
Abstract
Background
Social wasps in the subfamily Polistinae (Hymenoptera: Vespidae) have been important in studies of the evolution of sociality, kin selection, and within colony conflicts of interest. These studies have generally been conducted within species, because a resolved phylogeny among species is lacking. We used nuclear DNA microsatellite flanking sequences, mitochondrial COI sequence, and morphological characters to generate a phylogeny for the Polistinae (Hymenoptera) using 69 species.
Results
Our phylogeny is largely concordant with previous phylogenies at higher levels, and is more resolved at the species level. Our results support the monophyly of the New World subgenera of Polistini, while the Old World subgenera are a paraphyletic group. All genera for which we had more than one exemplar were supported as monophyletic except Polybia which is not resolved, and may be paraphyletic.
Conclusion
The combination of DNA sequences from flanks of microsatellite repeats with mtCOI sequences and morphological characters proved to be useful characters establishing relationships among the different subgenera and species of the Polistini. This is the first detailed hypothesis for the species of this important group.
Similar content being viewed by others

Background
A robust phylogenetic tree is of paramount importance as a beginning point for many kinds of evolutionary studies [1–4]. Understanding the evolutionary history of a character can allow us to tease apart factors important for adaptation to a specific environmental condition from those due to history [5, 6]. However, obtaining accurate phylogenetic relationships can sometimes be difficult. Historically, morphological traits were used in combination with the principles of cladistics [7, 8] to elucidate phylogenetic relationships. Morphological traits require specialists who can detect sometimes minute differences between species, and such expertise requires years of training.
Nucleic acid sequence information, primarily from mitochondrial DNA (mtDNA) has allowed us to make enormous strides towards resolving phylogenies during the last twenty years [9–15]. But these sequence regions have the disadvantage that they are often not evolving at ideal rates to reveal histories of desired traits. The branching pattern of gene trees and species trees are not necessarily overlapping in time and proceed concurrently. Also, evolution of mtDNA genes might not necessarily correspond to the speciation process [4]. Mitochondrial DNA genes are essentially single linkage groups and might not reveal the actual whole organism phylogeny [16, 17].
For nearly as long as sequence data have been available, there has been a heated debate about which kinds of data, molecular or morphological, provide the most accurate result when topologies obtained from different data sets are in conflict. Molecular data are undoubtedly considered by many to be more reliable than morphology (i.e., more likely to produce a "true" tree, e.g. [18]). It is clear that the perception of morphological data as inferior to molecular data also extends to value judgments on the quality of a particular morphological analysis based on how well the results of that analysis conform to the results of molecular analyses (e.g. [19]). Such standards are usually not applied to independent molecular analyses, which are also often in conflict with each other (e.g. [20]). No single particular data set may completely conform with our best estimate based on total evidence. The best estimator for a species phylogeny might therefore not necessarily depend on the sole use of one set of characters (morphological or molecular), but on a combination of different characters that can prove to be a better estimator of species trees [8, 13, 21–24].
Nuclear markers are independent from mitochondrial markers, and can be independent from each other if they are scattered through the genome. Nuclear genes represent a largely untouched resource for molecular systematics [25]. However, few nuclear genes, besides some rDNA sequences, have been used in phylogenetic studies [26]. The earliest molecular studies in Hymenoptera focused almost entirely on mitochontrial 16S rDNA [27–32]. More recently, the nuclear 28S rDNA gene has been used successfully for analyses of relationships within superfamilies, but with less success at higher taxonomic levels [33–36]. It has also been applied to social wasps [37], but its use is controversial [38]. Microsatellite loci are abundant, single copy, and widely distributed throughout the genome [39–41]. Few studies have used microsatellites to reconstruct phylogenies although they have been widely used for population genetic studies (e. g. [42–46]). Though their sequences are short and the repeat regions themselves are highly variable, sequence information from several microsatellite flanking regions can be combined for robust, informative and easily aligned characters that can help resolve difficult phylogenetic relationships. The single copy flanking sequences are much less variable than are numbers of repeats themselves, making them easier to align.
Here we present a phylogeny for a very important group of social wasps. Social insects of the order Hymenoptera constitute a highly diverged group, with cosmopolitan distributions. Because of the existence of different castes, division of labor, altruism, and a haplodiploid genetic system, these social insects represent a unique system to study kin selection [47–50]. The subfamily Polistinae (paper wasps) is the second largest of six subfamilies of the family Vespidae, and is entirely composed of social wasps. It is made up of four tribes (Polistini, Epiponini, Mischocyttarini and Ropalidiini), with 943 species ([51], unpublished). This group probably arose in the mid- to late Jurassic. Carpenter [51] has estimated that this group diverged from other Vespidae at the breakup of Gondwana, as much as 140 million years ago. A molecular study, which used microsatellite loci to establish the conservation and polymorphisms across different species of this subfamily, discovered interesting results for divergence times among species [52]. The divergence time for Polistes bellicosus and P. annularis was estimated to be between 10 to 80 million years. The divergence time for the entire tribe is larger than among the species of the genus Polistes and smaller than for the subfamilies of Vespidae (between 80 and 175 million years), and Vespinae and Polistinae probably split in the Mid Jurassic [52]. The tribe Polistini is the only one with a cosmopolitan distribution (Table 1). The genus Polistes was formerly subdivided into 12 different subgenera, either distributed in the New World (Fuscopolistes, Aphanilopterus, Palisotius, Epicnemius, Onerarius) or Old World (Gyrostoma, Stenopolistes, Nygmopolistes, Megapolistes, Polistella, Sulcopolistes and Polistes sensu stricto). More recently, seven of these subgenera have been synonymized [53], with one subgenus for the New World species (Aphanilopterus) and three for the Old World species (Gyrostoma, Polistella and Polistes sensu stricto). The New World subgenera were synonymized because two were found to be paraphyletic, a result not supported by the present study. The other three tribes, on the other hand, are either distributed in the New World (Epiponini, Mischocyttarini) or the Old World (Ropalidiini).
The subfamily Polistinae is characterized by two major behavioral groups, based on their mode of colony founding and mechanism of reproductive dominance [54, 55]. Independent founding is characterized by small, simply constructed nests without a protective paper envelope, founded by a single queen or few queens, without the assistance of workers [56, 57]. By contrast, swarm founding species have large colonies initiated by swarms consisting of multiple queens and a large number of workers [55].
The independent-founding group is made up of five genera: Polistes, Mischocyttarus, Belonogaster, Parapolybia, and some species of Ropalidia. Colonies are initiated by one or several inseminated egg-layers and no workers. Egg layers go through a solitary phase after insemination (few days to months). Soon after nest initiation, one fertilized female becomes the sole egg-layer. These groups of species are characterized by small colony sizes (rarely more than 50 adult wasps) and the lack of a protective envelope on the nest [56, 58, 59]. Reproductive dominance in this group is at least partly maintained through aggression by the queen.
The swarm-founding group is made up of 20 genera of the tribe Epiponini, the genus Polybioides, and some Ropalidia species. Colonies are initiated by a large number of workers and a small number of egg-layers. There is no colony phase without workers. Workers choose the site and built the nest while egg-layers wait. Swarm founding wasps characteristically have large colony sizes (usually over 50 adult wasps), and nests are often provided with a protective envelope. Reproductive dominance in this group is obtained through worker aggression on potential queens, and there is a tendency towards morphological castes in some but not most species [50, 55].
Many behavioral, ecological and evolutionary questions in this group could be asked in a phylogenetic framework including characteristics of nest structure [5, 60, 61], reproductive strategies [62–64], and solutions to potential conflicts of interest among colony members [57, 65, 66].
For the present study, we used a combination of nuclear (three microsatellite loci flanking regions and a coding system generated by the different repeat motifs, with priming sites largely conserved throughout the Polistinae subfamily; [52], adult morphological [2, 53, 67], larval [68–76], behavioral [77] and mitochondrial (COI) characters to obtain a better understanding of the relationships in this group. A study by Zhu et al. [78] used a subset of the characters and species presented in this study. The use of these characters was to reconstruct the evolution of microsatellite repeats within the Polistinae subfamily. For the completion of that study, the best tentative tree available at that time was used in order to establish the evolution of these microsatellite repeats in an explicitly phylogenetic perspective.
With this study we investigate two phylogenetic hypotheses. First, we wanted to test whether or not the subgenera of Polistes of Richards [79] are monophyletic. For this, we used 33 species of Polistes and 36 species in the outgroup, the Epiponini, Ropalidiini and Mischocyttarini. Second, we evaluated the phylogenetic relationships within and among the other three Polistinae tribes, Epiponini, Ropalidiini and Mischocyttarini, using the 33 species of the tribe Polistini as an outgroup.
Results
Polistini
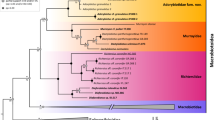
Morphological characters alone showed little resolution in the Polistini at the species level, instead supporting most of the subgenera as monophyletic – but not all (Fig. 1). New World species as a whole were monophyletic with a bootstrap support of 75% while Old World species were paraphyletic. Of the subgenera, Polistes sensu stricto, Megapolistes, Polistella, Epicnemius and Fuscopolistes were all supported as monophyletic with bootstraps of 64%, 95%, 74%, 63% and 61% respectively. But there was no resolution within these groups, and Aphanilopterus was not resolved as a group.

Bootstrap analysis of morphological characters with 1000 replications for the 33 species of the Polistini, using 36 species of Mischocyttarini, Ropalidiini, and Epiponini as outgroups.
The COI, and microsatellite combined characters supported the subgenera Megapolistes, Fuscopolistes, and Aphanilopterus with bootstraps of 93%, 93%, and 70% respectively (Figure 2). Both Polistella and Polistes sensu stricto were mostly supported at 50% and 66%, but each left out one species. There was also considerable resolution within the supported subgenera. Thus, the independent molecular and morphological trees offered resolution in both different and overlapping parts of the tree and, where they were resolved, were not in conflict.

Bootstrap analysis of the combined molecular characters (COI and microsatellite sequences) with 1000 replications for 33 species of the Polistini, using 36 species of Mischocyttarini, Ropalidiini, and Epiponini as outgroups.
The overall combined tree topology (Figure 3) is much more resolved than what was observed with the independent-character phylogenies alone (Figs. 1 and 2). The New World clade is monophyletic with 62% bootstrap support. Although the support for this same clade was 75% in the morphological characters tree (Fig. 1), the combined tree provides a much more resolved phylogeny at the species level. The clade for the Old World Polistes is paraphyletic. All the subgenera for which we have 2 or more exemplars are supported. The Old World subgenera, Megapolistes, Polistella, and Polistes sensu stricto are supported with bootstraps of 99%, 93% and 92% respectively. The New World subgenera, Fuscopolistes, Aphanilopterus and Epicnemius are supported with bootstraps of 99%, 96% and 70% respectively.

Bootstrap analysis of all characters with 1000 replications for the 33 species of the Polistini, using 36 species of Mischocyttarini, Ropalidiini, and Epiponini as outgroups.
Epiponini, Mischocyttarini, Ropalidiini
Morphological characters alone supported the Old World Ropalidiini and the New World Epiponini and Mischocyttarini with bootstraps of 64%, 85% and 91% respectively (Figure 4). All of the genera for which we had more than one species were also supported except for Polybia. There was little resolution within genera except for Brachygastra. However most genera were only represented by a few species.

Bootstrap analysis of morphological characters with 1000 replications for 36 of the species of Mischocyttarini, Ropalidiini, and Epiponini using 33 species of Polistini as outgroups.
The combined COI and microsatellite characters were largely uninformative on their own, though there was some grouping of epiponine species and resolution within the monophyletic Mischocyttarus (Figure 5).

Bootstrap analysis of molecular characters with 1000 replications for the 36 species of Mischocyttarini, Ropalidiini, and Epiponini using 33 species of Polistini as outgroups.
The combined tree is much more resolved than either separate analysis, and bootstrap supports are greater (Figure 6). Monophyly of Ropalidiini, Mischocyttarini, and Epiponini is supported at bootstrap levels of 83%, 95%, and 90% respectively. However the relationships between the tribes are not resolved. All genera for which we have more than one species are supported except for Polybia which is broken into three groups, among which relationships are not resolved (Figure 6). Relationships among genera within Ropalidiini and Epiponini are generally not well resolved. The basal genus of the Epiponini is unresolved.

Bootstrap analysis of all characters with 1000 replications for the 36 species of Mischocyttarini, Ropalidiini, and Epiponini using 33 species of Polistini as outgroups.
Discussion
Relationships in the Polistini
The combined character phylogeny is partly congruent with the earlier phylogeny proposed by Carpenter in 1996, though there are some differences (Figure 7). First, note that the earlier tree is not bootstrapped; Figure 1 is the bootstrapped tree for morphological characters, based on the present sample of species. Results from both studies on the monophyletic origin of the New and Old World Polistini subgenera only found support for the monophyletic origin of the New World Polistini (Fig. 3). In both studies, the Old World Polistini subgenera represent a paraphyletic group. However, the present study showed Polistes sensu stricto as the most basal subgenus (with a bootstrap support of 98%), in contrast to Carpenter [53], where it was the sister group of the New World subgenera (with a bootstrap support of 89%, Fig. 7). The present study found the sister group of the New World subgenera to be the clade of Polistella, Nygmopolistes, and Megapolistes. Several subgenera were absent from the present study (viz. Gyrostoma, Stenopolistes and Sulcopolistes) and it is possible that their absence affected character polarizations obtained within Polistes.

Comparison of the subgenera of Polistes phylogeny obtained by Carpenter's 1996 morphological study (Fig. 2.8) and the phylogeny obtained in the present study, including all morphological and molecular characters. Species included from each subgenus are listed in Table 1.
In contrast to Carpenter's [53] study, the present results support monophyly of the subgenus Aphanilopterus in the strict sense, and of the subgenus Epicnemius, albeit based on a smaller number of species.
Polistes sensu stricto is unique in the Polistinae for having social parasites, species that kill their host queen so her workers can rear the brood of the social parasite [12, 80–82]. The three social parasite species are monophyletic and most closely related to P. nimphus and P. dominulus, and less close to P. gallicus and P. biglumis [12, 82]. The present study agrees that the latter two species are sister species, less close to P. nimphus and P. dominulus, but this study does not put these two as sister species. The earlier study did not include morphological characters, and did not include either P. chinensis or P. marginalis which could account for the differences [82].
Relationships in the Epiponini, Mischocyttarini, Ropalidiini
The combined character phylogeny is largely congruent with the earlier phylogeny proposed by Carpenter in 1991 (Figure 8). Depending on the tribe, one or the other is more resolved, but not in major disagreement. Within the tribe Epiponini, of the polytomies observed in the Carpenter's 1991 topology, one was resolved: Agelaia and Angiopolybia are sister-groups (Fig. 8). The genus Polybia in the present study does not disagree with Carpenter et al.'s [67] conclusion that it was paraphyletic: in the present study it is unresolved, with P. (Formicicola) rejecta, P. (Trichinothorax) affinis and P. (Pedothoeca) emaciata separated from the main P. sensu stricto clade for the genus. Note that the present study is based on a broader sample of genera than in [67].

Comparison of the Polistinae subfamily phylogeny obtained by Carpenter's 1991 morphological study and the phylogeny obtained in the present study, including all morphological and molecular characters.
In a series of papers Strassmann and Queller argued that a form of worker control of sex ratios was general for the Epiponini [40, 83]. This form of worker control meant that colonies did not produce a new generation of queens until the colony had reduced in queen number to one or perhaps no queens. This is important to sociality because such a queen reduction before requeening maintains high relatedness within colonies. The species that they studied this cyclical oligogyny in were Parachartergus colobopterus, Protopolybia exigua, Brachygastra mellifica, Polybia emaciata and Polybia occidentalis [63, 83–86]. In addition West- Eberhard documented either cyclical oligogyny or some of its features in three additional genera, Agelaia, Synoeca and Metapolybia [87–89]. Taken together, these studies come from all of the four basal-most lineages of the Epiponini, and also cover 4 of the 6 lineages in our best sampled clade, the one containing Polybia (Figure 7). Thus, it seems they are justified in claiming the generality of this form of worker control for the tribe.
Importance of molecular and morphological characters
The inclusion of several genetic markers (mitochondrial and nuclear), improved the resolution of the trees obtained from morphology alone. The COI sequence alone did not resolve Polistes as a group; combining it with the microsatellites did. While COI and the microsatellite flanking regions did not resolve Polistella, adding the repeat regions did so. The use of microsatellite flanking regions and the presence/absence coding system for the repeated regions of the microsatellites proved to be useful in the resolution of some of the branches.
For the last fifteen years, a considerable rise in the use of molecular markers has been observed. The majority of the studies have exclusively included molecular data from mtDNA genes. Even when the information gathered for those studies has been informative, problems with the use of this type of molecular data (saturation synonymous sites with mtDNA; constrains imposed on RNA sites by secondary structure, length variation, etc.) have been considered [25, 90]. Unfortunately, few nuclear markers have been used in phylogenetic systematic studies in the past. The search for new, powerful, informative-single copy nuclear markers has increased our wealth of information in systematic studies. At the present time just a few more nuclear markers have been used for insect phylogenetic studies [26, 91]. More genes are needed for testing phylogenetic hypotheses. Meanwhile, the use of microsatellite flanking regions represents a valuable tool as nuclear single-copy markers. The wealth of microsatellite data is constantly growing across different taxa. The use of these regions will represent a powerful molecular tool in future studies.
Methods
Species and specimens used
We used a total of 69 species representing the four tribes of the subfamily Polistinae (Table 1). Specimens were either donated by colleagues, or were in our own collections. These specimens included 33 species from the tribe Polistini, 7 from the Mischocyttarini, 7 from the Ropalidiini and 22 from the Epiponini. We used the Polistini as the outgroup for the Epiponini+Mischocyttarini+Ropalidiini. We used the latter three tribes as the outgroup for the analysis of the Polistini.
Morphological and molecular characters
We used a total of 932 characters, including 95 morphological and behavioral characters (all morphological characters and character states from Carpenter's studies [2, 53, 67] examined on pinned specimens for this study; larval and behavioral characters and states from citations given in appendixes 1 and 2); a 488 base pair (bp) fragment of the mtDNA COI gene (appendix 3) [92]; a 311 bp fragment of single-copy nuclear flanking sequence of three microsatellite loci (Pbe216AAG [137 bp], Pbe269bAAG [75 bp], and, Pbe411AAT [99 bp] (appendix 4) [52, 93], and 38 characters representing the different trinucleotides in the repeat region (not their number; appendix 5 [78]). The repeat regions varied considerably across the four different tribes and across the different subgenera in the tribe Polistini. The coding system for the repeat region was based on presence or absence of each repeat motif and resulted in16 characters for Pbe216AAG, 15 characters for Pbe269bAAG, and 7 characters for Pbe411AAT. For example, the first four characters at Pbe216AAG were GGA, AAA, TAA, GCA, all substituted for AAG within the repeat region [78]. We did not count times a specific motif was repeated because this character is so variable its evolutionary history would be unclear.
DNA extraction, amplification and sequencing for COI, and microsatellites
We extracted DNA using protocol Strassmann I [94]. Prior to DNA extractions, we stored all specimens either at -80°C or in 100% ethanol at room temperature. PCR reactions followed protocols as in Wilcox et al. [95] for the mtDNA fragment, and Zhu et al. [78] for the microsatellite loci. Sequencing PCR products were completed directly with thermo sequenase radiolabeled 33P (Thermo Sequenace radiolabeled terminator cycle sequencing Kit, Amersham Life Science). DNA sequences were run on denaturing polyacrylamide gels, dried and exposed to X-ray film. All new DNA microsatellite sequences were deposited in Genbank under accession numbers AF119623-AF119660, AF120355-AF120455. COI sequences were deposited in Genbank under accession numbers AY382200-AY382265. The aligned DNA matrix is available at TreeBASE http://www.treebase.org/treebase/index.html, submission ID number SN1595.
Sequence alignment
The sequences for both mtDNA (COI) and for nuclear regions (microsatellite flanking regions) were aligned based on the criterion of maximum parsimony, using the program Malign, specifying a 3:1 change:gap ratio [96]. We occasionally adjusted alignments by eye when Malign gave poor results.
Systematic analysis techniques
We used Paup* 4.0b10 [97], Nona [98], and Winclada [99] to construct the phylogeny, with equal weight for all base-pair substitutions (including the mtDNA COI sequence, [100]). For the COI gene, the analysis was completed with the use of the three coding positions weighted equally and with the first and second coding positions only. The phylogenetic trees were not in conflict by excluding the third coding position.
We completed the phylogenetic analyses following the principles of maximum parsimony, maximum likelihood and distance analysis. As the overall topology was not in disagreement by applying different algorithms, we are only presenting the results obtained based on maximum parsimony. All parsimony analyses were run using approximate searches, due to the large number of species and characters. The runs consisted of 1000 replications of the parsimony ratchet [101] as implemented in Winclada, with 10% character reweighting and holding one tree; 500 replications at the "default amb-poly+" setting and 500 at "amb = poly-." Resulting trees were read into Nona and swapped on with "max*" (TBR branch swapping). Nona was also used to carry out 1000 random addition sequences as a check. Maximum parsimony trees were first estimated individually for each of the different data sets (morphological and molecular), and then in combination. Gaps were treated as missing characters. Bootstrap analyses with 1000 replications were also run for each data set. Paup* runs gave similar results as those with Winclada and Nona.
Table 2 shows the number of parsimoniously informative characters and constant characters for the each different set of data used in the study, for the 69 species used.
References
Eldredge N, Cracraft J: Phylogenetic Patterns and the Evolutionary Proces. 1980, Columbia University Press, NY
Carpenter JM: Phylogenetic Relationships and the Origin of Social Behavior in the Vespinidae. In: The social biology of Wasps. 1991, Cornell University Press, Ithaca, NY, 7-32.
Cameron SA, Mardulyn P: Multiple molecular data sets suggests independent origins of highly eusocial behavior in bees (Hymenoptera:Apinae). Syst Biol. 2001, 50: 194-214. 10.1080/10635150151125851.
Nichols R: Gene trees and species trees are not the same. Trends Ecol Evol. 2001, 16: 358-364. 10.1016/S0169-5347(01)02203-0.
Wenzel JW: Evolution of Nest Architecture. In: The social biology of Wasps. 1991, Cornell University Press, Ithaca, NY, 7-32.
Brooks DR, McLennan DA: Systematics, ecology, and behavior. Bioscience. 1995, 45: 687-
Hennig W: Phylogenetic Systematics. 1966, University of Illinois Press Urbana
Hillis DM, Wiens JJ: Molecules versus morphology in systematics. In: Phylogenetic Analysis of Morphological Data. 2000, Smithsonian Institution Press
Brown WM: Evolution of Animal Mitochondrial DNA. In: Evolution of Genes and proteins. 1983, Sinauer, Sunderland, MA, 62-88.
Cameron SA, Mardulyn P: Multiple origins of advanced eusociality in bees inferred from mitochondrial DNA sequences. Proc Natl Acad Sci. 1993, 90: 8687-8691.
Arévalo E, Davis SK, Sites JW: Mitochondrial DNA sequence divergence and phylogenetic relationships among eight chromosome races of the Sceloporus grammicus complex (Phrynosomatidae) in central Mexico. Syst Biol. 1994, 43: 387-418.
Choudhary M, Strassmann JE, Queller DC, Turillazzi S, Cervo R: Social parasites in polistine wasps are monophyletic: implications for sympatric speciation. Proc Biol Sci. 1994, 257: 31-35.
Moore WS: Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution. 1995, 49: 718-726.
Hillis DM, Moritz C, Mable BK: Molecular Systematics. 1996, Sinauer, Sunderland, MA
MTR Frost DR, Grant T, Titus TA: Phylogenetics of the lizard genus Tropidurus (Squamata: Tropiduridae: Tropidurinae): direct optimization, descriptive efficiency, and sensitivity analysis of congruence between molecular data and morphology. Mol Phyl Evol. 2001, 21: 352-71. 10.1006/mpev.2001.1015.
Richard M, Thorpe RS: Can microsatellites be used to infer phylogenies? Evidence from population affinities of the Western Canary Island lizard. Mol Phy Evol. 2001, 20:
Schmitz J, Ohme M, Suryobroto B, Zischler H: The colugo (Cynocephalus variegatus, Dermoptera): the primates' gliding sister. Molecular Biology and Evolution. 2002, 2308-2312.
Graur D: Molecular phylogeny and the higher classification of eutherian mammals. Trends Ecol Evol. 1993, 8: 141-147. 10.1016/0169-5347(93)90027-M.
Naylor GJ, Adams DC: Are the fossil data really at odds with the molecular data? Morphological evidence for cetartiodactyla phylogeny reexamined. Systematic Biology. 2001, 50: 444-453. 10.1080/106351501300318030.
Schultz TR, Engel MS, Ascher JS: Evidence for the origin of eusociality in the corbiculate bees (Hymenoptera: Apidae). J Kansas Entomol Soc. 2001, 74: 10-16.
Nei M: Molecular Evolutionary Genetics. 1987, Columbia University Press, NY
Nixon KC, Carpenter JM: On simultaneous analysis. Cladistics. 1996, 12: 221-241. 10.1006/clad.1996.0016.
Wiens JJ, Slingluff JL: How lizards turn into snakes: A phylogenetic analysis of body-form evolution in anguid lizards. Evolution. 2001, 55: 2303-2318.
Gatesy J, Amato G, Norell M, DeSalle R, Hayashi C: Combined support for wholesale taxic atavism in Gavialine crocodilians. Syst Biol. 2003, 52: 403-422. 10.1080/10635150309329.
Brower AVZ, DeSalle R: Patterns of mitochondrial versus nuclear DNA sequence divergence among nymphalid butterfies: the utility of wingless as a source of characters for phylogenetic inference. Insect Mol Biol. 1998, 7: 73-82. 10.1046/j.1365-2583.1998.71052.x.
Friedlander TP, Regier JC, Mitter C, Wagner DL: A nuclear gene for higher level phylogenetics: phosphoenolpyruvate carboxykinase tracks mesozoic-age divergences within Lepidoptera (Insecta). Mol Biol Evol. 1996, 13: 594-604.
Derr JN, Davis SK, Woolley JB, Wharton RA: Variation and the phylogenetic utility of the large ribosomal subunit of mitochondrial DNA from the insect order Hymenoptera. Mol Phyl Evol. 1992, 1: 338-341.
Derr JN, Davis SK, Woolley JB, Wharton RA: Reassessment of the 16S rRNA nucleotide sequence from members of the parasitic Hymenoptera. Mol Phyl Evol. 1992, 1: 338-341.
Dowton M, Austin AD: Molecular phylogeny of the insect order Hymenoptera: Apocritan relationships. Proc Natl Acad Sci. 1994, 91: 9911-9915.
Dowton M, Austin AD: Increased genetic diversity in mitochondrial genes is correlated with the evolution of parasitism in the Hymenoptera. J Mol Evol. 1995, 41: 958-965.
Dowton M, Austin AD, Antolin MF: Evolutionary relationships among the Braconidae (Hymenoptera: Ichneumonoidea) inferred from partial 16s rDNA gene sequences. Ins Mol Biol. 1998, 7: 129-150. 10.1046/j.1365-2583.1998.72058.x.
Kambhampati S, Volkl W, Mackauer M: Phylogenetic relationships among genera of Aphidiinae (Hymenoptera: Braconidae) based on DNA sequence of the mitochondrial 16S rRNA gene. Syst Entomol. 2000, 25: 437-445. 10.1046/j.1365-3113.2000.00129.x.
Whiting MF, Carpenter JC, Wheeler QD, Wheeler WC: The Strepsiptera problem: phylogeny of the holometabolous insect orders inferred from 18S and 28S ribosomal DNA sequences and morphology. Systematic Biology. 1997, 46: 1-68.
Giribet G, Rambla M, Carranza S, Bagun J, Riutort M, Ribera C: Phylogeny of the Arachnid Order Opiliones (Arthropoda) Inferred from a Combined Approach of Complete 18S and Partial 28S Ribosomal DNA Sequences and Morphology. Molecular Phylogenetics and Evolution. 1999, 11: 296-307. 10.1006/mpev.1998.0583.
Yeates DK, Wiegmann BM: Congruence and controversy: Toward a Higher-Level Phylogeny of Diptera. Annual Review of Entomology. 1999, 44-
Schulmeister S, Wheeler WC, Carpenter JM: Simultaneous analysis of the basal lineages of Hymenoptera (Insecta) using sensitivity analysis. Cladistics. 2002, 18: 455-484. 10.1016/S0748-3007(02)00100-7.
Schmitz J, Moritz RFA: Molecular phylogeny of Vespidae (Hymenoptera) and the evolution of sociality in wasps. Mol Phyl Evol. 1998, 9: 183-191. 10.1006/mpev.1997.0460.
Carpenter JM: On "Molecular Phylogeny of Vespidae (Hymenoptera) and the Evolution of Sociality in Wasps. Am Mus Novitat. 2003, 3389: 1-20. 10.1206/0003-0082(2003)389<0001:OMPOVH>2.0.CO;2.
Tautz D: Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res. 1989, 17: 6463-6470.
Queller DC, Strassmann JE, Hughes CR: Microsatellites and kinship. Trends Ecol Evol. 1993, 8: 285-10.1016/0169-5347(93)90256-O.
Choudhary M, Strassmann JE, Solis CR, Queller DC: Microsatellite variation in social insects. Biochem Genet. 1993, 31: 87-95.
Queller DC, Strassmann JE, Hughes CR: Genetic relatedness in colonies of tropical wasps with multiple queens. Science. 1988, 242: 1155-1157.
Estoup A, Scholl A, Pouvreau A, Solignac M: Monoandry and polyandry in bumble bees (Hymenoptera, Bombinae) as evidenced by highly variable microsatellites. Mol Ecol. 1995, 4: 89-93.
Bowcock AM, Ruiz-Linares A, Tomb\fohrde J, Minch E, Kidd JR, Cavalli-Sforza LL: High resolution of human evolutionary trees with polymorphic microsatellites. Nature. 1994, 368: 455-457. 10.1038/368455a0.
Angers B, Bernatchez L: Complex evolution of a salmonid microsatellite locus and its consequences in inferring allelic divergence from size information. Mol Biol Evol. 1997, 14: 230-238.
Franck P, Garnery L, Solignac M, Cornuet JM: The origin of West European subspecies of honeybees (Apis mellifera): New insights from microsatellite and mitochondrial data. Evolution. 1998, 52: 1119-1134.
Hamilton WD: The genetical evolution of social behaviour. J Theor Biol. 1964, 7: 1-16.
Pamilo P: Genetic relatedness and evolution of insect sociality. Behav Ecol Sociobiol. 1984, 15: 241-248.
Pamilo P, Crozier RH.: Evolution of Social Insect Colonies: Sex Allocation and Kin Selectio. 1996, Oxford University Press
Strassmann JE, Nguyen JS, Arévalo E, Cervo R, Zacchi F, Turillazzi S, Queller DC: Workers produce males only after queen death in Polistes gallicus, a Mediterranean social wasp. Journal of Evolutionary Biology. 2003, 16: 254-259. 10.1046/j.1420-9101.2003.00516.x.
Carpenter JM: Biogeographic patterns in the Vespidae (Hymenoptera), two views of Africa and South America. In: Biological relationships between Africa and South America. 1993, New Haven, CT: Yale University Press
Ezenwa VO, Peters JM, Hastings M, Zhu Y, Arévalo E, Seppä P, Pederson JS, Zacchi F, Queller DC, Strassmann JE: Ancient conservation of microsatellite loci in polistine wasps. Mol Phyl Evol. 1998, 9: 168-177. 10.1006/mpev.1998.0528.
Carpenter JM: Phylogeny and biogeography of Polistes. In Natural history and evolution of paper wasps. Edited by: Turillazzi S, West-Eberhard MJ. 1996, Oxford Univ Press
Jeanne RL: Evolution of social behavior in the Vespidae. Annu Rev Entomol. 1980, 25: 371-396. 10.1146/annurev.en.25.010180.002103.
Jeanne RL: The swarm-founding Polistinae. In: The social Biology of Wasps. 1991, Ithaca, NY: Cornell University Press
Gadagkar R: Belonogaster, Mischocyttarus, Parapolybia, and independent founding Ropalidia. In: The Social Biology of Wasps. Edited by: KRaR Matthews. 1991, Ithaca, NY: Cornell University Press, 149-190.
Keeping MG: Reproductive and worker castes in the primitively eusocial wasp Belonogaster petiolata (DeGeer) (Hymenoptera: Vespidae): evidence for pre-imaginal differentiation. J Insect Physiol. 2002, 48: 867-879. 10.1016/S0022-1910(02)00156-7.
Strassmann JE, Queller DC, Hughes CR: Constraints on independent nesting by Polistes foundresses in Texas. In: Chemistry and Biology of Scial Insects. Edited by: JEaH Rembold. 1987, Munich: Verlag J Peperny, 379-380.
Reeve HK: Polistes. In: The social Biology of Wasps. Edited by: KRaR Matthews. 1991, Ithaca, NY: Cornell University Press, 99-148.
Wenzel JW, Carpenter JM: Comparing methods: Adaptive traits and tests of adaptation. In: Phylogenetics and ecology. Edited by: PEaRI Vane-Wright. 1994, London: Academic Press, 79-101.
O'Donnell S, Jeanne RL: The nest as fortress: defensive behavior of Polybia emaciata, a mud-nesting eusocial wasp. J Insect Sci. 2002, 2:
Ezenwa VO, Henshaw M, Queller DC, Strassmann JE: Patterns of buzz running, a swarm initiation behaviour, in the neotropical wasp, Parachartergus colobopterus. Insect Soc. 1998, 45: 445-456. 10.1007/s000400050100.
Hastings MD, Queller DC, Eischen F, Strassmann JE: Kin selection, relatedness, and worker control of reproduction in a large-colony epiponine wasp, Brachygastra mellifica. Behav Ecol Sociobiol. 1998, 9: 573-581. 10.1093/beheco/9.6.573.
Herman RA, Queller DC, Strassmann JE: The role of queens in colonies of the swarm-founding wasp Parachartergus colobopterus. Anim Behav. 2000, 59: 841-848. 10.1006/anbe.1999.1385.
Arévalo E, Strassmann JE, Queller DC: Conflicts of interest in social insects: male production in two species of Polistes. Evolution. 1998, 52: 797-805.
Foster KR, Ratnieks FLW: Paternity, reproduction and conflict in vespine wasps: a model system for testing kin selection predictions. Behav Ecol Sociobiol. 2001, 50: 1-8. 10.1007/s002650100336.
Carpenter JM, Kojima J, Wenzel JW: Polybia, Paraphyly, and Polistine Phylogeny. Am Mus Novitat. 2000, 3298: 1-24.
Reid JA: On the classification of the larvae of the Vespidae. Trans R Entomol Soc London. 1942, 93: 285-331.
Dias Filho MM: Contribuição à morfologia de larvas de vespídeos sociais do Brasil (Hymenoptera, Vespidae). Rev Bras Entomol. 1975, 19: 1-36.
Wheeler GC, Wheeler J: Larvae of some eusocial bees and wasps. Contrib Sci. 1979, 321: 1-19.
Nelson JM: External morphology of Polistes (paper wasp) larvae in the United States. Melanderia. 1982, 38: 1-29.
Yamane S, Okazawa T: Mature larvae of some polistine wasps from Papua New Guinea and Fiji, with notes on larval characters of the Old World and Oceanian Polstinae (Hymenoptera: Vespidae). Rep Fac Sci Kagoshima Univ Earth Sci Biol. 1981, 65-75.
Kojima J, Yamane S: Systematic study of the mature larvae of Oriental polistine wasps (Hymenoptera: Vespidae) (1) Species of Ropalidia and Polistes from Sumatra and Java Islands. Rep Fac Sci Kagoshima Univ Earth Sci Biol. 1984, 17: 103-127.
Kojima J, Keeping MG: Larvae of Belonogaster juncea colonialis Kohl and B. petiolata (Degeer) (Hymenoptera: Vespidae). J Entomol Soc Sth Africa. 1985, 48: 233-239.
Kojima J: Descriptions of mature larvae of eight Australian Polistinae (Hymenoptera: Vespidae). J Aust Ent Soc. 1987, 26: 141-148.
Kojima J: Larvae of social wasps (Insecta: Hymenoptera, Vespidae). Nat Hist Bull Ibaraki Univ. 1998, 2: 7-227.
Wenzel JW: Application of the biogenetic law to behavioral ontogeny: A test using nest architecture in paper wasps. J Evol Biol. 1993, 6: 229-247.
Zhu Y, Queller DC, Strassmann JE: A phylogenetic perspective on sequence evolution in microsatellite loci. J Mol Evol. 2000, 50: 324-338.
Richards OW: The Social Wasps of the Americas, Excluding the Vespinae. London: British Museum (Natural History). 1978
Cervo R, Lorenzi MC, Turillazzi S: On the strategies of host nest invasion in three species of Sulcopolistes, social parasites of Polistes wasps. Actes Coll Insectes Soc. 1990, 6: 69-74.
Carpenter JM, Strassmann JE, Turillazzi S, Hughes CR, Solis CR, Cervo R: Phylogenetic relationships among paper wasp social parasites and their hosts (Hymenoptera: Vespidae: Polistinae). Cladistics. 1993, 9: 129-146. 10.1006/clad.1993.1008.
Carpenter JM: Phylogenetic relationships among European Polistes and the evolution of social parasitism (Hymenoptera:Vespidae, Polistinae). Mém Mus Natl Hist Nat. 1997, 173: 135-161.
Strassmann JE, Queller DC, Solís CR, Hughes CR: Relatedness and queen number in the Neotropical wasp, Parachartergus colobopterus. Anim Behav. 1991, 42: 461-70.
GK Strassmann JE, Queller DC, Hughes CR: Demographic and genetic evidence for cyclical changes in queen number in a neotropical wasp, Polybia emaciata. Am Nat. 1992, 140: 363-72. 10.1086/285417.
SJ Gastreich KR, Queller DC: Determinants of high genetic relatedness in the swarm-founding wasp, Protopolybia exigua. Ecology Ethology and Evolution. 1993, 5: 529-539.
Queller D, Strassmann J, Solís C, Hughes C, Moralez DeLoach D: selfish A strategy of social insect workers that promotes social cohesion. Nature. 1993, 365: 639-641. 10.1038/365639a0.
West-Eberhard M: Temporary queens in Metapolybia wasps: Nonreproductive helpers without altrusim. Science. 1978, 200: 441-443.
West-Eberhard M: Intragroup selection and the evolution of insect societies. In: Natural Selection and Social Behavior. Edited by: DT RD Alexander. 1981, New York: Chiron Press, 3-17.
West-Eberhard M: The genetic and social structure of polygynous social wasp colonies (Vespidae: Polistinae). In: Social Insects in the Environment: Proceedings of the 11th Interantional Congress of the IUSSI. Edited by: BM GK Veeresh, Viraktamath CA. 1990, New Delhi: Oxford and IBH, 254-255.
Palumbi SR: Rates of molecular evolution and the fraction of nucleotide positions free to vary. J Mol Evol. 1989, 29: 180-187.
Cho S, Mitchell A, Regier JC, Mitter C, Poole RW, Friedlander TP, Zhao S: A highly conserved nuclear gene for low-level phylogenetics: elongation factor 1a recovers morphology-based tree for heliothine moths. Mol Biol Evol. 1995, 12: 650-656.
Simon C, Frati F, Beckenback A, Crespi B, Liu H, Flook P.: Evolution, wighting and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am. 1994, 87: 651-701.
Strassmann JE, Barefield K, Solís CR, Hughes CR, Queller DC: Trinucleotide microsatellite loci for a social wasp, Polistes. Mol Ecol. 1997, 6: 97-100. 10.1046/j.1365-294X.1997.00158.x.
Strassmann JE, Solís CR, Peters JM, Queller DC: Strategies for finding and using highly polymorphic DNA microsatellite loci for studies of genetic relatedness and pedigrees. In: Molecular Methods in Zoology and Evolution. Edited by: JFaS Palumbi. 1996, New York, NY: Wiley, 163-180. 528–549
Wilcox TP, Hugg L, Zeh JA, Zeh DW: Mitochondrial DNA sequencing reveals extreme genetic differentiation in a cryptic species complex of neotropical pseudoscorpions. Mol Phyl Evol. 1997, 7: 208-216. 10.1006/mpev.1996.0388.
W Wheeler, Gladstein D: MALIGN. In: Book MALIGN. 1998, City: American Museum of Natural History, 2.7
Swofford DL: PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods). In: Book PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods). 2002, City: Florida State University, 4.0b10 for Macintosh
Goloboff PA: NONA. In: Book NONA. 1999, City: Program and documentation. Fundación e Instituto Miguel Lillo, version 2
Nixon KC: Winclada. In: Book Winclada. 2002, City: Published by the author, version 1.0
Källersjö M, Albert VA, Farris JS: Homoplasy Increases Phylogenetic Structure. Cladistics. 1999, 15: 91-93. 10.1006/clad.1999.0085.
Nixon K: The Parsimony Ratchet, a new method for rapid parsimony analysis. Cladistics. 1999, 15: 407-414. 10.1006/clad.1999.0121.
Acknowledgements
We are thankful to all our colleagues who donated specimens to the study (see Table 1). We also thank Barry Sullender for his assistance in the analysis of the characters and David Queller for his insightful comments. EA wants to thank Liam Donohoe for comments on early drafts of the manuscript. This material is based upon work supported by the National Science Foundation under Grant Nos. BIR-9612687, IBN-9808809, IBN-9900975, and DEB-9975351.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
EA carried out a portion of the molecular genetic studies, participated in the sequence alignment, analysis of the data, and drafted the manuscript. YZ carried out a portion of the molecular genetic studies and participated in the sequence alignment. JMC provided the morphological characters, participated in the analyses and writing. JES conceived of the study, and participated in its design, coordination and writing. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Arévalo, E., Zhu, Y., Carpenter, J.M. et al. The phylogeny of the social wasp subfamily Polistinae: evidence from microsatellite flanking sequences, mitochondrial COI sequence, and morphological characters. BMC Evol Biol 4, 8 (2004). https://doi.org/10.1186/1471-2148-4-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-4-8