
Abstract
Background
The outbreak of severe acute respiratory syndrome (SARS) caused a severe global epidemic in 2003 which led to hundreds of deaths and many thousands of hospitalizations. The virus causing SARS was identified as a novel coronavirus (SARS-CoV) and multiple genomic sequences have been revealed since mid-April, 2003. After a quiet summer and fall in 2003, the newly emerged SARS cases in Asia, particularly the latest cases in China, are reinforcing a wide-spread belief that the SARS epidemic would strike back. With the understanding that SARS-CoV might be with humans for years to come, knowledge of the evolutionary mechanism of the SARS-CoV, including its mutation rate and emergence time, is fundamental to battle this deadly pathogen. To date, the speed at which the deadly virus evolved in nature and the elapsed time before it was transmitted to humans remains poorly understood.
Results
Sixteen complete genomic sequences with available clinical histories during the SARS outbreak were analyzed. After careful examination of multiple-sequence alignment, 114 single nucleotide variations were identified. To minimize the effects of sequencing errors and additional mutations during the cell culture, three strategies were applied to estimate the mutation rate by 1) using the closely related sequences as background controls; 2) adjusting the divergence time for cell culture; or 3) using the common variants only. The mutation rate in the SARS-CoV genome was estimated to be 0.80 – 2.38 × 10-3 nucleotide substitution per site per year which is in the same order of magnitude as other RNA viruses. The non-synonymous and synonymous substitution rates were estimated to be 1.16 – 3.30 × 10-3 and 1.67 – 4.67 × 10-3 per site per year, respectively. The most recent common ancestor of the 16 sequences was inferred to be present as early as the spring of 2002.
Conclusions
The estimated mutation rates in the SARS-CoV using multiple strategies were not unusual among coronaviruses and moderate compared to those in other RNA viruses. All estimates of mutation rates led to the inference that the SARS-CoV could have been with humans in the spring of 2002 without causing a severe epidemic.
Similar content being viewed by others


Background
The earliest confirmed case of the severe acute respiratory syndrome (SARS) occurred in November, 2002 in the Guangdong province of China. Toward the end of the epidemic (as reported by July 31, 2003) there were 8,098 recognized cases in 31 countries or regions worldwide and 774 implicated deaths (WHO, http://www.who.int/csr/sars/country/table2003_09_23/en/). Due to an unprecedented international effort, the SARS coronavirus (SARS-CoV) was identified as the causal agent in late March 2003 and its first complete genomic sequences were published April 13, 2003 [1, 2]. One month later, SARS-like coronaviruses were found in palm civets and other animals in Guangdong, China, the first evidence of possible interspecies transmission of the virus [3]. The re-emergence of the isolated SARS cases in Asia in December, 2003 and in Anhui province and Beijing, China, in late April 2004, has confirmed a wide-spread conjecture that the SARS-CoV will likely be with humans for years to come. This re-emergence of SARS cases makes it legitimate to critically re-evaluate the time for the origin of the SARS-CoV.
There are 26 putative coding regions which cover about 98% of the 29.8-kb SARS-CoV genome. Approximately two-thirds of the genome are at the 5' side encoding the nonstructural proteins (orf1ab and orf1a) and one-third are at the 3' side encoding four structural proteins: spike glycoprotein (S), envelope (E), membrane (M), and nucleocapsid (N) [4]. The spike glycoprotein, especially its S1 subdomain, is responsible for binding to the specific receptor in the target cells [4, 5]. RNA polymerase and nsp1 genes are two major loci in orf1ab.
Estimating the mutation rate in RNA viruses and retroviruses is critical but also challenging for tracing their rapidly evolving paths. The rates estimated from the positive-strand ssRNA virus appear to be in a similar range (e.g., ~10-3 per site per year) from the negative-strand ssRNA virus, although a direct comparison is not possible because the mutation rates could be estimated from different regions or genes [6–15]. The estimated mutation rates in coronavirus, which SARS-CoV phylogenetically links to, are moderate to high compared to the others in the category of ssRNA viruses. For example, it was estimated to be 0.3 – 0.6 × 10-2 per site per year in the infectious bronchitis virus in a previous study [8]. However, the estimated mutation rate appears to have a wider range in the retrovirus [16–20]. More details are presented in the Discussion section.
How SARS-CoV evolves has important implications for both strategic planning in the prevention of SARS epidemics and development of a vaccine and antibodies. The mutation rate is among the most fundamental aspect of sequence evolution. If the pathogen evolves slowly, there will be a better chance for development of effective long lasting vaccines and successful treatment for patients from a particular geographic region will likely be effective for patients from other areas. On the other hand, if the pathogen (particularly the genes coding for major antigens) evolves rapidly, an effective strategy to prevent transmission of the SARS-CoV must be the top-priority, and an effective vaccine program may be problematic. The purpose of this study is to improve our understanding of the evolutionary mechanism in the SARS-CoV genome, and in particular to address the issues of the mutation rate and the time for the emergence of the SARS-CoV in the human population. We reported the estimated mutation rate in the SARS-CoV using the available complete genomic sequences whose clinical history either is certain or could be inferred.
Results
Mutation rate
The sources of the genomic sequences used in this study and the methods of estimating mutation rates are presented in the Methods section. The divergence time was inferred based on the information summarized in Figure 1. Table 1 shows the mutation rates estimated by three strategies. When the first strategy was used to adjust for sequencing errors and potential mutations in the cell culture, the mutation rate was estimated to be 0.80 – 2.38 × 10-3 nucleotide substitution per site per year using all the sequences not generated from mainland China, and 0.81 – 1.38 × 10-3 nucleotide substitution per site per year using the TOR2 and Urbani sequences only. When the second strategy was used, the mutation rate was estimated to be 0.74 – 1.62 × 10-3 nucleotide substitution per site per year, which is lower than that from using the first strategy. As expected, the mutation rate estimated using the third strategy was the lowest; 0.54 – 1.57 × 10-3 nucleotide substitution per site per year using the 11 sequences not generated from mainland China and 0.42 – 0.72 × 10-3 nucleotide substitution per site per year using the TOR2 and Urbani sequences only.

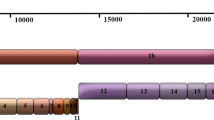
Clinical relations and estimated range of the divergence time among 16 SARS-CoV isolates. This figure is adapted from Figure 5 in [4]. Solid arrows indicate the certain SARS coronavirus transmission route and dashed lines indicate the uncertain route. SINxxxx denotes an unavailable primary contact of the Singaporean index patient (SIN2500). The numbers indicate a range of the diverged time (days) between two isolates.
Substitution rate in the coding regions
For all samples, the proportion of non-synonymous substitutions per non-synonymous site (Ka) was 0.63 × 10-3 and the proportion of synonymous substitutions per synonymous site (Ks) was 0.65 × 10-3, leading to Ka/Ks being 0.97. This ratio was 0.79 in the nonstructural region and 1.37 in the structural region. In particular, the values of Ka/Ks were 1.98 for nsp1 and 0.85 for S.
Table 2 shows the rates of nucleotide substitution in the coding regions of sequences. The overall rates of non-synonymous and synonymous substitutions were 1.16 – 3.30 × 10-3 and 1.67 – 4.67 × 10-3 per site per year, respectively. The non-synonymous rate was higher in the three genes E, M, and N, suggesting some of those mutations might increase antigenicity, although the number of mutations used to calculate these rates was small.
Time for the origin of SARS-CoV
The mutation rate estimated earlier allowed us to estimate the age of the most recent common ancestor (MRCA) of the sample, which should be about the same or more recent than the time for the origin of SARS-CoV. The phylogeny reconstructed by the neighbor-joining method with mid-point rooting or by maximum parsimony is overall consistent with the epidemic (Additional file 1). All the sequences from mainland China clustered together and separated from the remaining sequences, including those clinically related to the index patient A. GZ01 was distantly separated from other sequences. Assuming the MRCA is the root of the phylogeny, the age of the MRCA is then the divergence time between GZ01 and other sequences. Using the mutation rates estimated above, it is found that the MRCA could be alive at a time between March 28 and November 29, 2002 (strategy 1), between February 22 and October 3, 2002 (strategy 2), and even earlier (strategy 3). The most critical implication of these analyses is that it is entirely plausible that the MRCA of the sample could be alive as early as the spring of 2002.
Discussion
Some uncertainties in the quality of the sequence data and incomplete information from patient histories are two limiting factors of this study. The world-wide race to understand this novel virus has provided an unprecedented set of complete genome sequences of a pathogen in an interval of a few weeks, but likely side-effects of this race might be an elevated error rate in the released sequences and generating errors during the analysis. Among the 129 sequence variations reported [4], many were generated randomly by the algorithms during the alignment of the multiple sequences, therefore these should be removed or adjusted. The concern above has led us to wait until all the sequences used in this study have been significantly revised by their generators and to manually adjust the multiple-sequence alignment. Still some errors were unavoidable partly due to the intrinsic error rate of sequencing technology. For example among 18 common variations, 9 could not be uniquely assigned to the internal branches of the phylogeny. This incongruence is likely partially due to sequence errors. The existence of sequence errors can also be inferred by examining the ratio of transitional versus transversional changes. If nucleotide substitution occurs randomly, there are two transversional substitutions on average for each transitional substitution, and the ratio of transition to transversion should be 0.5. However, transition is generally favored over transversion in many organisms. For example, the ratio is approximately 2 in the human genome [21, 22]. The ratio has not been discussed extensively in the RNA viruses; however, it appears to be higher than that in the mammalian genomes based on the two previous reports of 3.7 in the influenza A virus [23] and 5.0 in the Marburg virus [24]. In this study, 60 transitional substitutions and 54 transversional substitutions were observed among the 16 sequences, thus the ratio was 1.1. The ratio in five sequences from mainland China was 0.9, considerably smaller than 2.2 which was observed in the other eleven sequences. This suggests that sequences from mainland China may be more erroneous than the other sequences. On the other hand, the ratio was 0.9 for the singleton variations, which was much lower than the ratio of 3.5 for the non-singleton variants. This further indicates that singletons were more problematic.
Because of the unknown level of errors in the sequences, a conservative approach to estimating the mutation rate was taken. Three strategies were used to reduce the effect of sequence errors, one being more aggressive than the other two. The mutation rates estimated by the first two strategies were quite similar. In the third strategy, all the variants unique to a given isolate were excluded. Such a strategy is very conservative because the amount of singletons is expected to be large in a rapid expanding environment (see below). Therefore the mutation rate was placed in the range of 0.80 – 2.38 × 10-3 nucleotide substitution per site per year based on the 11 sequences used. This rate, along with the rate of synonymous substitutions estimated in this study, is close to that recently reported using another approach [25]. In comparison to other coronaviruses, this rate is lower than that in the mouse hepatitis virus, similar to that in the transmissible gastroenteritis virus, but higher than that in the infectious bronchitis virus (Table 3) [6–8]. The estimated mutation rate is at the same order of magnitude as in other RNA viruses, for example, 2.3 × 10-3 nucleotide substitution per site per year in the influenza A viruses [12, 13]. The estimated mutation rate in HIV appears to have a wide range [16, 17]. It is likely that the mutation rate in the SARS-CoV is not higher than that in HIV. Therefore, the SARS-CoV is not an unusual coronavirus or RNA virus in terms of its speed of nucleotide changes. One of the challenging tasks, therefore, is to find those variations which led to the SARS-CoV being unique from other RNA viruses, especially coronaviruses, and how those variations changed the functionality and helped to transmit it to humans.
Nucleotide variation is distributed along the entire genome. Based on our alignment and the annotation in GenBank, 21 of the 26 open reading frames had the variations, including genes encoding polymerase, spike glycoprotein, envelope, membrane, and nucleocapsid protein. The estimated mutation rate suggests that approximately 2 to 6 new mutations will occur each month in a virus assuming the overall uniform mutation rate. However, the rate of the non-synonymous substitutions might vary during the course of the SARS-CoV evolution [25]. It was observed that there was an excess of mutations (and amino acid changes) in the external branches of the phylogeny of a large sample of the HA gene sequences of influenza A, which was partially caused by sampling bias [26]. From a population genetics standpoint, a large proportion of mutations should occur in the external branches when the infected hosts have rapidly increased. Therefore, one should not conclude that mutation rate is low because of a relatively small number of mutations in the internal branches [27]. Our analysis, even by a conservative estimation of mutation rate, indicates that the SARS-CoV population has already harbored a considerable amount of genetic diversity.
The emerging time of the SARS-CoV is of special importance in dissecting the origin of the virus as well as the dynamics of the epidemic. The time for the most recent common ancestor of the 16 isolates was estimated to be between February 2002 and November 2002. Although this is consistent with the date for the earliest known case of SARS and those estimated in other studies [25, 28], it also suggests that SARS-CoV could have been present longer than generally believed, that is, around November 2002. One possible scenario is that the SARS-CoV had already infected some people in the spring of 2002 but failed to cause epidemics; its spread was however suppressed in the summer (similar to the summer of 2003), and re-emerged around November to cause the epidemic in 2003. Given the current re-emergence of SARS cases, this scenario is becoming more likely. There were indeed some media reports of SARS-like symptoms of patients in the spring of 2002 although none have been convincingly confirmed. An alternative scenario is that the common ancestor of the SARS-CoV lived in the spring of 2002, but the host was animals. The recent finding of high sequence homology between the isolate from a newly emerged SARS case (December 16, 2003) and the isolates from the masked palm civets [29] makes civets as the primary suspect of reservoir for SARS-CoV.
Conclusions
The estimated mutation rate and the synonymous and non-synonymous substitution rates in the SARS-CoV genome were moderate compared to that in coronavirus and other RNA viruses, suggesting that the SARS-CoV is not an unusual coronavirus in terms of its speed of nucleotide or amino acid changes. Based on the mutation rates estimated in this study, the emerging time of the most recent common ancestor of the 16 isolates can be placed between February 2002 and November 2002. This suggests that the SARS-CoV could have been with humans as early as the spring of 2002 without causing a severe epidemic.
Methods
Sequence data
We obtained 16 complete genomic sequences from the NCBI website http://www.ncbi.nlm.nih.gov/. Among them, five sequences (BJ01-04 and GZ01) were obtained from the hosts collected in mainland China and the remaining sequences (TOR2, Urbani, CUHK-W1, CUHK-Su10, HKU-39849, five Singaporean sequences, and TW1) were from the hosts in other geographic regions. Detailed information of the sequences is shown in Table 4.
Sequence analysis
CLUSTAL X [30], a window-based user interface to the CLUSTAL W, was used to align the multiple sequences. The alignment was further manually examined and adjusted. All gene annotation information and nucleotide position designations in this study refer to the TOR2 sequence (GenBank accession ID: NC_004718). To avoid complications, only the single nucleotide variations were analyzed and all alignment gaps were excluded. This led to the identification of a total of 114 single nucleotide variations among all the sequences and an average of 18.2 nucleotide differences between two sequences.
The MEGA2 computer program [31] was used to calculate the pair-wise nucleotide differences. The resulting genetic distances were corrected by Jukes and Cantor's method [32]. The phylogeny of the sample was reconstructed using both neighbor-joining and maximum parsimony methods [31, 33].
Mutation rate can be estimated in principle by the number of nucleotide differences between two sequences divided by twice their divergent time, i.e., the time to their most recent common ancestor. Due to better documented contact histories, mutation rates were estimated only by the sequences whose hosts were not from mainland China, that is, sequences TOR2, Urbani, CUHK-W1, CUHK-Su10, HKU-39849, five Singaporean sequences, and TW1. First, the range of the divergence time between each pair of sequences was inferred based on information on infection history, reported strain isolation dates and sequence release dates (Additional file 2) [4, 34–36]. For example, the divergence time between isolates TOR2 and Urbani was estimated to be in the range of 34 to 58 days [35, 36]. Second, nucleotide difference between each pair of sequences was calculated with adjustments to reduce the effect of sequencing errors and potential mutations during cell culture. Three strategies were used. The first strategy was used to reduce the number of pair-wise nucleotide differences by the averaged number of nucleotide differences observed in five closely related Singaporean sequences [4]. This strategy effectively assumes that there is no real nucleotide difference among these five sequences so that their observed differences reflect the level of errors. The second strategy was used to reduce the pair-wise nucleotide difference by two and to add 7 days to the divergence time to account for cell culture time. This strategy assumes that the mutation rate during the cell culture is the same as that in the human host and that on average the sequencing error is one nucleotide per genome. In the third strategy, we excluded all the nucleotide variants which had been observed only once (singletons) among the 61 human SARS-CoV sequences reported in [25]. The rational is that non-singleton mutations observed in a sample are much less likely due to sequencing errors as well as mutations during the laboratory passage of virus. This strategy is apparently conservative and can be regarded as the lower bound of the mutation rate. Finally, the mutation rate per site per year was estimated by

where d ij is the genetic distance between sequence i and j, t ij is twice their divergence time (in number of days), and n is the number of sequences.
A mutation in a codon is non-synonymous (or non-silent) if it changes the amino acid, and is synonymous (silent) otherwise. The number of non-synonymous mutations per non-synonymous site (Ka) and the number of synonymous mutations per synonymous site (Ks) were computed using the method of Li, Wu, and Luo [37]. The non-synonymous and synonymous substitution rates were calculated using the divergence time as estimated above. Only the second strategy was applied to the rate estimation because the number of nucleotide differences used for the adjustment in the first strategy can not be separated for the non-synonymous and synonymous mutations.
References
Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL: The Genome sequence of the SARS-associated coronavirus. Science. 2003, 300: 1399-1404. 10.1126/science.1085953.
Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ: Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003, 300: 1394-1399. 10.1126/science.1085952.
Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JSM, Poon LLM: Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003, 302: 276-278. 10.1126/science.1087139.
Ruan Yi Jun, Wei Chia Lin, Ling Ai Ee, Vega Vinsensius B, Thoreau Herve, Se Thoe Su Yun, Chia Jer-Ming, Ng Patrick, Chiu Kuo Ping, Lim Landri: Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. The Lancet. 2003, 361: 1779-1785. 10.1016/S0140-6736(03)13414-9.
Lai MC, Holmes KV: In: Knipe DM, Howley, PM, eds. Fundamental Virology. 2001, Lippincott, PA, Williams & Wilkins, 4
Chen W, Baric RS: Function of a 5'-end genomic RNA mutation that evolves during persistent mouse hepatitis virus infection in vitro. J Virol. 1995, 69: 7529-7540.
Enjuanes L, Sune C, Gebauer F, Smerdou C, Camacho A, Anton IM, Gonzalez S, Talamillo A, Mendez A, Ballesteros ML, Sanchez C: Antigen selection and presentation to protect against transmissible gastroenteritis coronavirus. Vet Microbiol. 1992, 33: 249-262. 10.1016/0378-1135(92)90053-V.
Cavanagh D, Mawditt K, Adzhar A, Gough RE, Picault JP, Naylor CJ, Haydon D, Shaw K, Britton P: Does IBV change slowly despite the capacity of the spike protein to vary greatly?. Adv Exp Med Biol. 1998, 440: 729-734.
Lu L, Nakano T, Orito E, Mizokami M, Robertson BH: Evaluation of accumulation of hepatitis C virus mutations in a chronically infected chimpanzee: comparison of the core, E1, HVR1, and NS5b regions. J Virol. 2001, 75: 3004-3009. 10.1128/JVI.75.6.3004-3009.2001.
Nakao H, Okamoto H, Fukuda M, Tsuda F, Mitsui T, Masuko K, Iizuka H, Miyakawa Y, Mayumi M: Mutation rate of GB virus C/hepatitis G virus over the entire genome and in subgenomic regions. Virology. 1997, 233: 43-50. 10.1006/viro.1997.8615.
Villaverde A, Martinez MA, Sobrino F, Dopazo J, Moya A, Domingo E: Fixation of mutations at the VP1 gene of foot-and-mouth disease virus. Can quasispecies define a transient molecular clock?. Gene. 1991, 103: 147-153. 10.1016/0378-1119(91)90267-F.
Xu X, Cox NJ, Bender CA, Regnery HL, Shaw MW: Genetic variation in neuraminidase genes of influenza A (H3N2) viruses. Virology. 1996, 224: 175-183. 10.1006/viro.1996.0519.
Shu LL, Bean WJ, Webster RG: Analysis of the evolution and variation of the human influenza A virus nucleoprotein gene from 1933 to 1990. J Virol. 1993, 67: 2723-2729.
Krossoy B, Nilsen F, Falk K, Endresen C, Nylund A: Phylogenetic analysis of infectious salmon anaemia virus isolates from Norway, Canada and Scotland. Dis Aquat Organ. 2001, 44: 1-6.
Schrag SJ, Rota PA, Bellini WJ: Spontaneous mutation rate of measles virus: direct estimation based on mutations conferring monoclonal antibody resistance. J Virol. 1999, 73: 51-54.
Li WH, Tanimura M, Sharp PM: Rates and dates of divergence between AIDS virus nucleotide sequences. Mol Biol Evol. 1988, 5: 313-330.
Fu Yun-Xin: Estimating mutation rate and generation time from longitudinal samples of DNA sequences. Mol Biol Evol. 2001, 18: 620-626.
Muller-Trutwin MC, Corbet S, Tavares MD, Herve VM, Nerrienet E, Georges-Courbot MC, Saurin W, Sonigo P, Barre-Sinoussi F: The evolutionary rate of nonpathogenic simian immunodeficiency virus (SIVagm) is in agreement with a rapid and continuous replication in vivo. Virology. 1996, 223: 89-102. 10.1006/viro.1996.0458.
Mansky LM, Temin HM: Lower mutation rate of bovine leukemia virus relative to that of spleen necrosis virus. J Virol. 1994, 68: 494-499.
Braun MJ, Clements JE, Gonda MA: The visna virus genome: evidence for a hypervariable site in the env gene and sequence homology among lentivirus envelope proteins. J Virol. 1987, 61: 4046-4054.
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X: The sequence of the human genome. Science. 2001, 291: 1304-1351. 10.1126/science.1058040.
Zhao Z, Boerwinkle E: Neighboring-nucleotide effects on single nucleotide polymorphisms: a study of 2.6 million polymorphisms across the human genome. Genome Res. 2002, 12: 1679-1686. 10.1101/gr.287302.
Ferguson NM, Galvani AP, Bush RM: Ecological and immunological determinants of influenza evolution. Nature. 2003, 422: 428-433. 10.1038/nature01509.
Suzuki Y, Gojobori T: The origin and evolution of Ebola and Marburg viruses. Mol Biol Evol. 1997, 14: 800-806.
The Chinese SARS Molecular Epidemiology Consortium: Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004, 303: 1666-1669. 10.1126/science.1092002.
Bush RM, Smith CB, Cox NJ, Fitch WM: Effects of passage history and sampling bias on phylogenetic reconstruction of human influenza A evolution. Proc Natl Acad Sci USA. 2000, 97: 6974-6980. 10.1073/pnas.97.13.6974.
Brown Earl G, Tetro Jason A: Comparative analysis of the SARS coronavirus genome: a good start to a long journey. The Lancet. 2003, 361: 1756-1757. 10.1016/S0140-6736(03)13444-7.
Zeng F, Chow KY, Leung FC: Estimated timing of the last common ancestor of the SARS coronavirus. N Engl J Med. 2003, 349: 2469-2470. 10.1056/NEJM200312183492523.
Cyranoski D: Swift response greets return of SARS in China. Nature. 2004, 427: 89-
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25: 4876-4882. 10.1093/nar/25.24.4876.
Kumar S, Tamura K, Jakobsen IB, Nei M: MEGA2: Molecular Evolutionary Genetics Analysis software. 2001, Tempe, AZ, Arizona State University
Jakes TH, Cantor CR: Evolution of protein molecules. Mammalian Protein Metabolism. Edited by: Munro HN. 1969, New York, Academic Press, 21-132.
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4: 406-425.
Donnelly Christl A, Ghani Azra C, Leung Gabriel M, Hedley Anthony J, Fraser Christophe, Riley Steven, Abu-Raddad Laith J, Ho Lai-Ming, Thach Thuan-Quoc, Chau Patsy: Epidemiological determinants of spread of causal agent of severe acute respiratory syndrome in Hong Kong. The Lancet. 2003, 361: 1761-1766. 10.1016/S0140-6736(03)13410-1.
Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ: A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003, 348: 1953-1966. 10.1056/NEJMoa030781.
Poutanen SM, Low DE, Henry B, Finkelstein S, Rose D, Green K, Tellier R, Draker R, Adachi D, Ayers M, Chan AK, Skowronski DM, Salit I, Simor AE, Slutsky AS, Doyle PW, Krajden M, Petric M, Brunham RC, McGeer AJ: Identification of severe acute respiratory syndrome in Canada. N Engl J Med. 2003, 348: 1995-2005. 10.1056/NEJMoa030634.
Li WH, Wu CI, Luo CC: A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol Biol Evol. 1985, 2: 150-174.
Acknowledgements
This work was supported in part by grants from the National Institute of Health, Yunnan Province (2003C0001Z), and Yunnan University. ZZ was supported by a startup fund of the Virginia Commonwealth University. We thank two anonymous reviewers for valuable suggestions and Sara Barton and Jill Opalesky-Smith for improving the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
ZZ carried out the data collection and sequence alignment, participated in the data analysis, and wrote the manuscript. HL compared the mutation rate among RNA viruses. XW collected and estimated the dates of the isolates. YZ participated in the data analysis. KZ, YPZ, and EB participated in the design and coordination of the study. YXF conceived of the study, and participated in its design, coordination and writing. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Zhao, Z., Li, H., Wu, X. et al. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol Biol 4, 21 (2004). https://doi.org/10.1186/1471-2148-4-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-4-21