
Abstract
Background
The use of molecular genetic technologies for broodstock management and selective breeding of aquaculture species is becoming increasingly more common with the continued development of genome tools and reagents. Several laboratories have produced genetic maps for rainbow trout to aid in the identification of loci affecting phenotypes of interest. These maps have resulted in the identification of many quantitative/qualitative trait loci affecting phenotypic variation in traits associated with albinism, disease resistance, temperature tolerance, sex determination, embryonic development rate, spawning date, condition factor and growth. Unfortunately, the elucidation of the precise allelic variation and/or genes underlying phenotypic diversity has yet to be achieved in this species having low marker densities and lacking a whole genome reference sequence. Experimental designs which integrate segregation analyses with linkage disequilibrium (LD) approaches facilitate the discovery of genes affecting important traits. To date the extent of LD has been characterized for humans and several agriculturally important livestock species but not for rainbow trout.
Results
We observed that the level of LD between syntenic loci decayed rapidly at distances greater than 2 cM which is similar to observations of LD in other agriculturally important species including cattle, sheep, pigs and chickens. However, in some cases significant LD was also observed up to 50 cM. Our estimate of effective population size based on genome wide estimates of LD for the NCCCWA broodstock population was 145, indicating that this population will respond well to high selection intensity. However, the range of effective population size based on individual chromosomes was 75.51 - 203.35, possibly indicating that suites of genes on each chromosome are disproportionately under selection pressures.
Conclusions
Our results indicate that large numbers of markers, more than are currently available for this species, will be required to enable the use of genome-wide integrated mapping approaches aimed at identifying genes of interest in rainbow trout.
Similar content being viewed by others

Background
The use of molecular genetic technologies for broodstock management and selective breeding of aquaculture species is becoming increasingly more common with the continued development of genome tools and reagents for species of interest [1]. Rainbow trout are the most widely produced salmonid in the US, attracting significant interest due to their economic impacts as an aquaculture species and on sport fisheries, and as a model research organism for studies related to carcinogenesis, toxicology, comparative immunology, disease ecology, physiology and nutrition [2]. To this end several international laboratories have produced genetic maps for this species to aid in the identification of loci affecting phenotypes of interest. These maps primarily include amplified fragment length polymorphisms (AFLPs) and microsatellites [3–9] and have resulted in the identification of many quantitative/qualitative trait loci (QTL) affecting phenotypic variation in traits associated with albinism, disease resistance, temperature tolerance, sex determination, embryonic development rate, spawning date, condition factor and growth [10–19]. In spite of these efforts, the elucidation of the precise allelic variations and/or genes underlying phenotypic diversity has yet to be achieved in this species having low marker densities and lacking a whole genome reference sequence.
Experimental designs which integrate complex segregation analyses with linkage disequilibrium (LD) approaches facilitate the discovery of genes affecting important traits [20–22]. To this end, the extent of LD has been characterized for humans and several agriculturally important livestock species including cattle, sheep, chickens, and pigs. Farnir et al. [20] genotyped 284 autosomal microsatellite markers on 581 dutch black-and-white dairy cattle to construct a whole genome LD map (excluding the sex chromosomes) spanning 2702 cM. Estimations of LD between syntenic loci using Lewontin's normalized D' [23] revealed large blocks of LD spanning tens of centiMorgans including values of 50% for markers <5 cM and decaying to 16% for distances of 50 cM. The value of D' between non-syntenic loci was estimated to be 12%. Vallejo et al. [24] selected distantly related animals to quantify the level of genetic diversity in United States Holstein cattle. While only 23 Holstein bulls were genotyped with 54 microsatellite loci that spanned most of the autosomal genome, extensive LD was detected in the United States Holstein population in agreement with the findings of Farnir et al. [20]. In 2002, McRae et al. [25] observed similar results by genotyping 90 microsatellites from 10 chromosomes on 276 progeny from Coopworth sheep, estimating D' values of 34.3% for marker distances <60 cM, 12.4% for syntenic markers >60 cM, and 12.4% for non-sytenic loci. In 2005 Heifetz et al. [26] reported that significant LD was only observed for loci < 5 cM apart in a commercial layer chicken population. In 2006 Harmegnies et al. [27] characterized LD in two commercial pig populations of 33 and 44 unrelated individuals by genotyping 29 and 5 microsatellite markers on two chromosomes, respectively. Estimates of r2 (squared correlation coefficient) revealed significant LD only for loci < 1 cM apart. More recently, Du et al. [28] and McKay et al. [29] evaluated LD in pigs and cattle, respectively, by genotyping syntenic single nucleotide polymorphisms (SNPs) and observing significant LD for marker pairs < 3 cM apart for pigs and 0.5 Mb for cattle. All of these results indicate that large numbers of markers from high-density maps are required to identify genes of interest using whole genome association studies in these species.
The USDA/ARS National Center for Cool and Cold Water Aquaculture (NCCCWA) has established a breeding program for rainbow trout, the use of molecular genetic technologies in this program is expected to enhance capabilities for selective breeding of important aquaculture production traits. To this end we have worked within international collaborations to develop genomic tools and technologies for rainbow trout [5, 8, 9, 30–46] while initiating and characterizing our broodstock population with respect to genetic and phenotypic variation relevant to aquaculture production [47–57]. Our approaches include genetic linkage and LD mapping for the identification of QTL affecting traits of economic importance which will permit the development of marker/gene assisted selection strategies [58, 59] and eventually genomic selection [60]. Characterizing the extent of LD in the NCCCWA rainbow trout broodstock population will support the use of integrated mapping approaches and facilitate the identification of genes affecting traits of interest by determining the marker densities required to conduct genome association scans. The NCCCWA broodstock population is closely related to commercial germplasm [49], therefore our findings also have the potential to impact industry selective breeding programs. In addition to LD we have evaluated effective population size (Ne) in an effort to characterize the true breeding size of our population. This estimate of Ne should be considered when making decisions concerning selection pressure. To this end, we characterized extent of LD by genotyping 96 unrelated individuals with 49 markers spanning four chromosomes.
Results
Genotypes were obtained for a total of 49 microsatellites on chromosomes OMY13 (n = 6), OMY14 (n = 20), OMY17 (n = 8), and OMYSex (n = 15). Genotyping success rate was 95.5% with 82 to 96 animals scored for each marker. The number of alleles per marker, marker heterozygosity, PIC, Allelic Diversity, and exact tests for departure from HWE proportions are reported in Table 1. The number of alleles per marker averaged 10.6 and ranged from 3 to 24. Marker heterozygosities ranged from 28.1% to 94.8% with an average of 69.8%. Polymorphism Information Content and Allelic Diversity ranged from 0.385 to 0.936 and 0.425 to 0.94, with averages of 0.745 and 0.773, respectively. A total of 19 loci had significant departure from HWE at P < 0.05, consisting of 3 markers from OMY13, 9 from OMY14, 2 from OMY17, and 5 from OMYSex.
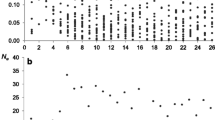
LD for syntenic loci was estimated for each individual chromosome and the four chromosomes combined (Figure 1), the genome-wide estimate for extent of significant LD (r2 > 0.25) was about 2 cM. The non-linear modeling of LD decline using the Sved (1971) equation indicates that the extent of significant LD decays sharply with physical distance. It also indicates that significant LD effects (i.e., r2 ≥ 0.25) are not expected beyond 5 cM.

Decline of linkage disequilibrium ( r2) with distance (recombination rate in Morgans) for chromosomes 13, 14, 17 and Sex. The estimates of r2 for pairs of markers were adjusted for experimental sample size 1/n, were n is the chromosome sample size (n = 192). The predicted LD value plot (filled non-linear curve) was estimated fitting the equation LD ij = 1/(1+kb j d ij )+e ij performing non-linear modeling with JMP® Genomics 3.1 (SAS Institute Inc., Carey, NC, 2007). Here, LD ij is the observed LD for marker pair i in chromosome j, d ij is the distance in Morgans for marker pair i in chromosome j, b j is the estimate of effective population size for chromosome j, and the constant k = 2 for sex chromosome and k = 4 for autosomes.
The LD for non-syntenic loci was estimated for each chromosome pairing and all pairings of non-syntenic loci, the proportion of pairings having (r2 > 0.25) is shown in Figure 2. Clearly, the proportion of significant LD (r2 ≥ 0.25) among non-syntenic loci was quite low

Proportion of markers pairs with significant extent of LD ( r2 ≥ 0.25). All marker pairs were evaluated in addition to pairwise combinations of all non-syntenic loci.
Effective population sizes were estimated for each chromosome and the four chromosomes as shown in Table 2. The estimated average effective population size was N e = 145 with an average N e /N ratio of 0.45.
Discussion
The extent of LD in the NCCCWA selective breeding program was evaluated by genotyping 49 microsatellites from four chromosomes on a total of 96 unrelated individuals from the 2005 (n = 43) and 2006 (n = 53) brood classes. As a result of an evolutionarily recent autopolyploid genome duplication event [61], many microsatellite markers for rainbow trout amplify two loci which can be scored independently and placed on genetic maps [4]. Although medium density genetic maps based on microsatellites including duplicated markers have been constructed [6, 9], they are problematic for use in population genetic analysis such as whole genome association studies as alleles from the two loci often have overlapping and/or identical allele sizes. Thus the number of loci available for use in whole genome association studies is much less than what is available for segregation analyses. Only evaluating single locus markers resulted in observation of a 6 cM region of OMY13 with 6 loci, a 108 cM region of OMY14 with 20 loci, a 55 cM region of OMY17 with 8 loci, and a 58 cM region of OMYSex with 15 loci. Overall, 227 cM of the 2927 cM genome was represented in this study. Of interest is the 55 cM region of OMY14 spanning the region from 75-130 cM where 6/7 loci depart from HWE. It is possible that these loci are under selection in the NCCCWA broodstock population which includes selective pressure for disease resistance and growth.
The level of LD between syntenic loci decayed rapidly at distances greater than 2 cM which is similar to observations of LD in other agriculturally important species including cattle [20], sheep [25], pigs [27, 28] and chickens [26]. However, significant LD was also observed up to 40 cM as reported by Farnir et al. [20] and McRae et al. [25] (Figure 1). The average proportion of significant LD (r2 > 0.25) between non-syntenic loci was under .005, however OMY13 and OMY17, which have a common ancestor chromosome resulting from the salmonid whole genome duplication event, showed a significantly higher proportion of significant LD.
We acknowledge that the LD range reported here may be up-biased because evidences for structure and admixture have been shown in the NCCCWA Broodstock population [62] and we used unrelated individuals from 2005 and 2006 brood years that are being improved for disease resistance and production traits, respectively, in the NCCCWA selective breeding program.
As the NCCCWA selective breeding program continues throughout successive generations, we expect that the effective population size will continue to decrease. Our estimate of Ne = 145 (Table 2) based on genome wide LD indicates that this population will respond well to high selection intensity as it has for a single generation of breeding for resistance to the causative agent of bacterial cold-water disease [50]. Using this estimate we can observe the effects of selection and identify correlations with the effects of inbreeding, usually first observed in reproductive success. However, the range of effective population size based on individual chromosomes was 75.51 - 203.35, possibly indicating that suites of genes on each chromosome are disproportionately under selection pressure. This also establishes the need to develop whole genome applications for these species for studies of LD. Although F-statistics have been used to estimate population genetic parameters [49], characterization of LD will also provide valuable information about population substructure and should be a factor in developing strategies aimed at the identification of associations between markers and QTL.
We chose to evaluate the extent of LD by genotyping representatives of each year class a single generation after the population was founded by mixing germplasm from multiple sources. Our resulting characterization of LD not only provides information about population structure, but serves to document the degree of genetic diversity used to initiate the broodstock program. We expect our estimates of Ne to be higher than populations with limited genetic diversity and no introduction of new germplasm in recent past generations.
The estimated N e /N ratio of 0.45 in this study is between the ranges of estimates reported in this species in previous studies [63–65]. Other studies in salmonids consistently reported that the variance in reproductive success is the key factor to reduce N e /N in salmon populations [64, 66].
Characterizing the extent and distribution of LD helps to determine the required marker density for LD mapping and genomic selection as they both require markers to be in LD with QTL. Our observation of significant syntenic LD at distances over 2 cM has implications for designing genome wide association studies in this population and on this species. The sex averaged map is roughly 3000 cM long; therefore 1500 markers are required to identify loci of interest. The male map having 2500 cM would require 1125 markers; the female map having 4300 cM would require 2150 markers. Currently about 1800 microsatellite markers are available for genome analyses in trout. However, we must take into consideration that: 1) roughly 33% are duplicated; 2) on average 70% are informative based on estimates of heterozygosity; and 3) these markers are not necessarily spaced at regular intervals throughout the genome.
Conclusions
To effectively conduct whole genome association studies the number of available markers for rainbow trout must be increased. Whereas genotyping microsatellites can be very expensive and time consuming process, it is likely that a marker system that enables high-throughput genotyping protocols such as single nucleotide polymorphisms will be the basis for LD mapping and genomic selection studies in the near future.
Methods
Germplasm
The NCCCWA rainbow trout selective breeding program was initiated in 2002 through 2004 with the acquisition of fish from Troutlodge, Inc. (Sumner, WA), the Donaldson strain from the University of Washington (Seattle, WA), the House Creek strain from the College of Southern Idaho (Twin Falls, ID), and the Shasta strain from the Ennis National Fish Hatchery, (Ennis, MT) [49]. To date, selective breeding for increased aquaculture production efficiency has been conducted by evaluating and selecting for growth traits on even years and resistance to challenge by the bacterial pathogen Flavobacterium psychrophilum on odd years [47, 50]. To identify individuals that could best represent the genetic variation contained within this broodstock population, we identified 96 unrelated individuals (no siblings or half-siblings) from the 2005 and 2006 brood year classes (n = 43 and 53, respectively) to represent the 144 and 177 fish that were actually used to produce select matings. The 2005 year class, part of our odd year selection for disease resistance, is the first generation after the 2003 founder population. The 2006 year class, part of our even year selection for growth, is the second generation after the founder population in 2002. However, a significant contribution of new germplasm was also introduced in 2004. Fin clips from anesthetized fish were collected for DNA extraction as outlined in a protocol approved by the NCCCWA IACUC, number 025-1-26-05. The extraction protocols followed the phenol-chloroform method described in Sambrook and Russell [67]. DNA samples were quantified by spectrophotometer (Beckman DU 640, Beckman Instruments, St. Louis, MO, USA) and diluted to a concentration of 12.5 ng/ul for PCR.
Genotyping
Microsatellite markers from four chromosomes (n = 49) were selected from the NCCCWA genetic map [9] for genotyping the DNA panel of 96 fish representing NCCCWA broodstock (Table 1). This included markers mapped to the sex chromosome (OMYSex), chromosome 14 having the largest linkage group in cM (OMY14), and paralogous regions of chromosomes 13 and 17 (OMY13, OMY17) as defined by duplicated loci resulting from an evolutionarily recent genome duplication event [4, 9, 61]. Only single locus markers were utilized. Markers were either genotyped using the tailed protocol [68] or by direct fluorescent labeling (with FAM, HEX, or NED) of the forward primer according to manufacturer protocols [[69] USA]. Primer pairs were obtained from commercial sources (forward primers labeled with FAM or HEX from Alpha DNA, Montreal, Quebec, Canada, or NED from ABI, Foster City, CA, USA). PCR reactions consisted of 12 μ l reaction volumes containing 12.5 ng DNA, 1.5-2.5 mM MgCl2, 1.0 μ M of each primer, 200 μ M of dNTPs, 1× manufacturer's reaction buffer and 0.5 units Taq DNA polymerase. Thermal cycling consisted of an initial denaturation at 95°C for 15 min followed by 30 cycles of 95°C for 1 min, annealing temperature for 45 s, 72°C extension for 45 s and a final extension at 72°C for 10 min. PCR products were visualized on agarose gels after staining with ethidium bromide. Markers were grouped in combinations of two or three markers based on differences in fluorescent dye color and amplicon size. Three μ l of each PCR product was added to 20 μ l of water, 1 μ l of the diluted sample was added to 12.5 μ l of loading mixture made up with 12 μ l of HiDi formamide and 0.5 of Genscan 400 ROX internal size standard. Samples were denatured at 95°C for 5 min and kept on ice until loading on an automated DNA sequencer ABI 3730 DNA Analyzer (ABI, Foster City, CA, USA). Output files were analyzed using GeneMapper version 3.7 (ABI, Foster City, CA, USA), formatted using Microsoft Excel and stored in Microsoft Access database.
Analysis
The initial dataset for analysis included marker genotype data for 49 microsatellite loci typed on 96 unrelated individuals. These marker genotype data were analyzed to estimate frequency of alleles per marker using the ALLELE procedure of software package SAS®, version 9.3.1 [70]. Within each marker, alleles with frequency ≤ 0.01 were merged to minimize the up biasing effect of rare alleles on LD estimates. Then, the marker alleles initially recorded in size of fragments were recoded into a consecutive-numbered allele system using the computer program RECODE [71]. This dataset with recoded marker genotypes were divided into files including marker loci corresponding to each of the four linkage groups analyzed in this study. Subsequently, these recoded marker genotype data were used in haplotype reconstruction and linkage disequilibrium analysis.
Haplotype reconstruction
For each linkage group, the most likely haplotype configuration for each individual was estimated using the software package PHASE, version 2.1, following procedures described by [72] and [73]. Briefly, this is a statistical method for inferring haplotypes from unphased genotype data for a sample of "unrelated" individuals from a population; population haplotype frequencies are assumed under Hardy-Weinberg equilibrium (HWE) but PHASE has proven robust to deviations from HWE, the effect of population structure and moderate amounts of recombination. The estimated haplotype data was formatted for subsequent analysis with the ALLELE procedure of the software package SAS®, version 9.3.1 [70]. The haplotype data was formatted into SAS ALLELE procedure format (option TALL for data input format) using a Java script (Haplo_2SAS.jar) written by Christopher Schmitt. This script is available for academic use upon request to RLV. This formatted dataset was used in linkage disequilibrium analysis.
Hardy-Weinberg equilibrium
The HWE analysis for each of the 49 loci typed in 96 unrelated individuals was performed using the ALLELE procedure of the software package SAS®, version 9.3.1 [70] using the option TALL for data input format. The input data were the most likely haplotypes reconstructed for each individual using the computer program PHASE and reformatted into SAS format as outlined above. The exact test for HWE for each microsatellite loci (exact P- value) was estimated using 10,000 permutations.
Linkage disequilibrium between syntenic loci
The LD analysis was performed between marker pairs within each linkage group using the ALLELE procedure of software package SAS®, version 9.3.1 [70]. We used the option TALL for data input format, and the HAPLO = GIVEN option which indicates that the haplotypes have been observed, and thus the observed haplotype frequencies were used in the LD test statistic and measures.
The linkage (or gametic) disequilibrium coefficient D uv between two alleles from marker loci M and N, respectively, was estimated using this expression [74],

Where p uv is probability that an individual receives the haplotype M u N v for marker loci M and N, p u is the probability of the u allele, and p v is the probability of the v allele.
The ALLELE procedure calculates the maximum likelihood estimates,  , of the LD coefficient between a pair of alleles at different markers. This procedure calculates an overall chi-square statistic to test that all of the D
uv
's between two markers are zero as follows,
, of the LD coefficient between a pair of alleles at different markers. This procedure calculates an overall chi-square statistic to test that all of the D
uv
's between two markers are zero as follows,

Which has (k-1) and (l-1) degrees of freedom for markers with k and l alleles, respectively. A Monte Carlo estimate of the exact P-value for testing the hypothesis was calculated by conditioning on the haplotype counts. The significance level is obtained by permuting the alleles at one locus to form 2 n new two-locus haplotypes. We used 10,000 permutations to estimate the exact P- values.
We used the squared correlation coefficient (r2) as the LD measure for each pair of alleles M u and N v located at loci M and N, respectively [75],

Where D = p11p12- p12p21 is the LD coefficient, which can be directly estimated using the observed haplotype frequencies  when using the option HAPLO = GIVEN with the ALLELE procedure of SAS®, version 9.3.1 [70]. Since these measures are designed for biallelic markers, the measures are calculated for each allele at locus M with each allele at locus N, where all other alleles at each locus are combined to represent one allele. Thus for each allele M
u
in turn,
when using the option HAPLO = GIVEN with the ALLELE procedure of SAS®, version 9.3.1 [70]. Since these measures are designed for biallelic markers, the measures are calculated for each allele at locus M with each allele at locus N, where all other alleles at each locus are combined to represent one allele. Thus for each allele M
u
in turn,  is used as the frequency of allele M
u
, and
is used as the frequency of allele M
u
, and  represents the frequency of "not M
u
"; similarly for each N
v
in turn,
represents the frequency of "not M
u
"; similarly for each N
v
in turn,  represents the frequency of allele N
v
, and
represents the frequency of allele N
v
, and  the frequency of "not N
v
."
the frequency of "not N
v
."
Linkage disequilibrium decay with distance
The decline of linkage disequilibrium with distance (recombination rate in Morgans) was estimated by fitting the following equation [76],

Where  is the observed LD for marker pair i in chromosome j, the constant k = 2 for sex chromosome and k = 4 for autosomes, d
ij
is the recombination rate from two-point linkage analysis for marker pair i in chromosome j, b
j
is the estimate of effective population size for chromosome j, and e
ij
is a random residual. The estimates of r2 for pairs of markers were adjusted for experimental sample size
is the observed LD for marker pair i in chromosome j, the constant k = 2 for sex chromosome and k = 4 for autosomes, d
ij
is the recombination rate from two-point linkage analysis for marker pair i in chromosome j, b
j
is the estimate of effective population size for chromosome j, and e
ij
is a random residual. The estimates of r2 for pairs of markers were adjusted for experimental sample size  , where n is the chromosome sample size (n = 192). We performed the non-linear modeling with JMP® Genomics 3.1 (SAS Institute Inc., Carey, NC, 2007).
, where n is the chromosome sample size (n = 192). We performed the non-linear modeling with JMP® Genomics 3.1 (SAS Institute Inc., Carey, NC, 2007).
Linkage disequilibrium between nonsyntenic loci
We estimated LD adjusted for experimental sample size (r2 - ) between pairs of non-syntenic loci using the ALLELE procedure of software package SAS®, version 9.3.1 [70]. As input data, we used marker genotype data with alleles recoded into a consecutive-numered system (non-phased marker genotype data). In the analysis, we used the option HAPLO = EST which indicates that the maximum likelihood estimates of the haplotype frequencies are used to calculate the LD test statistic as well as the LD measures. For LD estimation among non-syntenic loci, we used the ALLELE procedure options ALLELEMIN = GENOMIN = HAPLOMIN = 0.01. This last statement ensures that only alleles, genotypes, and haplotypes with frequency ≥ 0.01 are used in the LD analysis.
Effective population size
The analysis was based on the known relationship between LD as measured by r2 (squared correlation of allele frequencies at a pair of loci) and effective population size N e ,

Where c is the recombination rate between the microsatellite loci and n is the experimental sample size. The constant α = 1 in the absence of mutation [76] and α = 2 if mutation is taken into account [77–79]. The constant k was set to k = 2 for sex chromosome and k = 4 for autosomes.
Given the formulae described in the linkage disequilibrium section, and knowing r2 and c, we estimated N e for each chromosome by fitting this nonlinear regression model,

Where is the observed LD (adjusted for chromosome sample size n) for marker pair i in chromosome j, c
ij
is the recombination rate from two-point linkage analysis for marker pair i in chromosome j. The parameter β
j
is the estimator of effective population size for chromosome j where  . The parameters α
j
and β
j
were estimated iteratively using non-linear modeling with JMP® Genomics 3.1 (SAS Institute Inc., Carey, NC, 2007).
. The parameters α
j
and β
j
were estimated iteratively using non-linear modeling with JMP® Genomics 3.1 (SAS Institute Inc., Carey, NC, 2007).
References
Liu ZJ, Ed: Aquaculture Genome Technologies. 2007, Ames, Iowa: Blackwell Publishing
Thorgaard GH, Bailey GS, Williams D, Buhler DR, Kaattari SL, Ristow SS, Hansen JD, Winton JR, Bartholomew JL, Nagler JJ: Status and opportunities for genomics research with rainbow trout. Comp Biochem Physiol B Biochem Mol Biol. 2002, 133: 609-646. 10.1016/S1096-4959(02)00167-7.
Young WP, Wheeler PA, Coryell VH, Keim P, Thorgaard GH: A detailed linkage map of rainbow trout produced using doubled haploids. Genetics. 1998, 148: 839-850.
Sakamoto T, Danzmann RG, Gharbi K, Howard P, Ozaki A, Khoo SK, Woram RA, Okamoto N, Ferguson MM, Holm LE: A microsatellite linkage map of rainbow trout (Oncorhynchus mykiss) characterized by large sex-specific differences in recombination rates. Genetics. 2000, 155: 1331-1345.
Nichols KM, Young WP, Danzmann RG, Robison BD, Rexroad CE, Noakes M, Phillips RB, Bentzen P, Spies I, Knudsen K: A consolidated linkage map for rainbow trout (Oncorhynchus mykiss). Anim Genet. 2003, 34: 102-115. 10.1046/j.1365-2052.2003.00957.x.
Guyomard R, Mauger S, Tabet-Canale K, Martineau S, Genet C, Krieg F, Quillet E: A type I and type II microsatellite linkage map of rainbow trout (Oncorhynchus mykiss) with presumptive coverage of all chromosome arms. BMC Genomics. 2006, 7: 302-10.1186/1471-2164-7-302.
Phillips RB, Nichols KM, DeKoning JJ, Morasch MR, Keatley KA, Rexroad C, Gahr SA, Danzmann RG, Drew RE, Thorgaard GH: Assignment of rainbow trout linkage groups to specific chromosomes. Genetics. 2006, 174: 1661-1670. 10.1534/genetics.105.055269.
Danzmann RG, Cairney M, Davidson WS, Ferguson MM, Gharbi K, Guyomard R, Holm LE, Leder E, Okamoto N, Ozaki A: A comparative analysis of the rainbow trout genome with 2 other species of fish (Arctic charr and Atlantic salmon) within the tetraploid derivative Salmonidae family (subfamily: Salmoninae). Genome. 2005, 48: 1037-1051. 10.1139/g05-067.
Rexroad CE, Palti Y, Gahr SA, Vallejo RL: A second generation genetic map for rainbow trout (Oncorhynchus mykiss). BMC Genet. 2008, 9: 74-10.1186/1471-2156-9-74.
Leder EH, Danzmann RG, Ferguson MM: The candidate gene, Clock, localizes to a strong spawning time quantitative trait locus region in rainbow trout. J Hered. 2006, 97: 74-80. 10.1093/jhered/esj004.
Martinez V, Thorgaard G, Robison B, Sillanpaa MJ: An application of Bayesian QTL mapping to early development in double haploid lines of rainbow trout including environmental effects. Genet Res. 2005, 86: 209-221. 10.1017/S0016672305007871.
Nichols KM, Broman KW, Sundin K, Young JM, Wheeler PA, Thorgaard GH: Quantitative trait loci x maternal cytoplasmic environment interaction for development rate in Oncorhynchus mykiss. Genetics. 2007, 175: 335-347. 10.1534/genetics.106.064311.
O'Malley KG, Sakamoto T, Danzmann RG, Ferguson MM: Quantitative trait loci for spawning date and body weight in rainbow trout: testing for conserved effects across ancestrally duplicated chromosomes. J Hered. 2003, 94: 273-284. 10.1093/jhered/esg067.
Perry GM, Danzmann RG, Ferguson MM, Gibson JP: Quantitative trait loci for upper thermal tolerance in outbred strains of rainbow trout (Oncorhynchus mykiss). Heredity. 2001, 86: 333-341. 10.1046/j.1365-2540.2001.00838.x.
Perry GM, Ferguson MM, Sakamoto T, Danzmann RG: Sex-linked quantitative trait loci for thermotolerance and length in the rainbow trout. J Hered. 2005, 96: 97-107. 10.1093/jhered/esi019.
Robison BD, Wheeler PA, Sundin K, Sikka P, Thorgaard GH: Composite interval mapping reveals a major locus influencing embryonic development rate in rainbow trout (Oncorhynchus mykiss). J Hered. 2001, 92: 16-22. 10.1093/jhered/92.1.16.
Zimmerman AM, Evenhuis JP, Thorgaard GH, Ristow SS: A single major chromosomal region controls natural killer cell-like activity in rainbow trout. Immunogenetics. 2004, 55: 825-835. 10.1007/s00251-004-0645-6.
Ozaki A, Sakamoto T, Khoo S, Nakamura K, Coimbra MR, Akutsu T, Okamoto N: Quantitative trait loci (QTLs) associated with resistance/susceptibility to infectious pancreatic necrosis virus (IPNV) in rainbow trout (Oncorhynchus mykiss). Mol Genet Genomics. 2001, 265: 23-31. 10.1007/s004380000392.
Nakamura K, Ozaki A, Akutsu T, Iwai K, Sakamoto T, Yoshizaki G, Okamoto N: Genetic mapping of the dominant albino locus in rainbow trout (Oncorhynchus mykiss). Mol Genet Genomics. 2001, 265: 687-693. 10.1007/s004380100464.
Farnir F, Coppieters W, Arranz JJ, Berzi P, Cambisano N, Grisart B, Karim L, Marcq F, Moreau L, Mni M: Extensive genome-wide linkage disequilibrium in cattle. Genome Res. 2000, 10: 220-227. 10.1101/gr.10.2.220.
Kruglyak L: Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet. 1999, 22: 139-144. 10.1038/9642.
Meuwissen TH, Karlsen A, Lien S, Olsaker I, Goddard ME: Fine mapping of a quantitative trait locus for twinning rate using combined linkage and linkage disequilibrium mapping. Genetics. 2002, 161: 373-379.
Lewontin RC: The detection of linkage disequilibrium in molecular sequence data. Genetics. 1995, 140: 377-388.
Vallejo RL, Li YL, Rogers GW, Ashwell MS: Genetic diversity and background linkage disequilibrium in the North American Holstein cattle population. J Dairy Sci. 2003, 86: 4137-4147.
McRae AF, McEwan JC, Dodds KG, Wilson T, Crawford AM, Slate J: Linkage disequilibrium in domestic sheep. Genetics. 2002, 160: 1113-1122.
Heifetz EM, Fulton JE, O'Sullivan N, Zhao H, Dekkers JC, Soller M: Extent and consistency across generations of linkage disequilibrium in commercial layer chicken breeding populations. Genetics. 2005, 171: 1173-1181. 10.1534/genetics.105.040782.
Harmegnies N, Farnir F, Davin F, Buys N, Georges M, Coppieters W: Measuring the extent of linkage disequilibrium in commercial pig populations. Anim Genet. 2006, 37: 225-231. 10.1111/j.1365-2052.2006.01438.x.
Du FX, Clutter AC, Lohuis MM: Characterizing linkage disequilibrium in pig populations. Int J Biol Sci. 2007, 3: 166-178.
McKay SD, Schnabel RD, Murdoch BM, Matukumalli LK, Aerts J, Coppieters W, Crews D, Dias Neto E, Gill CA, Gao C: Whole genome linkage disequilibrium maps in cattle. BMC Genet. 2007, 8: 74-10.1186/1471-2156-8-74.
Coulibaly I, Gharbi K, Danzmann RG, Yao J, Rexroad CE: Characterization and comparison of microsatellites derived from repeat-enriched libraries and expressed sequence tags. Anim Genet. 2005, 36: 309-315. 10.1111/j.1365-2052.2005.01305.x.
Gahr SA, Rexroad CE, Rise ML, Hunt P, Koop B: A survey of expressed sequence tags from the rainbow trout (Oncorhynchus mykiss) pituitary. Anim Biotechnol. 2007, 18: 213-230. 10.1080/10495390701337335.
Koop BF, Schalburg KRv, Leong J, Walker N, Lieph R, Cooper GA, Robb A, Beetz-Sargent M, Holt RA, Moore R: A salmonid EST genomic study: genes, duplications, phylogeny and microarrays. BMC Genomics. 2008, 9: 545-10.1186/1471-2164-9-545.
Palti Y, Danzmann RG, Rexroad CE: Characterization and mapping of 19 polymorphic microsatellite markers for rainbow trout (Oncorhynchus mykiss). Anim Genet. 2003, 34: 153-156. 10.1046/j.1365-2052.2003.00965_7.x.
Palti Y, Fincham MR, Rexroad CE: Characterization of 38 polymorphic microsatellite markers for rainbow trout (Oncorhynchus mykiss). Molecular Ecology Notes. 2002, 2: 449-452. 10.1046/j.1471-8286.2002.00274.x.
Palti Y, Gahr SA, Hansen JD, Rexroad CE: Characterization of a new BAC library for rainbow trout: evidence for multi-locus duplication. Anim Genet. 2004, 35: 130-133. 10.1111/j.1365-2052.2004.01112.x.
Ramachandra RK, Salem M, Gahr S, Rexroad CE, Yao J: Cloning and characterization of microRNAs present during early embryonic development in rainbow trout (Oncorhynchus mykiss). BMC Developmental Biology. 2008, 8: 41-10.1186/1471-213X-8-41.
Rexroad CE, Coleman RL, Gustafson AL, Hershberger WK, Killefer J: Development of rainbow trout microsatellite markers from repeat enriched libraries. Mar Biotechnol (NY). 2002, 4: 12-16. 10.1007/s10126-001-0058-6.
Rexroad CE, Coleman RL, Hershberger WK, Killefer J: Rapid communication: Thirty-eight polymorphic microsatellite markers for mapping in rainbow trout. J Anim Sci. 2002, 80: 541-542.
Rexroad CE, Coleman RL, Hershberger WK, Killefer J: Eighteen polymorphic microsatellite markers for rainbow trout (Oncorhynchus mykiss). Anim Genet. 2002, 33: 76-78. 10.1046/j.1365-2052.2002.0742d.x.
Rexroad CE, Coleman RL, Martin AM, Hershberger WK, Killefer J: Thirty-five polymorphic microsatellite markers for rainbow trout (Oncorhynchus mykiss). Anim Genet. 2001, 32: 317-319. 10.1046/j.1365-2052.2001.0730b.x.
Rexroad CE, Lee Y, Keele JW, Karamycheva S, Brown G, Koop B, Gahr SA, Palti Y, Quackenbush J: Sequence analysis of a rainbow trout cDNA library and creation of a gene index. Cytogenet Genome Res. 2003, 102: 347-354. 10.1159/000075773.
Rexroad CE, Palti Y: Development of Ninety-Seven Polymorphic Microsatellite Markers for rainbow trout (Oncorhynchus mykiss). Transactions of the American Fisheries Society. 2003, 132: 1214-1221. 10.1577/T02-086.
Rexroad CE, Rodriguez MF, Coulibaly I, Gharbi K, Danzmann RG, Dekoning J, Phillips R, Palti Y: Comparative mapping of expressed sequence tags containing microsatellites in rainbow trout (Oncorhynchus mykiss). BMC Genomics. 2005, 6: 54-10.1186/1471-2164-6-54.
Rodriguez MF, Gahr SA, Rexroad CE, Palti Y: Rapid microsatellite detection from rainbow trout (Oncorhynchus mykiss) bacterial artificial chromosome using PCR screening method. Marine Biotechnology. 2006, 8: 346-350. 10.1007/s10126-005-5064-7.
Salem M, Kenney B, Rexroad CE, Yao J: Development of a 37K High-density oligo-nucleotide microarray for rainbow trout. Journal of Fish Biology. 2007, 72: 2187-2206. 10.1111/j.1095-8649.2008.01860.x.
Phillips RB, Nichols KM, DeKoning JJ, Morash MR, Keatley KA, Rexroad CE, Danzmann RG, Drew RE, Thorgaard GH: Assignment of Rainbow Trout Linkage Groups to Specific Chromosomes. Genetics. 2006, 174: 1661-1670. 10.1534/genetics.105.055269.
Lankford S, Weber G: Associations between plasma growth hormone, insulin-like growth factor-1 and cortisol, with stress responsiveness and growth performance in a selective breeding program for rainbow trout (Oncorhychus mykiss). North American Journal of Aquaculture. 2006, 68: 151-159. 10.1577/A05-014.1.
Silverstein J: Using genetic variation to understand control of feed intake in fish. Fish Physiology and Biochemistry. 2004, 27: 173-178. 10.1023/B:FISH.0000032724.36866.ce.
Silverstein JT, King T, Rexroad CE: Genetic Variation Measured by Microsatellites Among Three Strains of Domesticated Rainbow Trout. Aquaculture Research. 2004, 35: 40-48. 10.1111/j.1365-2109.2004.00979.x.
Silverstein JT, Vallejo R, Palti Y, Leeds T, Rexroad CE, Welch T, Wiens G, Ducrocq V: Rainbow trout resistance to bacterial cold-water disease is moderately heritable and is not adversely correlated with growth. Journal of Animal Science. 2009, 87: 860-867. 10.2527/jas.2008-1157.
Vallejo RL, Rexroad CE, Silverstein JT, Janss LL, Weber GM: Evidence of major genes affecting stress response in rainbow trout using Bayesian methods of complex segregation analysis. J Anim Sci. 2009, 87: 3490-3505. 10.2527/jas.2008-1616.
Silverstein J, Hostuttler M, Blemings K: Strain differences in feed efficiency measured as residual feed intake in rainbow trout. Aquaculture Research. Aquaculture Research. 2005, 36: 704-711. 10.1111/j.1365-2109.2005.01278.x.
Hershberger W, Hostuttler M: Variation in Time to First Cleavage in Rainbow Trout Oncorhynchus mykiss Embryos: A Major Factor in Induction of Tetraploids. Journal of the World Aquaculture Society. 2005, 36: 96-102.
Silverstein J: Relationships among feed intake, feed efficiency and growth in juvenile rainbow trout (Oncorhynchus mykiss). North American Journal of Aquaculture. 2006, 68: 168-175. 10.1577/A05-010.1.
Weber G, Silverstein J: Evaluation of a stress response for use in a selective breeding program for improved growth and disease resistance in rainbow rout (oncorhynchus mykiss). North American Journal of Aquaculture. 2006, 69: 69-79. 10.1577/A05-103.1.
Weber G, Vallejo R, Lankford S, Silverstein J, Welch T: Cortisol Response to a Crowding Stress: Heritability and Association with Disease Resistance to Yersinia ruckeri in Rainbow Trout. North American Journal of Aquaculture. 2008, 70: 425-433. 10.1577/A07-059.1.
Hadidi S, Glenney G, Welch T, Silverstein J, Wiens G: Spleen Size Predicts Resistance of Rainbow Trout to Flavobacterium psychrophilum Challenge. Journal of Immunology. 2008, 180: 4156-4165.
Dekkers JC: Commercial application of marker- and gene-assisted selection in livestock: strategies and lessons. J Anim Sci. 2004, 82 (E-Suppl): E313-328.
Soller M, Beckman J: Genetic polymorphism in varietal identification and genetic improvement. Theoretical and Applied Genetics. 1983, 67: 25-33. 10.1007/BF00303917.
Meuwissen TH, Hayes BJ, Goddard ME: Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001, 157: 1819-1829.
Allendorf FW, Thorgaard GH: Tetraploidy and the evolution of salmonid fishes. Evolutionary Genetics of Fishes. Edited by: Turner BJ. 1984, New York: Plenum Press, 1-46.
Johnson NA, Rexroad CE, Hallerman EM, Vallejo RL, Palti Y: Development and evaluation of a new microsatellite multiplex system for parental allocation and management of rainbow trout (Oncorhynchus mykiss) broodstocks. Aquaculture. 2007, 266: 53-62. 10.1016/j.aquaculture.2007.02.054.
Araki H, Waples RS, Ardren WR, Cooper B, Blouin MS: Effective population size of steelhead trout: influence of variance in reproductive success, hatchery programs, and genetic compensation between life-history forms. Mol Ecol. 2007, 16: 953-966. 10.1111/j.1365-294X.2006.03206.x.
Ardren WR, Kapuscinski AR: Demographic and genetic estimates of effective population size (Ne) reveals genetic compensation in steelhead trout. Mol Ecol. 2003, 12: 35-49. 10.1046/j.1365-294X.2003.01705.x.
Heath DD, Busch C, Kelly J, Atagi DY: Temporal change in genetic structure and effective population size in steelhead trout (Oncorhynchus mykiss). Mol Ecol. 2002, 11: 197-214. 10.1046/j.1365-294X.2002.01434.x.
Hedrick P: Large variance in reproductive success and the Ne/N ratio. Evolution. 2005, 59: 1596-1599.
Sambrook J, Russell D: Molecular cloning: a laboratory manual. 2001, Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press, 3
Boutin-Ganache I, Raposo M, Raymond M, Deschepper CF: M13-tailed primers improve the readability and usability of microsatellite analyses performed with two different allele-sizing methods. Biotechniques. 2001, 31: 24-26.
De La Vega FM, Dailey D, Ziegle J, Williams J, Madden D, Gilbert DA: New generation pharmacogenomic tools: a SNP linkage disequilibrium Map, validated SNP assay resource, and high-throughput instrumentation system for large-scale genetic studies. Biotechniques. 2002, 48-50. 52, 54, Suppl
SAS: SAS 9.1.3 Help and Documentation. 2007, Cary, NC: SAS Institute Inc
RECODE. [http://watson.hgen.pitt.edu/register/docs/recode.html]
Stephens M, Smith NJ, Donnelly P: A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001, 68: 978-989. 10.1086/319501.
Stephens M, Donnelly P: A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003, 73: 1162-1169. 10.1086/379378.
Weir BS: Genetic Data Analysis II: Methods for Discrete Population Genetic Data. 1996, Sunderland, MA: Sinauer Associates Inc. Publishers
Devlin B, Risch N: A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics. 1995, 29: 311-322. 10.1006/geno.1995.9003.
Sved JA: Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor Popul Biol. 1971, 2: 125-141. 10.1016/0040-5809(71)90011-6.
Hill WG: Linkage disequilibrium among multiple neutral alleles produced by mutation in finite population. Theor Popul Biol. 1975, 8: 117-126. 10.1016/0040-5809(75)90028-3.
Weir BS, Hill WG: Effect of mating structure on variation in linkage disequilibrium. Genetics. 1980, 95: 477-488.
McVean GA: A genealogical interpretation of linkage disequilibrium. Genetics. 2002, 162: 987-991.
Acknowledgements
We are grateful to Roseanna Long and Kristy Shewbridge for contributing their technical expertise to this project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
CR participated in design, marker selection, genotyping, and drafted the manuscript, RV participated in design, performed the calculations for LD and effective population size. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rexroad, C.E., Vallejo, R.L. Estimates of linkage disequilibrium and effective population size in rainbow trout. BMC Genet 10, 83 (2009). https://doi.org/10.1186/1471-2156-10-83
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-10-83