformatics support and organized collection of Brahman samples, CG provided bioinformatics support, HM organized the collection of Japanese Black samples, PS provided bioinformatics support, ZW provided statistical support, CVT made intellectual and bioinformatics contributions, JW made intellectual contributions and help in the manuscript preparation, JT participated in study design, provided Angus, Limousin and Holstein samples and helped draft the manuscript, SSM intellectual contributions. All authors read and approved the final manuscript.
Electronic supplementary material
12863_2007_553_MOESM1_ESM.xls
Additional file 1: Genoprob summary. Proportion of genotypes reaching a given level of confidence where pGmx is the probability that the indicated genotype is correct and oGmx is the probability that the phase order of the two alleles of the genotype is correct. (XLS 86 KB)
{kind=link}
12863_2007_553_MOESM5_ESM.png
{kind=link}
Additional file 5: Average r2 values for inter-marker distances of 100–500 kb for each breed and chromosome. (PNG 78 KB)
12863_2007_553_MOESM6_ESM.pdf
Additional file 6: Pedigrees for each breed. Pedigrees for each breed include a Unique animal IDs, sex of each animal and relationship of each animal genotyped. All genotyped animals were shaded in gray. (PDF 104 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
McKay, S.D., Schnabel, R.D., Murdoch, B.M. et al. Whole genome linkage disequilibrium maps in cattle. BMC Genet 8, 74 (2007). https://doi.org/10.1186/1471-2156-8-74
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-8-74