
Abstract
Background
The size of the vertebrate eye and the retina is likely to be controlled at several stages of embryogenesis by mechanisms that affect cell cycle length as well as cell survival. A mutation in the zebrafish out of sight (out) locus results in a particularly severe reduction of eye size. The goal of this study is to characterize the outm233mutant, and to determine whether mutations in the out gene cause microphthalmia in humans.
Results
In this study, we show that the severe reduction of eye size in the outm233mutant is caused by a mutation in the zebrafish gdf6a gene. Despite the small eye size, the overall retinal architecture appears largely intact, and immunohistochemical studies confirm that all major cell types are present in outm233retinae. Subtle cell fate and patterning changes are present predominantly in amacrine interneurons. Acridine orange and TUNEL staining reveal that the levels of apoptosis are abnormally high in outm233mutant eyes during early neurogenesis. Mutation analysis of the GDF6 gene in 200 patients with microphthalmia revealed amino acid substitutions in four of them. In two patients additional skeletal defects were observed.
Conclusions
This study confirms the essential role of GDF6 in the regulation of vertebrate eye size. The reduced eye size in the zebrafish outm233mutant is likely to be caused by a transient wave of apoptosis at the onset of neurogenesis. Amino acid substitutions in GDF6 were detected in 4 (2%) of 200 patients with microphthalmia. In two patients different skeletal defects were also observed, suggesting pleitrophic effects of GDF6 variants. Parents carrying these variants are asymptomatic, suggesting that GDF6 sequence alterations are likely to contribute to the phenotype, but are not the sole cause of the disease. Variable expressivity and penetrance suggest a complex non-Mendelian inheritance pattern where other genetic factors may influence the outcome of the phenotype.
Similar content being viewed by others

Background
Microphthalmia, anophthalmia, and chorioretinal coloboma are ocular malformations that affect 1 in 3000-4000 individuals [1–3]. In microphthalmia and anophthalmia, one or both eyes are abnormally small or clinically absent. Colobomata are clefts caused by absent eye tissue, due to a failure of the optic fissure to close. Colobomata are frequently grouped with microphthalmia and anophthalmia, as they are often associated with a reduction of eye size. The aetiology of these ocular malformations is complex, and a wide variation is seen in phenotypic expression. Recessive, dominant and X-linked modes of inheritance have been described, but often sporadic and non-Mendelian inheritance patterns are seen [4, 5]. In addition, a variety of environmental factors may be causative in certain cases, including viral infection, such as rubella, irradiation and drug intake in pregnancy.
Mutations in the transcription factor SOX2 are the most prevalent monogenic cause of microphthalmia and anophthalmia identified to date [6]. Other genes include the transcription factors PAX6, OTX2, CHX10 and RAX [7–10]. More recently, mutations in three members of the transforming growth factor-β (TGF-β) superfamily (BMP4; GDF6, also known as BMP13; and GDF3) have been associated with microphthalmia/anophthalmia [11–15]. Members of the TGF-β superfamily of secretory signaling molecules play essential roles in embryonic development [16, 17]. Members of this superfamily regulate cell proliferation and apoptosis, and play important roles in various processes such as the creation of dorsal-ventral axes in the embryo, specification of the neural crest, bone formation, and organogenesis [18–20].
A segmental deletion encompassing the GDF6 gene, and several amino acid substitutions in GDF6 have been identified in patients with ocular anomalies, including coloboma and microphthalmia [13–15]. In addition, a chromosomal rearrangement and several amino acid substitutions in GDF6 have been detected in patients with Klippel-Feil syndrome, a complex skeletal disorder characterized by congenital fusion of the cervical spine, and in patients with other skeletal defects [21, 14]. Gdf6 is expressed in the dorso-temporal retina, and morpholino (MO) knockdown experiments of Gdf6 in zebrafish and Xenopus result in reduced eye size or even the absence of optic lobes [13, 22]. As these experiments were performed using antisense compounds, it is likely that this variability reflects imperfections of the morpholino knockdown approach. In Xenopus, and to a lesser extent in zebrafish, this phenotype is accompanied by a disorganization of retinal layering [22, 13]. In addition, the presence of skeletal anomalies (curled or kinked tails) was observed in a fraction of MO-treated zebrafish embryos. The loss of Gdf6 in homozygous knockout mice causes abnormalities in joint, ligament and cartilage formation, and variable ocular phenotypes [23, 14]. Finally, a small eye phenotype in the zebrafish mutant dark halfs327was attributed to a nonsense mutation in the gdf6a gene [24]. The authors of this work show that gdf6a establishes dorsal-ventral positional information in the retina and controls the formation of the retinotectal map.
Given conflicting results of studies in different vertebrate model systems, the role of GDF6 in eye development merits further investigation. We performed analysis of the gdf6a gene in the zebrafish outm233mutant, characterized by a severe reduction of eye size [25, 26]. We found that the outm233mutation eliminates the initiation codon in the gdf6a gene, disrupting the function of this gene. Despite the reduction of eye size, retinal lamination is normal in most mutant animals, and the optic nerve appears largely unaffected. The small eye size phenotype in outm233is associated with a wave of apoptosis during early stages of development. Other than eye size reduction, no obvious defects are observed in the external appearance of outm233mutants, including its craniofacial skeleton. outm233mutant homozygotes frequently survive to adulthood and display a variable reduction of size. Other than eye defects, we have not observed obvious abnormalities in the appearance of swimming behavior of outm233mutant homozygotes. To investigate the role of GDF6 in human eye defects, we screened the GDF6 gene in 200 patients suffering from microphthalmia, anophthalmia or related abmormalities, and detected amino acid substitutions in four of them.
Results
The out of sightm233phenotype
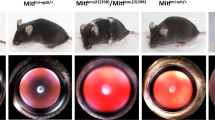
The out of sight (out)m233mutant was identified in a large-scale ENU mutagenesis screen [25, 26]. Out of several small eye mutants that were isolated in this experiment, outm233displayed the most severe phenotype already distinguishable during day 2 of development [26] (Figure 1). Pigmentation appears to be darker in outm233, compared to its wild-type siblings at 5 dpf, but no obvious defects are observed in other organs, and some mutant fish differentiate the swim bladder. The majority of mutant individuals die between 5 and 10 dpf, and only a small fraction of animals survive to several weeks of age. These individuals have grossly normal body shapes, but feature very small eyes and in the most extreme cases are anophthalmic (Figure 1E, F). Histological analysis indicates that both the retina and the lens are grossly reduced in size in outm233mutants (Figure 1). Although the retina is reduced in size by ~50% at 5 dpf, in most mutant individuals its overall architecture appears grossly intact.

Phenotype of the zebrafish out of sight mutant. (A - B) Lateral views of wild-type zebrafish (A) and of the outm233mutant (B) at 36 hpf, 48 hpf (a1 and b1), 72 hpf (a2 and b2) and 96 hpf (a3 and b3). The outm233mutant is characterized by a severe reduction in eye size. No obvious defects are observed in other organs. (C - D) Transverse plastic sections of wild-type (C) and outm233mutant (D) retinae at 36 hpf, 4 dpf (c1 and d1) and 5 dpf (c2 and d2). Although the retina and lens are grossly reduced in size in outm233mutants, their overall architectures appear largely intact. (E - F) An anophthalmic outm233homozygous mutant at ca. 2 months of age. An enlargement of the head region is shown in (F). (G - H) A wild-type individual (G) and its microphthalmic outm233sibling (H) at 2.5 months of age. In A, B, and E-H, anterior is to the left.
Positional cloning of the outm233mutation
Mapping analysis revealed that the out locus is located on chromosome 16 between markers Z6293 and Z8819 (Figure 2). This region contains around 41 annotated genes, including gdf6a (radar, LOC566470). Sequence analysis of gdf6a in the outm233mutant identified a c.1A > G transition affecting the start codon (p.Met1Val). This is the first ATG in the gdf6a transcript, and is preceded by a termination codon 42 bp upstream, confirming that this is the translation start site. The first ATG following the affected start codon is located four base pairs downstream (c.5A), and if used would result in a shift in the open reading frame. However, that ATG might not be used as a translation start site since it is not embedded in a Kozak consensus sequence. Another translation start site that is predicted to be functional by NetStart [27] is located at position c.223A, and its use would produce a protein in the correct reading frame but lacking the first 74 amino acids, including the signal peptide.

Positional cloning of the zebrafish out of sight mutation. (A) The out locus maps to chromosome 16 between markers Z6293 and Z8819. Sequence analysis of the gdf6a gene from this interval identified a mutation affecting its start codon (c.1A > G, p. Met1Val) in mutant (C) but not wild-type (B) individuals. SP = signal peptide, ASD = active signaling domain. (D, E) The overexpression of wild-type (E) but not mutant (D) gdf6a causes ventral eye defects. Shown are lateral (left panels) and ventro-lateral (right panels) views of zebrafish larvae at 3 dpf. Anterior is to the left, asterisks indicate the lens. (F) The frequency of ventral eye defects, following the overexpression of wild-type or mutant gdf6a in four independent experiments, as indicated on the horizontal axis (1 through 4). Numbers above the bar graph indicate sample sizes.
To confirm that its defects are indeed responsible for the outm233mutant phenotype, we overexpressed the wild-type gdf6a in the progeny of crosses between outm233heterozygotes. This was accomplished by injecting into embryos a DNA construct containing gdf6a under control of a heat-shock promoter. A mutant gdf6a gene that contains the p.Met1Val substitution was used in these experiments as a negative control. The overexpression of the wild-type gdf6a produced a significant decrease (p = 0.003, chi square) in the frequency of phenotypically mutant animals from ca. 20% (34/171) to 8% (13/154). At the same time, we observed that ca. 9% (14/155) of animals featured one small and one normal size eye. This phenotype was entirely absent in animals treated with the control mutant construct (0/171). We interpret the one eye phenotype as a partial rescue of outm233eye size defect.
To further test whether the p.Met1Val substitution affects gene function, we overexpressed wild-type and mutant Gdf6 in zebrafish using a heat-inducible promoter. In agreement with previous studies [24], the overexpression of the wild-type Gdf6 construct frequently results in ventral eye defects. In contrast to that, the overexpression of the mutant gene produces ventral eye abnormalities with a much lower frequency (Figure 2D-F). These data are in agreement with the results of outm233rescue experiments, and indicate that the p.Met1Val substitution affects gene function. Based on these results, we conclude that mutation in the gdf6a gene is responsible for small eye phenotype in the outm233mutant strain.
Evaluation of cell fate in outm233retinae
To develop a better understanding of the mutant phenotype, we evaluated the number of retinal cells by counting cell nuclei on sections through wild-type and gdf6a/outm233retinae at 36 and 96 hpf (Figure 3). At 36 hpf, the number of retinal cells is not statistically different in outm233mutants, compared to the wild-type. At 96 hpf, cell numbers in all cell layers examined are reduced in mutant embryos, compared to the wild type (p < 0.01 for all cell layers, Student's t-test).

The number of cells in wild-type and outm233mutant retinae. (A - B) Hoechst staining of 4-μm plastic sections through the retina of wild-type (A) and outm233(B) embryos at 36 hpf. (C) Diagram showing the number of nuclei that were counted in the dorsal, ventral, and entire retina. At 36 hpf, cell numbers in mutant embryos are not significantly different from these in wild-type siblings. Cells were counted on sections from 7 and 4 embryos for wild-type and mutant, respectively. (D - E) Hoechst staining of 4-μm plastic sections through the retina of wild-type (D) and outm233(E) embryos at 96 hpf. (F) Diagram showing the number of nuclei that were counted in the ganglion cell layer (GCL), in the inner nuclear layer (INL) and in the photoreceptor layer (PRCL). By 96 hpf, cell numbers in all these three layers are reduced in mutant embryos, compared to the wild type (for GCL, p < 0.01; for INL and PRCL, p < 0.001; t-test). For both wild-type and mutant, cells were counted on sections from 6 embryos. Asterisks indicate the lens, and arrowheads the optic nerve.
To determine whether all major cell classes are present in outm233retinae, sections through wild-type and mutant larvae were immunolabeled at 2-6 dpf using several antibodies, including anti-carbonic anhydrase for Müller glia [28]; anti-parvalbumin, anti-serotonin, and anti-GABA for subsets of amacrine cells [29]; anti-Pax6 and anti-HuC/HuD for ganglion and amacrine cells [30, 31]; anti-Zpr1 for red/green cone photoreceptor cells; anti-rod opsin for rod cells [32, 33]; and anti-neurolin for ganglion cells [33] (Figure 4, and data not shown). These experiments demonstrated that all cell classes assayed are present in outm233embryos in relatively normal proportions, however, some of them display aberrant patterns. For example, in ca. ~50% of outm233embryos, we observed the absence of Zpr1-positive cells in some regions of the photoreceptor cell layer (Figure 4). This defect frequently correlates with the mislocalization of parvalbumin-positive cells (Figure 4F, compare to the wild type in E). The most interesting defect is present in GABA-positive amacrine cells. On the majority of sections, the number of GABA-positive neurons is increased in the retinal periphery and decreased in the center of the retina (arrowheads in Figure 4N, compare to M, graph in 4O). We have not observed obvious defects in the ganglion cell layer or in Muller glia. The optic nerve displays grossly normal appearance (Figure 4B), and glial cells extend apical processes into the photoreceptor cell layer (Figure 4L, inset).

Cell fate in wild-type and outm233retinae. (A-B) Transverse sections through the retina stained with anti-neurolin antibody (red) in wild-type (A) and outm233(B) embryos. The ganglion cell layer and the optic nerve display grossly normal appearance at 56 hpf. Sections were counterstained with YoPro (green). (C-D). Anti-parvalbumin antibody (red) stains a subset of amacrine cells on transverse sections through the retinae of wild-type (C) and outm233mutant (D) embryos. A displacement of some amacrine cells towards and occasionally into the photoreceptor cell layer is observed in approximately 50% of outm233embryos (arrowhead in D). (E - F) Anti-paravalbumin and Zpr1 double immunostaining (both in green) in wild-type (E) and in outm233mutant (F) embryos. In approximately 50% of the embryos, Zpr1-positive cells are absent in the dorsal and/or ventral regions of the photoreceptor layer (arrowheads in F). (G - H) Images of whole embryos at 42.5 hpf. Ganglion cells are visualized with brn3c-GFP transgene in wild-type (G) and outm233mutant (H) embryos. Anterior is to the left. (I -J) The Zpr1 antibody stains red and green cones (green signal) while anti-serotonin antibody labels serotonin-positive amacrine cells (red) in wild-type (I) and outm233mutant (J) embryos. (K-L) Anti-carbonic anhydrase antibody (red) stains Müller glia in wild-type (K) and outm233mutant (L) embryos. Arrowhead in the inset indicates the apical processes of the Muller glia, which terminate at the outer limiting membrane. (M - N) Anti-GABA antibody (green) stains a subset of amacrine cells in wild-type (M) and outm233mutant (N) embryos. On the majority of sections, the number of GABA-positive neurons is increased in the retinal periphery of outm233homozygotes (arrowheads). (O) Graph showing the frequency of GABA-positive cells in the central retina. The number of cells per an arbitrary unit of distance if provided. Fewer GABA-positive cells are observed in the mutant, compared to the wild type (p < 0.01, t-test). Blue dots represent sections. As some sections have the same frequency of GABA-positive cells, the number of dots does not equal the number of sections. n ≥ 4 embryos for both wild-type and mutant. Asterisks mark the lens. (C-F) and (I-N) show retinae at 5 dpf.
Evaluation of cell proliferation and apoptosis during outm233eye development
A reduction of organ size can be caused, among other reasons, by changes in the cell cycle, including a premature differentiation of proliferative precursor cells, or increased cell death [34, 35]. An increase in the length of G1, S, or G2 phase, for example, would result in a decrease in the frequency of cells positive for phospho-H3, a marker of mitosis. Conversely, an increase in M-phase length would result in an increased occurrence of phospho-H3 positive cells. If, on the other hand, retinal precursor cells are differentiating prematurely, then neurolin, a marker of ganglion cells, the first retinal neurons to become postmitotic [36], is likely to be expressed earlier in the mutant, compared to the wild-type retina. The immunolabeling of mutant and wild-type embryos at 36 hpf with anti-phospho-histone H3 and anti-neurolin antibodies revealed that the levels of phospho-H3 histone-positive (i.e. mitotic) nuclei relative to all nuclei, and the appearance of neurolin-positive ganglion cells in mutant retinae are normal (Figure 5, and data not shown). To further confirm that the timing of neurogenesis is not affected in outm233mutants, we crossed the Tg(pou4f3:gap43-GFP) transgene into the outm233mutant background. This transgenic line expresses GFP in the ganglion cell layer by 42 hpf [37]. We have not observed consistent differences in the onset of GFP expression between outm233mutants and their wild-type siblings (n > 20 for both wild-type and mutant; Figure 4H, and data not shown). This indicates that the timing of ganglion cell neurogenesis is unlikely to be defective in outm233embryos. Based on anti-phospho-H3 staining, it also appears that the length of the M-phase, relative to the rest of the cell cycle is unchanged in the mutant. We cannot, however, exclude the possibility that the overall cell cycle length is affected in outm233animals.

Cell proliferation in the out of sight retina. (A - B) Immunolabeling of transverse cryosections through the retinae of wild-type (A) and outm233mutant (B) embryos with anti-phospho-histone H3 (red) antibodies at 36 hpf. Sections were counterstained with YoPro (green). (C) Graph illustrating the numbers of phospho-H3 histone-positive nuclei on sections through wild-type and outm233mutant retinae as indicated. (D) Graph showing the ratio of phospho-H3-positive cells to all cells. No differences between wild-type and outm233embryos were identified. In (C - D), black dots represent sections. As some sections have the same number of phospho-H3-positive cells, the number of dots does not equal the number of sections. n ≥ 10 embryos and ≥ 15 sections for both wild-type and mutant samples. Asterisks indicate the lens, and arrowheads the optic nerve. Dorsal is up.
Reduced eye and lens size in mutant embryos may also be due to increased apoptotic removal of retinal cells. To test this possibility, apoptotic cells in wild-type and mutant embryos were visualized by staining with acridine orange (26 hpf, not shown) or TUNEL (30, 48, 72, and 120 hpf). The levels of apoptosis are abnormally high in outm233mutant eyes during early neurogenesis. Apoptosis in the lens, a normal event in wild-type zebrafish at 24-25 hpf [38, 39], persists in mutant embryos. At 30 hpf, an obvious increase of apoptosis is seen in the lens and the retina of mutants, compared to the wild type (Figure 6). Apoptosis persists in and around the lens region in the mutant at 48 hpf. By 76 hpf, on the other hand, there is hardly any apoptotic activity visible in the mutant eye, and a significantly larger amount of apoptosis occurs in the wild-type retina (Figure 6). At 5dpf, few apoptotic cells are detectable both in wild-type and mutant animals. These results suggest that the small eye phenotype is caused by a wave of apoptosis in the mutant eye.

Apoptosis in outm233mutant embryos. (A - H) Transverse cryosections through the zebrafish eye. Apoptosis is detected using the TUNEL assay (red signal). Levels of apoptosis are abnormally high in outm233mutant (E-H) eyes during early neurogenesis. At 30 hpf, there is an increase of apoptosis in the lens and the retina of mutants (E) compared to the wild-type (A). Apoptosis persists in and around the lens region at 48 hpf (F, compare to B). By 76 hpf, hardly any apoptosis is visible in the mutant eye (G), and a significantly increased amount of apoptosis occurs in the wild-type (C) retina, compared to the mutant. At 5 dpf, hardly any apoptotic cells are found in both the wild type (D) and the mutant (H). (I) The amount of cell death is quantitated. The average number of apoptotic cells per section is provided. Arrowheads point to the lens. n ≥ 9 sections from at least 3 embryos for each time point both for wild-type and mutant samples.
Mutation analysis of the GDF6 gene in patients with microphthalmia
A cohort of 200 unrelated probands with coloboma, microphthalmia and/or anophthalmia was screened for mutations in the GDF6 gene. Three heterozygous synonymous changes were identified in 11 patients (c.93C > T/p.Ser31Ser, c.852C > G/p.Ser284Ser, and c.936G > C/p.Ser312Ser). A heterozygous transition (c.955G > A) leading to an amino acid substitution (p.Ala319Thr) was identified in a patient (ID178) with isolated unilateral microphthalmia. This variant was not detected on 362 control chromosomes.
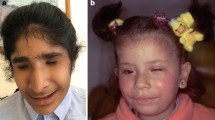
In three unrelated patients (ID66, ID136 and 792-535), we identified a heterozygous transition (c.746C > A) leading to an amino acid change (p.Ala249Glu) in the GDF6 prodomain (Figure 7). Besides microphthalmia, patient ID66 has malformed ossicles. Patient ID136 has been diagnosed with cystic fibrosis due to mutations in the CFTR gene, microphthalmia, cleft palate, hemivertebrae, hemifacial hypoplasia, a ventriculoperitoneal shunt for hydrocephalus, talipes, growth hormone deficiency and a small pituitary gland. The third patient, 792-535, has a half-sister who was also diagnosed with microphthalmia. The half-sister does not carry the p.Ala249Glu variant, while the carrier father does not have microphthalmia (Figure 7C).

GDF6 analysis in patients with microphthalmia, anophthalmia and coloboma. (A) Two different heterozygous amino acid changes, p.Ala249Glu and p.Ala319Thr, were detected in four unrelated individuals. (B) The sequence of the heterozygous p.Ala249Glu variant in patient 792-535. (C) The affected half-sib of patient 792-535 does not carry the p.Ala249Glu variant, while it was detected in the unaffected father.
Discussion
In this study, we characterized the zebrafish outm233mutant strain and identified a mutation in the gdf6a gene affecting the translation start site (p.Met1Val). The size of the retina and the lens are grossly reduced in gdf6a/outm233mutants, but the overall retinal architecture appears largely intact. This is in contrast to other small eye size phenotypes in zebrafish, which often exhibit retinal defects in lamination or retinal degeneration [40, 26, 41]. Despite eye size reduction, all cell classes that we assayed for are present in outm233mutants, although some display variable abnormalities. In ~50% of outm233embryos we observed partial absence of the photoreceptor cell layer in the dorsal and/or ventral regions of the retina. In the regions of photoreceptor absence, we observed a displacement of amacrine cells towards and occasionally into the photoreceptor cell layer. Another interesting defect is present in GABA-positive neurons. These cells are present in abnormally large numbers at the retinal margin at 6 dpf. As neurogenesis continues at the margin of the retina throughout the lifetime of the organism and the quantity of GABA-positive cells decreased in the central retina, this phenotype is most likely transient. As postmitotic GABA-positive neurons near the marginal zone become older, their excess is most probably eliminated by apoptosis.
Our studies suggest that a major contributing factor to the small eye and lens size is deregulated apoptosis. Apoptosis is a normal event during the development of the zebrafish retina, normally occurring around 24 to 25 hpf [38]. The increased apoptosis in the outm233mutant is evident at 26 hpf and persists until at least 48 hpf, suggesting that retinal precursor cells which normally contribute to the neural retina and lens are being eliminated through apoptosis, which results in a reduction of cell numbers in all retinal layers by 72 hpf.
While this manuscript was in preparation, the small eye size in another zebrafish mutant (dark halfs327), was attributed to a nonsense mutation in the gdf6a gene [24]. The authors of this work show that gdf6a is necessary to induce dorsal fate in the retina. Loss of gdf6a prevents specification of the retinal ganglion cells with dorsal identity and prevents innervation of the ventral tectum. Gdf6a activates the expression of known dorsal markers (bmp4, tbx5, tbx2b, efnb2) and represses the expression of ventral fate determinant, vax2. Similar to our observations in the outm233mutant, the retina of the dark halfs327homozygotes features a transient increase in cell death [24]. Lamination defects have not been reported.
Previous studies have examined the effect of GDF6 loss of function on eye development by morpholino antisense oligonucleotide (MO) knockdown in Xenopus and zebrafish [13, 22]. A striking reduction of eye size was observed in Xenopus embryos [22]. In zebrafish, MO injection resulted in highly variable ocular anomalies including ventral colobomata, persistent dorsal-retinal groove, microphthalmia, and even anophthalmia [13]. The retina was disorganized after MO knockdown in Xenopus and zebrafish, and immunolabeling with anti-XAP-1 and anti-islet-1 antibodies in Xenopus did not detect any photoreceptor, ganglion or amacrine cells [13, 22]. This is in contrast to our findings in the outm233mutant, where retinal lamination is largely normal and all cell classes are present.
MO knockdown of gdf6a in zebrafish recently identified the presence of skeletal anomalies (curled or kinked tails), although their prevalence was much lower than that of ocular anomalies [14]. Axial abnormalities were also seen in Xenopus tadpoles following morpholino injection [21]. In these studies, MO knockdown of gdf6a did not always lead to a mutant phenotype, as significantly reduced levels of correctly spliced gdf6a mRNA were identified in phenotypically unaffected morphant embryo pools [14]. In contrast to these findings, other than eye size reduction, no obvious defects are observed in the external appearance of outm233mutants, including its craniofacial skeleton. The external morphology of dark halfs327mutants was also inconspicuous, with the exception of smaller eyes [24]. Similarly, we also did not observe any defects in circulation and axial vasculature in the outm233mutant, although these were reported after MO-knockdown of gdf6a in zebrafish [13, 42, 43]. The differences observed between MO-injected embryos and the outm233and dark halfs327mutant strains might be attributed to a toxic effect of the MOs or to non-specific interference of anti-gdf6a morpholinos with other genes. Non-target related phenotypes including neuronal cell death have been reported for 18% of MOs, even when relatively low MO concentrations are used [44]. Mistargeting by MOs could be caused by the simultaneous inactivation of an essential gene with serendipitous homology, or perhaps by some unexpected chemical contaminants found in a small fraction of MO syntheses [44]. Such nonspecific effects of morpholino knockdown experiments can be usually excluded via rescue experiments [45, 46]. In the case of gdf6, rescue experiments are difficult to perform as the overexpression of this gene results in the ventralization of the entire embryo [22, 21]. An alternative explanation for the discrepancy could be that the outm233mutant may be a hypomorph that retains some residual gdf6a function. This, however, seems unlikely since the outm233mutation disrupts the translation start site, which will either abolish protein synthesis (if the ATG at position c.5A is used) or produce a protein lacking the signal peptide (if the ATG at position c.233A is used). Such a truncated protein is unlikely to be transported to the endoplasmic reticulum and Golgi for posttranslational modifications, which include two proteolytic cleavage steps[47]. Another defect in the zebrafish gdf6a gene was previously reported to result in a short body axis, a reduction of head structures, and death by 48 hpf [48]. That mutant, however, carries a large deletion of many genes, which could explain the more severe phenotype.
In a previous study, a segmental deletion encompassing the GDF6 gene was identified in a patient with bilateral chorioretinal colobomata [13]. The patient exhibited multiple developmental defects, including neurodevelopmental impairment, bilateral soft-tissue syndactyly of the toes, and an atrial septal defect. Besides GDF6, 30 other genes are also lost in the deletion, which could contribute to the range of developmental defects seen in this patient.
Mutations in GDF6 have also been recently reported in patients with Klippel-Feil syndrome, a complex skeletal disorder characterized by congenital fusion of vertebrae within the cervical spine [21]. A chromosomal inversion segregated with the disease in a 5-generation family exhibiting an autosomal dominant inheritance pattern. Besides fusion of the vertebral bodies and laminae, the affected family members had scoliosis of the thoracic and lumbar spine, fusion of the carpal and tarsal joints, and restricted elbow movement. No DNA loss was evident at the inversion boundaries, and no coding genes appeared to be disrupted. The proximal inversion breakpoint was located 623 kb 3'of GDF6. The authors suggest that the disease is associated with GDF6, perhaps by disruption of long-range enhancer elements, since the skeletal anomalies in this family correspond with the phenotype observed in Gdf6 knockout mice [23]. In these mice, defects in joint, ligament, and cartilage formation were seen in the wrist, the ankle, the middle ear, and the coronal suture between bones in the skull [23]. Analysis of GDF6 in 127 individuals with Klippel-Feil syndrome identified two missense variants, p.Ala249Glu and p.Leu289Pro [21]. No ocular defects were described in these patients [21]. The two variants were not identified on 708 control chromosomes [21].
In a recent study, 489 patients with ocular anomalies and 81 patients with vertebral segmentation defects were screened for GDF6 mutations [14]. The authors identified four amino acid substitutions associated with ocular phenotypes, two with a skeletal phenotype, and one alteration (p.Ala249Glu) was identified in 3 probands who either had ocular or skeletal phenotypes. These variants were not detected on at least 366 control chromosomes. For three variants, segregation analysis showed the presence of the alteration in an unaffected parent indicating incomplete penetrance. The effect of two amino acid substitutions, p.Ala249Glu and p.Lys424Arg, were evaluated with SOX9 reporter gene assays and Western blot analysis. Mutant GDF6 was less effective in activating SOX9 reporter gene activity compared to wild-type GDF6. By Western blot analysis it was shown that the level of secreted mature GDF6 was reduced with p.Ala249Glu and p.Lys424Arg [14]. Screening of 50 patients with ocular anomalies recently identified GDF6 mutations in four (8%) patients[15]. Three of these patients carried the p.Ala249Glu variant.
In this study, we identified two different amino acid substitutions, p.Ala249Glu and p.Ala319Thr, in four (2%) of 200 patients with ocular malformations. Although both variants are conserved among several species, neither is conserved in zebrafish gdf6a. Therefore it was not possible to perform rescue experiments in the outm233mutant to determine the pathogenic potential of these variants. Interestingly, the p.Ala249Glu variant has been associated both with Klippel-Feil syndrome and with ocular anomalies [21, 14, 15]. The variant was detected on 8 of a total of 1478 chromosomes of patients with ocular malformations [13–15], and not on a total of 1074 control chromosomes [14, 21] (Fisher's exact test, p-value 0.01), strongly suggesting that it is pathogenic. Parents carrying this variant are asymptomatic, suggesting that GDF6 sequence alterations are likely to contribute to the phenotype, but are not the sole cause of the disease. This is confirmed by the fact that the affected sib of one of the patients in our study does not carry the p.Ala249Glu variant, indicating that other factors contribute to the disease. Besides microphthalmia, one patient in our study carrying the p.Ala249Glu variant has malformed ossicles. Interestingly, middle ear defects were also observed in Gdf6-/-mice, suggesting that the p.Ala249Glu variant could contribute to the ocular and middle ear defects in this patient. Finally, another microphthalmic patient in our study who carries the p.Ala249Glu variant has several skeletal anomalies, including cleft palate, hemivertebrae and talipes. Microphthalmia in combination with cleft lip and palate have recently been described in another patient carrying the p.Ala249Glu variant [15], suggesting that the extra-ocular defects might also be attributed to the p.Ala249Glu variant.
Conclusions
This study confirms the essential role of GDF6 in the regulation of vertebrate eye size. The reduced eye size in the zebrafish gdf6a/outm233mutant is likely to be caused by a transient wave of apoptosis at the onset of neurogenesis. Amino acid substitutions in GDF6 were detected in four patients with microphthalmia. In two patients different skeletal defects were observed, suggesting pleitrophic effects of p.Ala249Glu. Variable expressivity and penetrance suggest a complex non-Mendelian inheritance pattern where other genetic factors may influence the outcome of the phenotype.
Methods
Animals
The maintenance and breeding of zebrafish strains and staging of embryonic development were performed as described previously [49, 50]. All animal protocols were approved by the MEEI and Tufts University animal care committees. Embryos were observed using an Axioscope microscope (Zeiss, Thornwood, NY) or a Leica (Deerfield, IL) MZ12 dissecting microscope. Images were recorded with digital cameras, and processed using Adobe Photoshop software. The outm233mutant strain was originally recovered in a large-scale N-ethyl-N-nitrosurea (ENU) mutagenesis screen [25, 26]. To visualize ganglion cells, we used the Tg(pou4f3:gap43-GFP) trangenic line [37].
Histology
Zebrafish larvae were raised to the desired age and fixed in 4% paraformaldehyde (PFA) in PBST overnight at 4°C. Embedding, sectioning, and staining were performed as described previously [51, 52]. Sections were examined using an Axioscope Microscope (Zeiss) and images were recorded using an AxioCam digital camera (Zeiss).
Positional cloning
A map cross was set up between heterozygous G0 carriers of the outm233allele (AB genetic background) and wild-type WIK strain homozygotes. To determine the position of the out locus in the genome, we used a panel of 96 F2 diploid embryos obtained via incrossing of F1 animals.
Bulk segregant mapping analysis was performed by the Mutant Mapping Facility at the University of Louisville on DNA isolated from the mutant embryos http://www.biochemistry.louisville.edu/zfmapping/index.html. Positional candidate genes were selected based on the zebrafish genome sequence (Ensembl Database, Sanger Center UK, and UCSC Genome Browser, University of California at Santa Cruz). Search for mutations was performed by RT-PCR of RNA isolated from wild-type and outm233embryos, and by PCR on genomic DNA. PCR products were purified using the QIAquick PCR purification kit (Qiagen) and analyzed by direct sequencing. The effect of the mutation, which affects the translation start site, was evaluated with NetStart 1.0 [27].
Rescue and overexpression experiments
The wild-type gdf6a gene was previously cloned into the pNG6 vector under the control of a heat-shock promoter [24]. The p.Met1Val mutation was introduced by site-directed mutagenesis of the wild-type gdf6a construct with phusion DNA polymerase (Finnzymes, Espoo, Finland). The wild-type and mutant constructs were injected into zebrafish embryos at the one-cell stage using standard approaches [53]. Expression was activated via a 1 hour heat shock at the 12 somite stage. The eye phenotype was evaluated at 3 and 5 dpf.
Staining of nuclei
To count cells in retinal layers, wild-type and mutant embryos were fixed in 4% PFA at 36 and 96 hpf, and embedded in JB-4 resin (Polysciences), according to the manufacturer's instructions. Subsequently, 4-μm sections were collected on microscope slides, immersed in Hoechst 33258 solution (Molecular Probes; 1 mg/ml in PBS) for 10 min, and washed in PBS for 1 h. Alternatively, YoPro (Molecular Probes) was diluted 1:5,000 in PBST and used to stain frozen sections for 10 min. Sections were analyzed using UV illumination.
Analysis of cell death
Two methods were used to evaluate apoptotic cell death: acridine orange staining and TUNEL. To apply the first method, embryos were dechorionated, and placed in 5 mg/ml acridine orange (acridinium chloride hemi-zinc chloride; Sigma) in E3 medium [54] for 30 min. Embryos were then washed in E3 medium and viewed using UV illumination on an upright microscope. For TUNEL detection of cell death, zebrafish larvae were fixed in 4% PFA, cryoprotected in sucrose, and cryosectioned at 14 μm. Following two washes, 10 min each, in 50 mM PBS (pH 7.4), sections were treated with Proteinase K (Roche Applied Sciences, Indianapolis, IN) for 10 min at the concentration of 2 μg/ml in 50 mM Tris/HCl buffer, pH 8. Subsequently, sections were rinsed 3 times in 50 mM Tris/HCl buffer, incubated with 70% ethanol/30% acetic acid solution at -20°, washed 5 times, 5 min each, in 50 mM Tris/HCl buffer, and treated with blocking solution, containing (v/v in PBST) 10% normal goat serum, and 0.5% Triton X-100 (30 min, room temperature). Finally, sections were incubated with TUNEL reaction mixture (Roche) at 37°for several hours according to manufacturer's instructions. To detect apoptotic nuclei, sections were rinsed in 50 mM PBS for 10 min, coverslipped, and analyzed on a Leica SP2 confocal microscope equipped with a 40x lens. Digital images were processed with Adobe Photoshop Software and used to obtain counts of apoptotic cells.
Immunohistochemistry
Antibody staining was performed on frozen sections as described in previous publications [51, 52, 29]. The following primary antibodies and dilutions were used: rabbit anti-phospho-H3 histone (1:200, Upstate Biotechnology); mouse monoclonal anti-neurolin (1:25, Zebrafish International Resource Center); rabbit polyclonal anti-carbonic anhydrase (1:250, gift of Paul Linser, Whitney Laboratory, St. Augustine, FL); mouse monoclonal anti-parvalbumin (1:250, Chemicon); mouse monoclonal anti-HuC/HuD (1:20, Molecular Probes); mouse monoclonal Zpr1 (1:250, Zebrafish International Resource Center); rabbit polyclonal anti-serotonin (1:250, Sigma); rabbit polyclonal anti-tyrosine hydroxylase (1:250, Chemicon); rabbit polyclonal anti-GABA (1:500, Sigma); and rabbit polyclonal anti-PAX6 (1:200, Covance). The anti-serotonin and anti-Pax6 antibodies required citrate treatment prior to blocking in normal goat serum [29]. After staining, sections were imaged using Leica SP2 confocal microscope equipped with a 40x lens. Digital images were processed with Adobe Photoshop Software.
Patient samples and mutation analysis
Informed consent was obtained from all participants in this study in accordance with the local Ethic Boards in the respective institutions where they were examined. Blood samples were collected, and genomic DNA was isolated by standard protocols. The coding region and splice junctions of the two exons of GDF6 were screened in 200 patients with coloboma, microphthalmia or anophthalmia by automated sequencing [21]. PCR products were treated with ExoSAP-IT (USB) or purified with 96-wells NucleoFast purification plates (Machery-Nagel) before automated sequencing. All mutations and polymorphisms were confirmed by a second round of PCR amplification.
References
Lowry RB, Kohut R, Sibbald B, Rouleau J: Anophthalmia and microphthalmia in the Alberta Congenital Anomalies Surveillance System. Can J Ophthalmol. 2005, 40: 38-44.
Morrison D, FitzPatrick D, Hanson I, Williamson K, van H V, Fleck B, Jones I, Chalmers J, Campbell H: National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet. 2002, 39: 16-22. 10.1136/jmg.39.1.16.
Shaw GM, Carmichael SL, Yang W, Harris JA, Finnell RH, Lammer EJ: Epidemiologic characteristics of anophthalmia and bilateral microphthalmia among 2.5 million births in California, 1989-1997. Am J Med Genet A. 2005, 137: 36-40.
Gregory-Evans CY, Williams MJ, Halford S, Gregory-Evans K: Ocular coloboma: a reassessment in the age of molecular neuroscience. J Med Genet. 2004, 41: 881-891. 10.1136/jmg.2004.025494.
Verma AS, Fitzpatrick DR: Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007, 2: 47-10.1186/1750-1172-2-47.
Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van HV, Fitzpatrick DR: Mutations in SOX2 cause anophthalmia. Nat Genet. 2003, 33: 461-463. 10.1038/ng1120.
Ferda PE, Ploder LA, Yu JJ, Arici K, Horsford DJ, Rutherford A, Bapat B, Cox DW, Duncan AM, Kalnins VI, Kocak-Altintas A, Sowden JC, Traboulsi E, Sarfarazi M, McInnes RR: Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat Genet. 2000, 25: 397-401. 10.1038/78071.
Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL: PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet. 1994, 7: 463-471. 10.1038/ng0894-463.
Ragge NK, Brown AG, Poloschek CM, Lorenz B, Henderson RA, Clarke MP, Russell-Eggitt I, Fielder A, Gerrelli D, Martinez-Barbera JP, Ruddle P, Hurst J, Collin JR, Salt A, Cooper ST, Thompson PJ, Sisodiya SM, Williamson KA, Fitzpatrick DR, van H V, Hanson IM: Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet. 2005, 76: 1008-1022. 10.1086/430721.
Voronina VA, Kozhemyakina EA, O'Kernick CM, Kahn ND, Wenger SL, Linberg JV, Schneider AS, Mathers PH: Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum Mol Genet. 2004, 13: 315-322. 10.1093/hmg/ddh025.
Bakrania P, Efthymiou M, Klein JC, Salt A, Bunyan DJ, Wyatt A, Ponting CP, Martin A, Williams S, Lindley V, Gilmore J, Restori M, Robson AG, Neveu MM, Holder GE, Collin JR, Robinson DO, Farndon P, Johansen-Berg H, Gerrelli D, Ragge NK: Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am J Hum Genet. 2008, 82: 304-319. 10.1016/j.ajhg.2007.09.023.
Ye M, Berry-Wynne KM, sai-Coakwell M, Sundaresan P, Footz T, French CR, Abitbol M, Fleisch VC, Corbett N, Allison WT, Drummond G, Walter MA, Underhill TM, Waskiewicz AJ, Lehmann OJ: Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum Mol Genet. 2010, 19: 287-298. 10.1093/hmg/ddp496.
Asai-Coakwell M, French CR, Berry KM, Ye M, Koss R, Somerville M, Mueller R, van H V, Waskiewicz AJ, Lehmann OJ: GDF6, a novel locus for a spectrum of ocular developmental anomalies. Am J Hum Genet. 2007, 80: 306-315. 10.1086/511280.
Asai-Coakwell M, French CR, Ye M, Garcha K, Bigot K, Perera AG, Staehling-Hampton K, Mema SC, Chanda B, Mushegian A, Bamforth S, Doschak MR, Li G, Dobbs MB, Giampietro PF, Brooks BP, Vijayalakshmi P, Sauve Y, Abitbol M, Sundaresan P, van H V, Pourquie O, Underhill TM, Waskiewicz AJ, Lehmann OJ: Incomplete penetrance and phenotypic variability characterize Gdf6-attributable oculo-skeletal phenotypes. Hum Mol Genet. 2009, 18: 1110-1121. 10.1093/hmg/ddp008.
Gonzalez-Rodriguez J, Pelcastre EL, Tovilla-Canales JL, Garcia-Ortiz JE, mato-Almanza M, Villanueva-Mendoza C, Espinosa-Mattar Z, Zenteno JC: Mutational screening of CHX10, GDF6, OTX2, RAX and SOX2 genes in 50 unrelated microphthalmia-anophthalmia-coloboma (MAC) spectrum cases. Br J Ophthalmol. 2010, 94: 1100-1104. 10.1136/bjo.2009.173500.
Hogan BL: Bone morphogenetic proteins in development. Curr Opin Genet Dev. 1996, 6: 432-438. 10.1016/S0959-437X(96)80064-5.
Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, Hewick RM, Wang EA: Novel regulators of bone formation: molecular clones and activities. Science. 1988, 242: 1528-1534. 10.1126/science.3201241.
Gordon KJ, Blobe GC: Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008, 1782: 197-228.
Xiao YT, Xiang LX, Shao JZ: Bone morphogenetic protein. Biochem Biophys Res Commun. 2007, 362: 550-553. 10.1016/j.bbrc.2007.08.045.
Reddi AH: BMPs: from bone morphogenetic proteins to body morphogenetic proteins. Cytokine Growth Factor Rev. 2005, 16: 249-250. 10.1016/j.cytogfr.2005.04.003.
Tassabehji M, Fang ZM, Hilton EN, McGaughran J, Zhao Z, de Bock CE, Howard E, Malass M, Donnai D, Diwan A, Manson FD, Murrell D, Clarke RA: Mutations in GDF6 are associated with vertebral segmentation defects in Klippel-Feil syndrome. Hum Mutat. 2008, 29: 1017-1027. 10.1002/humu.20741.
Hanel ML, Hensey C: Eye and neural defects associated with loss of GDF6. BMC Dev Biol. 2006, 6: 43-10.1186/1471-213X-6-43.
Settle SH, Rountree RB, Sinha A, Thacker A, Higgins K, Kingsley DM: Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev Biol. 2003, 254: 116-130. 10.1016/S0012-1606(02)00022-2.
Gosse NJ, Baier H: An essential role for Radar (Gdf6a) in inducing dorsal fate in the zebrafish retina. Proc Natl Acad Sci USA. 2009, 106: 2236-2241. 10.1073/pnas.0803202106.
Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, Stainier DY, Zwartkruis F, Abdelilah S, Rangini Z, Belak J, Boggs C: A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996, 123: 37-46.
Malicki J, Neuhauss SC, Schier AF, Solnica-Krezel L, Stemple DL, Stainier DY, Abdelilah S, Zwartkruis F, Rangini Z, Driever W: Mutations affecting development of the zebrafish retina. Development. 1996, 123: 263-273.
Pedersen AG, Nielsen H: Neural network prediction of translation initiation sites in eukaryotes: perspectives for EST and genome analysis. Proc Int Conf Intell Syst Mol Biol. 1997, 5: 226-233.
Peterson RE, Fadool JM, McClintock J, Linser PJ: Muller cell differentiation in the zebrafish neural retina: evidence of distinct early and late stages in cell maturation. J Comp Neurol. 2001, 429: 530-540. 10.1002/1096-9861(20010122)429:4<530::AID-CNE2>3.0.CO;2-C.
Avanesov A, Dahm R, Sewell WF, Malicki JJ: Mutations that affect the survival of selected amacrine cell subpopulations define a new class of genetic defects in the vertebrate retina. Dev Biol. 2005, 285: 138-155. 10.1016/j.ydbio.2005.06.009.
Ekstrom P, Johansson K: Differentiation of ganglion cells and amacrine cells in the rat retina: correlation with expression of HuC/D and GAP-43 proteins. Brain Res Dev Brain Res. 2003, 145: 1-8. 10.1016/S0165-3806(03)00170-6.
Macdonald R, Wilson SW: Distribution of Pax6 protein during eye development suggests discrete roles in proliferative and differentiated visual cells. Dev Genes Evol. 1997, 206: 363-369. 10.1007/s004270050065.
Larison KD, Bremiller R: Early onset of phenotype and cell patterning in the embryonic zebrafish retina. Development. 1990, 109: 567-576.
Schmitt EA, Dowling JE: Comparison of topographical patterns of ganglion and photoreceptor cell differentiation in the retina of the zebrafish, Danio rerio. J Comp Neurol. 1996, 371: 222-234. 10.1002/(SICI)1096-9861(19960722)371:2<222::AID-CNE3>3.0.CO;2-4.
Hidalgo A, ffrench-Constant C: The control of cell number during central nervous system development in flies and mice. Mech Dev. 2003, 120: 1311-1325. 10.1016/j.mod.2003.06.004.
Leevers SJ, McNeill H: Controlling the size of organs and organisms. Curr Opin Cell Biol. 2005, 17: 604-609. 10.1016/j.ceb.2005.09.008.
Hu M, Easter SS: Retinal neurogenesis: the formation of the initial central patch of postmitotic cells. Dev Biol. 1999, 207: 309-321. 10.1006/dbio.1998.9031.
Xiao T, Roeser T, Staub W, Baier H: A GFP-based genetic screen reveals mutations that disrupt the architecture of the zebrafish retinotectal projection. Development. 2005, 132: 2955-2967. 10.1242/dev.01861.
Dahm R, Schonthaler HB, Soehn AS, van MJ, Vrensen GF: Development and adult morphology of the eye lens in the zebrafish. Exp Eye Res. 2007, 85: 74-89. 10.1016/j.exer.2007.02.015.
Lang RA: Apoptosis in mammalian eye development: lens morphogenesis, vascular regression and immune privilege. Cell Death Differ. 1997, 4: 12-20. 10.1038/sj.cdd.4400211.
Allende ML, Amsterdam A, Becker T, Kawakami K, Gaiano N, Hopkins N: Insertional mutagenesis in zebrafish identifies two novel genes, pescadillo and dead eye, essential for embryonic development. Genes Dev. 1996, 10: 3141-3155. 10.1101/gad.10.24.3141.
Fadool JM, Brockerhoff SE, Hyatt GA, Dowling JE: Mutations affecting eye morphology in the developing zebrafish (Danio rerio). Dev Genet. 1997, 20: 288-295. 10.1002/(SICI)1520-6408(1997)20:3<288::AID-DVG11>3.0.CO;2-4.
Crosier PS, Kalev-Zylinska ML, Hall CJ, Flores MV, Horsfield JA, Crosier KE: Pathways in blood and vessel development revealed through zebrafish genetics. Int J Dev Biol. 2002, 46: 493-502.
Hall CJ, Flores MV, Davidson AJ, Crosier KE, Crosier PS: Radar is required for the establishment of vascular integrity in the zebrafish. Dev Biol. 2002, 251: 105-117. 10.1006/dbio.2002.0794.
Ekker SC, Larson JD: Morphant technology in model developmental systems. Genesis. 2001, 30: 89-93. 10.1002/gene.1038.
Omori Y, Malicki J: oko meduzy and related crumbs genes are determinants of apical cell features in the vertebrate embryo. Curr Biol. 2006, 16: 945-957. 10.1016/j.cub.2006.03.058.
Tsujikawa M, Malicki J: Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron. 2004, 42: 703-716. 10.1016/S0896-6273(04)00268-5.
Williams LA, Bhargav D, Diwan AD: Unveiling the bmp13 enigma: redundant morphogen or crucial regulator?. Int J Biol Sci. 2008, 4: 318-329.
Delot E, Kataoka H, Goutel C, Yan YL, Postlethwait J, Wittbrodt J, Rosa FM: The BMP-related protein radar: a maintenance factor for dorsal neuroectoderm cells?. Mech Dev. 1999, 85: 15-25. 10.1016/S0925-4773(99)00026-X.
Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF: Stages of embryonic development of the zebrafish. Dev Dyn. 1995, 203: 253-310.
Malicki J, Jo H, Wei X, Hsiung M, Pujic Z: Analysis of gene function in the zebrafish retina. Methods. 2002, 28: 427-438. 10.1016/S1046-2023(02)00262-1.
Malicki J: Development of the retina. Methods Cell Biol. 1999, 59: 273-299. full_text.
Pujic Z, Malicki J: Mutation of the zebrafish glass onion locus causes early cell-nonautonomous loss of neuroepithelial integrity followed by severe neuronal patterning defects in the retina. Dev Biol. 2001, 234: 454-469. 10.1006/dbio.2001.0251.
Avanesov A, Malicki J: Approaches to study neurogenesis in the zebrafish retina. Methods Cell Biol. 2004, 76: 333-384. full_text.
Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DA, Odenthal J, van Eeden FJ, Jiang YJ, Heisenberg CP, Kelsh RN, Furutani-Seiki M, Vogelsang E, Beuchle D, Schach U, Fabian C, Nusslein-Volhard C: The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 1996, 123: 1-36.
Acknowledgements
The authors are grateful to Dr. Zac Pujic for help with the initial characterization of the out mutant phenotype, to Krysta Voesenek for assisting the cloning experiments, to Dr. Paul Linser for providing the anti-carbonic anhydrase antibody, and to Dr. Herwig Baier for providing the pNG6-gdf6a expression construct. This work was supported by NEI R21 award EY018427 to JM. AIdH was supported by the Netherlands Organisation for Scientific Research (916.56.160) and a Ter Meulen Fund Stipend of the Royal Dutch Academy of Arts and Sciences. EIT is supported by an unrestricted grant from Research to Prevent Blindness. The Anophthalmia Registry at Albert Einstein Medical Center is funded by the Albert B. Millett Memorial Fund, A Mellon Mid-Atlantic Charitable Trust, Rae S. Uber Trust, A Mellon Mid-Atlantic Charitable Trust and Gustavus and Louis Pfeiffer Research Foundation. NR is supported by an Academy of Medical Sciences/The Health Foundation Senior Surgical Scientist Fellowship and by grants from VICTA (Visually Impaired Children Taking Action) and the late Ernest Henri Polak Trust.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AIdH carried out the molecular genetic studies and drafted the manuscript. JB and PK carried out the histological and immunohistochemical studies. TB, EIT, NKR, and AS provided patient samples. JM conceived of the study, participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
den Hollander, A.I., Biyanwila, J., Kovach, P. et al. Genetic defects of GDF6 in the zebrafish out of sight mutant and in human eye developmental anomalies. BMC Genet 11, 102 (2010). https://doi.org/10.1186/1471-2156-11-102
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-11-102