
Abstract
Background
A back curvature defect similar to kyphosis in humans has been observed in swine herds. The defect ranges from mild to severe curvature of the thoracic vertebrate in split carcasses and has an estimated heritability of 0.3. The objective of this study was to identify genomic regions that affect this trait.
Results
Single nucleotide polymorphism (SNP) associations performed with 198 SNPs and microsatellite markers in a Duroc-Landrace-Yorkshire resource population (U.S. Meat Animal Research Center, USMARC resource population) of swine provided regions of association with this trait on 15 chromosomes. Positional candidate genes, especially those involved in human skeletal development pathways, were selected for SNP identification. SNPs in 16 candidate genes were genotyped in an F2 population (n = 371) and the USMARC resource herd (n = 1,257) with kyphosis scores. SNPs in KCNN2 on SSC2, RYR1 and PLOD1 on SSC6 and MYST4 on SSC14 were significantly associated with kyphosis in the resource population of swine (P ≤ 0.05). SNPs in CER1 and CDH7 on SSC1, PSMA5 on SSC4, HOXC6 and HOXC8 on SSC5, ADAMTS18 on SSC6 and SOX9 on SSC12 were significantly associated with the kyphosis trait in the F2 population of swine (P ≤ 0.05).
Conclusions
These data suggest that this kyphosis trait may be affected by several loci and that these may differ by population. Carcass value could be improved by effectively removing this undesirable trait from pig populations.
Similar content being viewed by others


Background
Spine curvature defects have been reported in swine herds for the past three decades [1–5]. These defects have been classified as lordosis and/or kyphosis phenotypes. These reports describe this condition as a defect that can be detected visually in the live animal. We have detected a skeletal defect in both a commercial F2 and a resource herd of swine where the curvature of the spine is readily detectable in the animal carcass, but not easily identifiable externally on the live animal.
Prior literature has attributed these skeletal defects to environmental conditions rather than to genetic factors [1–5]. However more recently, Holl et al. [6] showed that the skeletal condition detected in the swine herds in this study has a genetic component with heritability estimates of 0.30 in a F2 population of animals and 0.32 in the USMARC resource population. Holl et al. [6] further reported that age, sex, number of ribs, number of lumbar vertebrae, number of nipples, carcass length and hot carcass weight were not significantly associated with the kyphosis phenotype.
Since the pig kyphosis condition is a moderately heritable trait, the purpose of this study was to identify positional candidate genes in chromosomal regions found to be associated with this kyphosis condition and to ultimately to eliminate this defect and improve carcass quality. The candidate genes evaluated in this study were: cadherin 7 (CDH7) and cerberus 1 homolog (CER1) on SSC1, cartilage oligomeric matrix protein (COMP), potassium intermediate/small conductance calcium-activated channel, subfamily N, member 2 (KCNN2) and solute carrier family 26 member 2 (SLC26A2) on SSC2, son of sevenless homolog 1 (SOS1) on SSC3, proteasome (prosome, macropain) subunit, alpha type, 5 (PSMA5) and Lix1 homolog (mouse)-like (LIX1L) on SSC4, homeobox C8 (HOXC8) on SSC5, ADAM metallopeptidase with thrombospondin type 1 motif, 18 (ADAMTS18), ryanodine receptor (RYR1) and lysyl hydroxylase (PLOD1) on SSC6, cullin 7 (CUL7) and runt-related transcription factor 2 (RUNX2) on SSC7, SRY-box 9 (SOX9) on SSC12 and histone acetyltransferase (monocytic leukemia) 4 (MYST4) on SSC14 (Table 1).
Causative mutations for human back defects have been identified in several of these genes (Table 1). SLC26A2 is mutated in diastrophic dysplasia with scoliosis [7]. Pseudoachondroplasia with symptoms of vertebral anomalies can be caused by mutations in COMP [8]. The over expression of HOXC8 has been shown to cause cartilage defects [9]. Mutations in RYR1 have been associated with the human conditions spondylocostal dysostosis (autosomal recessive) and minicore myopathy with ophthalmoplegia, both of which include curvature of the spine [10, 11]. PLOD1 mutations are responsible for the kyphoscoliotic subtype of Ehlers-Danlos syndrome VIA (EDS VIA) [12]. ADAMTS18 was recently identified in a genome-wide association study as a bone mass candidate gene for human ethnic groups [13]. Mutations in CUL7 cause 3 M Syndrome in which one symptom is tall vertebral bodies [14]. Cleidocranial dysplasia that can include the clinical features of scoliosis or vertebral malformation is caused by mutations in the transcription factor RUNX2/CBFA1 [15]. Mutations in SOX9 have been correlated to a campomelic dysplasia with skeletal anomalies [16]. CER1 has recently been associated with bone mass density and fracture in Southern Chinese women, as well as in normal bone development or metabolism in the mouse [17].
Results
Whole genome association in USMARC resource population
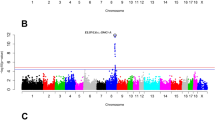
A whole genome association was performed to identify chromosomal regions of interest related to the kyphosis phenotype. Markers (170 SNPs and 28 microsatellites) were located across the pig genome (Additional file 1). Whole genome association tests of these markers in the USMARC resource population of animals (n = 1,257) revealed 35 markers with association on 15 different chromosomes (Table 2). Seven of these regions, located on SSC1, 2, 5, 7, 12, and 14, contained more than one marker within a 10 cM region with significant association; the others contained one SNP or microsatellite with association with reportable significance (P < 0.05; Table 2). Because the purpose of the association was to identify regions associated with kyphosis, only nominal P-values were used to investigate these regions for candidate genes.
Candidate genes in several regions could be identified by location, gene function and/or possible role in a human skeletal disorder. Candidate genes that were identified and selected for this study in these regions included: CDH7 and CER1 on SSC1; COMP, KCNN2 and SLC26A2 on SSC2; PSMA5 and LIX1L on SSC4; SOS1 on SSC3; HOXC8 on SSC5; PLOD1, RYR1 and ADAMTS18 on SSC6; CUL7 and RUNX2 on SSC7; SOX9 on SSC12; and MYST4 on SSC14. Table 1 describes the locations of these candidate genes in both the pig and the human and their role, if any, in human back curvature disorders.
Positional candidate gene SNP associations in the USMARC population
SNPs were identified in candidate genes for genotyping assays by sequencing twelve animals of 8 different sire lines with all 4 of the kyphosis category scores represented. When possible, the coding regions of these genes were sequenced to identify potential mutations. A total of 195 SNPs were detected in the 16 candidate genes. The markers and their genomic positions are shown in Additional file 2.
Of the SNPs identified in the candidate genes, 70 were genotyped in the USMARC resource population. Association analysis using MTDFREML detected 10 SNPs located in KCNN2, RYR1, PLOD1, and MYST4 with significant association (P ≤ 0.05) with kyphosis (Table 3). The most significant markers (P ≤ 0.005) were in PLOD1 (markers 65451_236 and 65458_588).
Six synonymous SNPs were discovered in the coding regions of PLOD1; these were located in exons 1, 5, 7, and 11. Another SNP was also identified in the 3' untranslated region of the gene (Additional file 2). Of the seven SNPs in the PLOD1 coding region, six were genotyped in the USMARC resource population of animals and two were analyzed (Table 3). Four were excluded from the analysis due to high numbers of genotyping errors. One of the coding SNP located in exon 11 of PLOD1, marker 65451_67, was significant for the kyphosis defect (P = 0.01; Table 3). Five additional markers located in introns 1, 11 and 14 of PLOD1 were significant for association with kyphosis in this population of animals. Two of these markers were significant at P < 0.005; they were markers 65451_236 in intron 11 (P = 0.00032) and marker 65458_588 in intron 14 (P = 0.0025; Table 3). The estimated additive effects of markers 65451_67, 65451_236, and 65458_588 were 0.13, 0.151, and 0.083, respectively.
Positional candidate gene SNP associations in the F2 population
Forty-seven SNPs in candidate genes were recombined into assay groups and genotyped across the F2 population of animals. These assays included SNPs that were significant in the USMARC population of animals. Nine were significantly associated (P ≤ 0.05) with the kyphosis defect (Table 4). These SNPs were located in CER1, CDH7, ADAMTS18, PSMA5, HOXC6, HOXC8 and SOX9. The most significant markers (P ≤ 0.005) were in CDH7 (marker 65672_206), HOXC6 and HOXC8 (markers 44017_483 and 44360_103, respectively) and SOX9 (marker 65525_208).
Twelve SNPs in the SOX9 gene that were genotyped in the USMARC resource population of animals were not significantly associated with kyphosis. Three SOX9 SNPs (markers 65517_61, 65525_208 and 65525_389) were genotyped in the F2 population of animals and all were significant for the back defect (P < 0.03; Table 4). Marker 65517_61 is located in the 5'UTR of the gene and the other two markers are located in intron 2. The effects of these SNPs on the kyphosis phenotype ranged from ± 0.0682 to ± 0.0881 (Table 4).
The SNP associated with kyphosis in CER1 (marker 65678_443) is a polymorphism responsible for a nonsynonymous amino acid alteration in exon 1 (Table 4). In total, three polymorphisms were identified in the coding regions of CER1. Two SNPs were identified in exon 1 of CER1; a nonsynonymous alteration responsible for an Arg to His change at amino acid 24 in the human sequence (marker 65678_419) and a nucleotide change causing a Phe to Ser amino acid change at position 32 (marker 65678_443). In addition, a silent base change was identified in the codon for Pro at amino acid 120 (Additional file 2). Marker 65678_443 was significant for kyphosis in the F2 population of swine (P = 0.034) and displayed an estimated effect of ± 0.131 (Table 4). This SNP was not associated with the back defect in the resource population.
A joint analysis of the markers genotyped in the two swine populations resulted in four markers that were significant. Markers 65451_236, 65458_515, 65517_61 and 65525_208 in PLOD1 and SOX9 were associated with nominal P of 0.02, 0.001, 0.02, and 0.05, respectively (data not shown).
The number of significant SNPs (P < 0.05) detected in these populations (14% in the resource population and 19% in the F2 population) exceeded the chance expectation of 5% (Tables 3 and 4). The FDR based on P = 0.05 for the resource population of swine is 0.35 and 0.26 for the F2 population of swine, suggesting that 35% of the associations detected are expected due to chance in the resource population and 26% were expected due to chance in the F2 population. The FDR based on P = 0.01 is 0.23 for the resource population and 0.094 for the F2 population.
A correction for multiple testing was applied to the SNPs significant for kyphosis. Markers were corrected using a Bonferroni calculation by multiplying the P-values by the number of SNPs genotyped in each population. One marker in PLOD1 remained significant (P = 0.02) in the resource population and one marker in HOXC8 remained significant (P = 0.05) in the F2 population (Tables 3 and 4).
Discussion
The kyphosis detected in the two swine populations evaluated in this paper and in Holl et al. [6], does not seem to be the same as the kyphosis conditions that were previously reported for swine in the literature [1–5]. It is difficult to detect the kyphosis defect in the live animal in the populations presented here and the curvature does not appear to be the result of hemivertibrae. The vertebrae in our swine herds appear to be of typical rectangular shape and curvature of the spine does not appear to be the result of ossification defects (data not shown).
The kyphosis condition presented in this paper also appears to differ from the human scoliosis condition, Scheuermann's disease. Vertebrae in subjects with Scheuermann's are curved due to ossification deficiencies that result in a wedge shape, which does not seem to be present in these swine herds. In addition, the mode of inheritance of Scheuermann's suggests an autosomal dominant heritability with complete penetrance in males and incomplete penetrance in females [18], while sex is not significantly associated with the condition presented here [6].
In humans, kyphosis is often one of a multitude of symptoms of a disorder. For example, kyphosis can be a symptom in Rett Syndrome, but the disease is characterized as a neurodevelopmental disorder and is detected almost exclusively in girls. Other symptoms include regression of higher functions, loss of speech, stereotypical movement of the hands, microcephaly, and mental retardation [19, 20]. Another example is Larsen Syndrome [21], caused by mutations in filamin B, where individuals can display kyphosis; however, other symptoms of the disease include dislocation of the knees, flat facial features, and cylindrical-shaped fingers. These herds of swine do not display neurological, vocalization loss, facial and/or developmental defects; the only noticeable symptom has been the spinal curvature.
While there are differences between known human back curvature defects, there are also some similarities. This condition displays a genetic mode of inheritance and a similar physical S curvature of the spine in all animals evaluated. In addition, the kyphosis condition described in the Duroc-Landrace-Yorkshire resource population was not detectable in neonatal animals (data not shown). Neonatal animals (n = 30) of parents known to pass along this kyphosis defect were harvested at 110 days of gestation and radiographed to determine whether the kyphosis condition could be identified early in these animals. The kyphosis was not detected in any of the neonatal animals examined. This condition may be similar to back curvature defects in humans that do not present until childhood or adolescence.
Several chromosomal regions were identified with a whole genome association analyses (Table 2). These regions were investigated further for genes that may be involved in the back defect via bone development pathways or known involvement with human spine defects. The development of vertebrae occurs through the reorganization of the somites that are formed on both sides of the midline neural tube during embryonic development [22]. The somites are reorganized into sclerotomes that ultimately give rise to the vertebrae. There are three phases of somite development during embryogenesis which includes a membraneous phase, a chondrification phase and an ossification phase [22]. Somite patterning for vertebrae specification is determined by the expression of the HOX gene family along the craniocaudal axis [22].
Two SNPs in the HOXC cluster of genes were identified as significant in the F2 population of animals (Table 4). The HOX genes are transcription factor clusters that control the positional architecture of an organism's body layout. Each HOX cluster (i.e., HOXA, HOXB, HOXC and HOXD) is located on a different chromosome and the genes in each cluster are thought to be the result of tandem duplications [23]. The number of the HOX gene describes the location of its anteroposterior axis expression with smaller gene numbers expressed closer to the head of the animal and larger numbers are expressed further down the spinal column. The HOXC8 and HOXD8 genes are involved in patterning the lower thoracic and lumbar vertebrae [24]. One of the significant SNP in this study is located in HOXC8, which is normally expressed during embryogenesis to reorganize the somites [22, 24] and also in chondrocytes [9]. When overexpressed in chondrocytes, HOXC8 causes cartilage defects characterized by an abundance of immature, proliferating chondrocytes. The normal function of HOXC8 is to act on skeletal development by influencing the maturation of chondrocytes along the differentiation pathway [9].
SNPs in SOX9 were significant in the F2 population. SOX9 is a master transcriptional regulator of chondrogenesis [25]. Mutations have been identified in SOX9 that cause haploinsufficiency and result in the human clinical condition campomelic dysplasia (CMD1). CMD1 is a skeletal condition often associated with XY sex reversal. Clinical phenotypes of this disorder include bowing and angulation of the long bones, short stature and anomalies of vertebrae and the spine [16]. SOX9 has also been shown to bind to regulatory regions of RUNX2 repressing its activity in vitro [26]. In CMD1 cartilage with heterozygous SOX9 mutations, RUNX2 expression was upregulated due to loss of repression [26]. We also evaluated RUNX2 in this study; however, we did not detect a significant association between the swine kyphosis condition in the two populations tested. Additional SNPs in the region of RUNX2 will need to be examined to rule out involvement of RUNX2 in this phenotype.
In addition to genes that may be involved with vertebral development pathways, genes that play a role in bone structure or bone growth were also investigated. Bone consists of osteoblasts that form bone, osteoclasts that remove bone tissue and osteocytes that are mature osteoblasts no longer capable of forming bone [27]. In addition, bone includes other fibers and compounds that help strengthen the bones; these consist mainly of collagen and minerals. The function of collagen is to give the bone tensile strength to allow the bone to withstand stretching forces [28]. The processes of producing, growing and maintaining bone and vertebrae are controlled by multiple genes and pathways.
PLOD1 SNPs were significantly associated with the back defect in the resource population of swine examined in this study. PLOD1 is an enzyme that forms hydroxylysine on collagen. Over 20 different mutations in the PLOD1 gene are responsible for Ehlers-Danlos Syndrome VIA (EDS VIA) [12], which is the kyphoscoliosis type of this connective tissue disorder. These mutations include a large duplication, point mutations that alter the amino acid sequence, mutations that result in a truncated protein, and splice site mutations that cause exons to be skipped [12]. Lysyl hydroxylase deficiency results in the common symptoms of muscle hypotonia, newborn joint laxity and kyphoscoliosis.
We did not identify any potentially detrimental mutations in the coding regions of PLOD1 or SOX9; however, we did discover several SNPs located close to intron junctions in PLOD1. It is also possible that in swine, the mutations in these genes may be located in promoter regions responsible for gene expression. While the majority of the human mutations in PLOD1 seem to involve a large duplication or tend to be located in the coding regions of the gene, the phenotypic expression of the disorder is also much more severe than the phenotype we have seen in these swine populations. Alternatively, it is also possible that we have not identified the correct positional candidate gene for kyphosis in these regions and that the SNPs in this study are simply in LD with the causative genes.
Conclusions
A total of 19 markers in 9 positional candidate genes were significantly associated with the skeletal defect; however, none of the same markers were detected in both the F2 and the USMARC resource populations of animals. These data suggest that the skeletal defect found in these swine herds is under polygenic control and that these populations are not segregating the same variation in these regions. Thus, there are likely to be additional SNPs in these and other genes that likely play a role in the kyphosis phenotype in swine. These data indicate that to eliminate this condition using genetic selection will involve screening SNPs in multiple genes and developing more predictive markers. The significant markers account for about 1% of the phenotypic variation in kyphosis present in these two commercial-like populations of swine. However, these markers must be evaluated in other commercial populations of swine with kyphosis before being used for genetic selection.
Methods
Animal care
Animals were fed ad libitum a corn-soybean meal grower ration of 18% protein from 8 to 13 wk, 16% from 13 to 16 wk of age and 15% protein after 16 wk of age [29]. Diets were formulated to meet or exceed National Research Council recommendations [30]. All animal procedures were reviewed and approved by the U.S. Meat Animal Research Center Animal Care and Use Committee and procedures for handling pigs complied with those specified in the Guide for the Care and Use of Agricultural Animals in Agricultural Research and Teaching [31].
F2 research and USMARC resource populations and phenotyping
The F2 population of swine was produced from 4 F1 boars and 50 F1 females. The F1 parents were reciprocal crosses between purebred Duroc and Landrace pigs and were raised indoors at a commercial production site under commercial production conditions. At approximately 114 kg, pigs were harvested at a commercial facility where they were processed [32]. These animals were phenotyped at USMARC 24 hours after harvest.
USMARC resource animals are from a multigenerational population developed by mating Yorkshire-Landrace composite females to either Duroc or Landrace boars selected from the industry. Twelve boars of each breed were mated to Yorkshire-Landrace females (n = 220) and further developed by reciprocally mating female descendants to either a Duroc or Landracesired boar. Subsequent inter se matings were random except that matings of the same sire line were avoided [33]. All animals were housed in indoor facilities on concrete floors and plastic slats. These animals were harvested at the USMARC abattoir and phenotypes were collected as described in Holl et al. [6]. Sows (n = 398) were harvested after first or second parity; the other animals were market age hogs. Briefly, subjective back scores were given for each spine evaluated in the split carcass. One score was given per animal by one trained individual per population of swine. Four categories were used: normal (0), mild (1), moderate (2), and severe (3). Mild was defined as a slight convex curvature of vertebrae. Moderate was defined as a convex curvature in the vertebrae and spinal cord. Severe was defined as an extreme convex curvature of the vertebrae and spinal cord [6]. Of the animals phenotyped in F2 and USMARC populations, 75.6 and 68.9% were normal (0), 11.1 and 23.3% were mild (1), 11.1 and 6.2% were moderate (2), and 2.2 and 1.5% were severe (3), respectively [6].
SNP discovery
Primer pairs for amplification of candidate genes were designed from available porcine EST, genomic, BAC-end, and BAC clone sequences deposited in GenBank using Primer 3 [34] (code available at http://frodo.wi.mit.edu/primer3/). Primer sets were obtained from IDT (Integrated DNA Technologies, Coralville, IA). PCR was performed in a Dyad PTC-0220 DNA engine (MJ Research Inc., Watertown, MA) using 0.5 U HotStar Taq polymerase (Qiagen, Valencia, CA); 1 × supplied buffer; 1.5 mM MgCl2; 200 μM dNTPs; 0.8 μM each primer; and 50-100 ng genomic DNA of 12 animals representing 8 different sire lines with all of the kyphosis scores represented, in 25 μl reactions. Five microliters of the PCR reaction was electrophoresed in 1.5% agarose gels to determine quality of amplification, and the remainder was prepared for sequencing after treatment with 0.1 U exonuclease I (USB, Cleveland, OH). Sequencing reactions were precipitated with 58% isopropanol and sequenced with an ABI 3730 capillary sequencer (Applied Biosystems, Foster City, CA). Bases were called with Phred and assembled into contigs with Phrap. Polymorphisms were identified using Polyphred and assessed using Consed http://www.phrap.org.
Genotyping analysis
Multiplex assays for use in the Sequenom MASSARRAY® system were designed using MASSARRAY® Assay Design software v3.1 (Sequenom, San Diego, CA). Each amplification primer had a 10-base tag added to ensure that the amplification primer masses were outside the range of the allele masses. Amplicon lengths were approximately 120 bp. Reaction conditions were performed as suggested by Sequenom iPLEX chemistry.
Whole genome association analyses
The resource population of animals (n = 1,257) was genotyped for 198 markers (28 microsatellites and 170 SNPs) dispersed throughout the genome to find chromosomal regions with significant association with the kyphosis phenotype. A single marker association was performed using the programs GenoProb and MTDFREML [35, 36]. The model included fixed effects of sex and harvest date, random additive polygenic effects, and SNP were fitted as a covariate (e.g., number of copies of an allele). Regressors for additive and dominance effects of each SNP were calculated using genotypic probabilities generated by allelic peeling algorithms in GenoProb [35] as described by Kuehn et al. [37]. Variance components were estimated using MTDFREML [36]. Each SNP was fitted separately due to potential multi-colinearity between regressions of closely linked markers. Nominal significance values were computed.
SNP association analysis (USMARC resource population)
The genotypic data and pedigree from the resource population of animals (n = 1,623) were analyzed using GenoProb and MTDFREML with the same animal model described above for the whole genome association analysis.
SNP association analysis (F2 population)
The genotypic data from the F2 population (n = 371) of animals were analyzed for association with the skeletal defect using Model 1 of the QTL Association option of Mendel 8.0.1 [38]. The program utilizes full pedigree and the model included the covariates (or predictors) of age and sex, and the contemporary groups (or household effects) of year and season. Covariance classes analyzed were additive, dominant and environmental.
False Discovery Rate (FDR)
The FDR was calculated for each population of animals with the following equation:
References
Done SH, Gresham ACJ: Lordosis and kyphosis ("humpy-back") in pigs. Pig J. 1998, 33: 134-141.
Done SH, Potter RA, Courtenay A, Peissel K: Lordosis and kyphosis ("humpy-back") in pigs: A second type of the condition associated with hemivertebrae. Pig J. 1999, 44: 148-153.
Lahrmann KH, Hartung K: Causes of kyphotic and lordotic curvature of the spine with cuneiform vertebral deformation in swine. Berl Munch Tierarztl Wochenschr. 1993, 106: 127-132.
Nielsen LWD, Hogendal P, Arnbjerg J, Jensen HE: Kyphosis in pigs. Proceedings of the 21st Meeting of the European Society of Veterinary Pathology: 10 September 2003; Dublin, Ireland;. 2003, 132-
Penny RHC, Walters JR: A "humpy-backed" syndrome of pigs. Vet Ann. 1986, 26: 128-135.
Holl JW, Rohrer GA, Shackelford SD, Wheeler TL, Koohmaraie M: Estimates of genetic parameters for kyphosis in two crossbred swine populations. J Anim Sci. 2008, 86: 1765-1769. 10.2527/jas.2007-0628.
Hastbacka J, Kerrebrock A, Mokkala K, Clines G, Lovett M, Kaitila I, de la Chapelle A, Lander ES: Identification of the Finnish founder mutation for diastrophic dysplasia (DTD). Eur J Hum Genet. 1999, 7: 664-670. 10.1038/sj.ejhg.5200361.
Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FFB, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere M, Lawler J: Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nature Genet. 1995, 10: 325-329. 10.1038/ng0795-325.
Yueh YG, Gardner DP, Kappen C: Evidence for regulation of cartilage differentiation by the homeobox gene HOXC-8. PNAS. 1998, 95: 9956-9961. 10.1073/pnas.95.17.9956.
Turnpenny PD, Bulman MP, Frayling TM, Abu-Nasra TK, Garrett C, Hattersley AT, Ellard S: A gene for autosomal recessive spondylocostal dysostosis maps to 19q13.1-q13.3. Am J Hum Genet. 1999, 65: 175-182. 10.1086/302464.
Jungbluth H, Zhou H, Hartley L, Halliger-Keller B, Messina S, Longman C, Brockington M, Robb SA, Straub V, Voit T, Swash M, Ferreiro A, Bydder G, Sewry CA, Muller C, Muntoni F: Minicore myopathy with ophthalmoplegia caused by mutations in the ryanodine receptor type 1 gene. Neurology. 2005, 65: 1930-1935. 10.1212/01.wnl.0000188870.37076.f2.
Giunta C, Randolph A, Steinmann B: Mutation analysis of the PLOD1 gene: an efficient multistep approach to the molecular diagnosis of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA). Mol Genet Me tab. 2005, 86: 269-276. 10.1016/j.ymgme.2005.04.014.
Xiong DH, Liu XG, Guo YF, Tan LJ, Wang L, Sha BY, Tang ZH, Pan F, Yang TL, Chen XD, Lei SF, Yerges LM, Zhu XZ, Wheeler VW, Patrick AL, Bunker CH, Guo Y, Yan H, Pei YF, Zhang YP, Levy S, Papasian CJ, Xiao P, Lundberg YW, Recker RR, Liu YZ, Liu YJ, Zmuda JM, Deng HW: Genome-wide association and follow-up replication studies identified ADAMTS18 and TGFBR3 as bone mass candidate genes in different ethnic groups. Am J Hum Genet. 2009, 84: 388-398. 10.1016/j.ajhg.2009.01.025.
Huber C, Dias-Santagata D, Glaser A, O'Sullivan J, Brauner R, Wu K, Xu X, Pearce K, Wang R, Uzielli ML, Dagoneau N, Chemaitilly W, Superti-Furga A, Dos Santos H, Negarbane A, Morin G, Gillessen-Kaesbach G, Hennekam R, Van der Burgt I, Black GC, Clayton PE, Read A, Le Merrer M, Scambler PJ, Munnich A, Pan ZQ, Winter R, Cormier-Daire V: Identification of mutations in CUL7 in 3-M syndrome. Nature Genet. 2005, 37: 1119-1124. 10.1038/ng1628.
Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JH, Owen MJ, Mertelsmann R, Zabel BU, Olsen BR: Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997, 89: 773-779. 10.1016/S0092-8674(00)80260-3.
Foster JW, Dominguez-Steglich MA, Guioli S, Kwok C, Weller PA, Stevanovic M, Weissenbach J, Mansour S, Young ID, Goodfellow PN, Brook JD, Schafer AJ: Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994, 372: 525-530. 10.1038/372525a0.
Tang PLF, Cheung C-L, Sham PC, McClurg P, Lee B, Chan S-Y, Smith DK, Tanner JA, Su AI, Cheah KSE, Kung AWC, Song Y-: Genome-wide haplotype association mapping in mice identifies a genetic variant in CER1 associated with BMD and fracture in Southern Chinese women. J Bone Miner Res. 2009, 24: 1013-1021. 10.1359/jbmr.081258.
Axenovich TI, Zaidman AM, Zorkoltseva1 IV, Kalashnikova EV, Borodin PM: Segregation analysis of Scheuermann disease in ninety families from Siberia. Am J Med Genet. 2001, 100: 275-279. 10.1002/ajmg.1290.
Shahbazian MD, Young JI, Yuva-Paylor LA, Spencer CM, Antalffy BA, Noebels JL, Armstrong DL, Paylor R, Zoghbi HY: Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002, 35: 243-254. 10.1016/S0896-6273(02)00768-7.
Zappella M, Meloni I, Longo I, Hayek G, Renieri A: Preserved speech variants of the Rett syndrome: molecular and clinical analysis. Am J Med Genet. 2001, 104: 14-22. 10.1002/ajmg.10005.
Johnston CE, Birch JG, Daniels JL: Cervical kyphosis in patients who have Larsen syndrome. J Bone Joint Surg. 1996, 78A: 538-545. 22
Dias MS: Normal and abnormal development of the spine. Neurosurg Clin N Am. 2007, 18: 415-429. 10.1016/j.nec.2007.05.003.
Ferraiuolo MA, Rousseau M, Carol Miyamoto C, Shenker S, Wang XQD, Nadler M, Blanchette M, Dostie J: The three-dimensional architecture of Hox cluster silencing. Nucleic Acids Res.
Van den Akker E, Fromental-Ramain C, de Graaff W, Le Mouellic H, Brulet P, Cambon P, Deschamps J: Axial skeletal patterning in mice lacking all paralogous group 8 Hox genes. Development. 2001, 128: 1911-1921.
Akiyama H: Control of chondrogenesis by the transcription factor SOX9. Mod Rheumatol. 2008, 18: 213-219. 10.1007/s10165-008-0048-x.
Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, Lee B: Dominance of SOX9 function over RUNX2 during skeletogenesis. PNAS. 2006, 103: 19004-19009. 10.1073/pnas.0605170103.
Nijweidi PJ, Feyen JHM: Cells of bone: Proliferation, differentiation, and hormonal regulation. Physiol Rev. 1986, 66: 855-886.
Viguet-Carrin S, Garnero P, Delmas PD: The role of collagen in bone strength. Osteoporos Int. 2006, 17: 319-336. 10.1007/s00198-005-2035-9. 27
Klindt J, Thallman RM, Wise T: Effects of sire line, sire, and sex on plasma urea nitrogen, body weight, and backfat thickness in offspring of Duroc and Landrace boars. J Anim Sci. 2006, 84: 1323-1330.
NRC: Nutrient Requirements of Swine. 1998, Natl. Acad. Press, Washington, DC, 10th rev
FASS: Guide for Care and Use of Agricultural Animals in Agricultural Research and Teaching. 1999, Fed. Animal Science Society, Savoy, IL, 1
Rohrer GA, Thallman RM, Shackelford S, Wheeler T, Koohmaraie M: A genome scan for loci affecting pork quality in a Duroc-Landrace F2 population. Anim Genet. 2006, 37: 17-27. 10.1111/j.1365-2052.2005.01368.x.
Lindholm-Perry AK, Rohrer GA, Holl JW, Shackelford SD, Wheeler TL, Koohmaraie M, Nonneman D: Relationships among calpastatin single nucleotide polymorphisms, calpastatin expression and tenderness in pork longissimus. Anim Genet. 2009, 40: 713-721. 10.1111/j.1365-2052.2009.01903.x.
Rozen S, Skaletsky HJ: Primer3 on the WWW for general users and for biologist programmers. Methods in Mol Biol. Edited by: Krawetz S, Misener S. 2000, Totowa NJ: Humana Press, Bioinformatics Methods and Protocols, 365-386.
Thallman RM: User's Manual for GenoProb Version 2.000. 2002, USDA-ARS, U.S. Meat Animal Research Center, Clay Center, NE
Boldman KG, Kriese LA, Van Vleck LD, Van Tassel CP, Kachman SD: A manual for the use of MTDFREML. A set of programs to obtain estimates of variance and covariances (Draft). 1995, USDA, ARS, Lincoln, NE
Kuehn LA, Rohrer GA, Nonneman DJ, Thallman RM, Leymaster KA: Detection of single nucleotide polymorphisms associated with ultrasonic backfat depth in a segregating Meishan × White Composite population. J Anim Sci. 2007, 85: 1111-1119. 10.2527/jas.2006-704.
Lange K, Cantor R, Horvath S, Perola M, Sabatti C, Sinsheimer J, Sobel E: Mendel Version 4.0: A complete package for the exact genetic analysis of discrete traits in pedigree and population data sets. Am J Hum Genet. 2001, 69 (Supplement): A1886-
Tang PL, Cheung CL, Sham PC, McClurg P, Lee B, Chan SY, Smith DK, Tanner JA, Su AI, Cheah KS, Kung AW, Song YQ: Genome-wide haplotype association mapping in mice identifies a genetic variant in CER1 associated with BMD and fracture in southern Chinese women. J Bone Miner Res. 2009, 24: 1013-1021. 10.1359/jbmr.081258.
Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo T, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, Kucherlapati RS: Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nature Genet. 2007, 39: 70-74. 10.1038/ng1926.
Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W, Ustaszewska A, Martin J: Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nature Genet. 2007, 39: 75-79. 10.1038/ng1939.
Meyers SN, Rogatcheva MB, Larkin DM, Yerle M, Milan D, Hawken RJ, Schook LB, Beever JE: Piggy-BACing the human genome II. A high-resolution, physically anchored, comparative map of the porcine autosomes. Genomics. 2005, 86: ,739-52. 10.1016/j.ygeno.2005.04.010.
Munoz G, Ovilo C, Estelle J, Silio L, Fernandez A, Rodriguez C: Association with letter size of new polymorphisms on ESR1 and ESR2 genes in a Chinese-European pig line. Genet Sel Evol. 2007, 39: 195-206. 10.1186/1297-9686-39-2-195.
Ciobanu DC, Bastiaansen JW, Lonergan SM, Thomsen H, Dekkers JC, Plastow GS, Rothschild MF: New alleles in calpastatin gene are associated with meat quality traits in pigs. J Anim Sci. 2004, 82: 2829-2839.
Kennes YM, Murphy BD, Pothier F, Palin MF: Characterization of swine leptin (LEP) polymorphisms and their association with production traits. Anim Genet. 2001, 32: 215-218. 10.1046/j.1365-2052.2001.00768.x.
Campbell EMG, Nonneman D, Rohrer GA: Fine mapping a quantitative trait locus affecting ovulation rate in swine on chromosome 8. J Anim Sci. 2003, 81: 1706-1714.
Acknowledgements
The authors are grateful for the excellent technical expertise provided by Sue Hauver, Kris Simmerman and Patty Beska and for the administrative assistance received from Donna Griess and Sherry Kluver. Mention of trade name proprietary product or specified equipment does not constitute a guarantee or warranty by the USDA and does not imply approval to the exclusion of other products that may be suitable. USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
ALP carried out the molecular genetic studies, participated in the design of the study and drafted the manuscript.
GAR collected the phenotypes on the USMARC resource population, conceived the study and participated in the project design and performed statistical analyses.
JWK, LAK and JWH performed statistical analysis of genotypic data.
SDS and TLW identified the kyphosis condition in the commercial population of animals and collected and provided phenotypic data.
DJN conceived the study, participated in design and coordination and helped draft the manuscript.
All authors read and approved the manuscript.
Electronic supplementary material
12863_2010_859_MOESM1_ESM.XLSX
Additional file 1: Genome-wide association of 198 microsatellite and SNP markers for kyphosis in swine. Primer sequences and association analysis for 198 SNPs and microsatellites located across the swine genome [46]. (XLSX 35 KB)
12863_2010_859_MOESM2_ESM.XLSX
Additional file 2: SNPs identified in candidate genes for kyphosis in swine. Primer sequences, location and dbSNP numbers for SNPs identified in candidate genes. Minor allele frequencies are provided for SNPs that were genotyped. (XLSX 37 KB)
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lindholm-Perry, A.K., Rohrer, G.A., Kuehn, L.A. et al. Genomic regions associated with kyphosis in swine. BMC Genet 11, 112 (2010). https://doi.org/10.1186/1471-2156-11-112
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-11-112