
Abstract
Background
Of the c. 450 families of flowering plants, only two are left "unplaced" in the most recent APG classification of angiosperms. One of these is the Apodanthaceae, a clade of c. 19 holoparasitic species in two or three genera occurring in North and South America, Africa, the Near East, and Australia. Because of lateral gene transfer between Apodanthaceae and their hosts it has been difficult to infer the family's true closest relatives.
Results
Here we report a phylogenetic analysis of 16 accessions representing six species of Apodanthaceae from the United States, Chile, Iran, and Australia, using the mitochondrial matR gene and the nuclear 18S gene. Data matrices include 190 matR sequences from up to 95 families in 39 orders of flowering plants and 197 18S sequences from 101 families representing the 16 orders of rosids. Analyses were performed at the nucleotide and at the amino acid level. Both gene trees agree with angiosperm phylogenies found in other studies using more genes. Apodanthaceae and the seven families of the order Cucurbitales form a clade with 100% bootstrap support from matR and 56% from 18 S. In addition, the Apodanthaceae and Cucurbitales matR gene sequences uniquely share two non-synonymous codon changes and one synonymous change, as well as a codon insertion, already found by Barkman et al. (2007).
Conclusions
Apodanthaceae belong in the Cucurbitales with which they share inferior ovaries, parietal placentation and a dioecious mating system, traits that are ancestral in Cucurbitales and which can now be interpreted as possible synapomorphies of an enlarged order Cucurbitales. The occurrence of Apodanthaceae in the Americas, Africa, the Near East, and Australia, and their adaptation to distantly related host species in the Fabaceae and Salicaceae suggest a long evolutionary history.
Similar content being viewed by others


Background
Among the c. 450 families of angiosperms [1] are about 11 parasitic lineages [2] of which some have completely lost the ability to photosynthesize. Such non-photosynthetic parasites grow embedded within the host tissues (as endoparasites) and emerge only during sexual reproduction [3]. Molecular-phylogenetic studies in recent years have revealed the sister groups of most parasitic plants [e. g. [2, 4–10]. Only the Apodanthaceae and the likewise holoparasitic Cynomoriaceae have not yet been placed with confidence, and the Apodanthaceae are also the only unplaced family-level clade in the latest classification of flowering plants [1, 11]. Before the advent of large-scale molecular sequencing and until about 2004, Apodanthaceae were considered to belong in the order Rafflesiales, together with Rafflesiaceae, Cytinaceae and Mitrastemonaceae [12, 13], mainly based on few or solitary, rather large to very large flowers with a single set of tepals that are commonly united into a conspicuous, corolloid calyx.
The aerial portions of Apodanthaceae consist of very short flowering shoots, each with a single flower that bursts out of the host's cortex during development [13]. Flowers are less than a centimeter across. The family contains three genera: the neotropical Apodanthes with about five species from Panama throughout South America to Uruguay and Argentina; the disjunctly distributed Pilostyles with c. nine species in the southern United States, Mexico, Panama, Venezuela, and Brazil, one species in Iran and Syria, and three in southwestern Australia [14]; and the African Berlinianche (a genus name that has never been validly published) with one or two species in Angola, southern Congo, Zambia, and Zimbabwe, Malawi, and Tanzania. Both species of "Berlinianche" were originally described in Pilostyles, and seven of the species of Pilostyles and Apodanthes have been moved between these genera, suggesting that the generic distinctions are not clear-cut. Apodanthes is supposed to have deciduous perianth segments, while Pilostyles has persistent perianth segments. The three genera also overlap in their host preferences, with Apodanthes parasitizing the Fabaceae Adesmia and the Salicaceae Casearia and Xylosma, and Pilostyles and Berlinianche parasitizing a range of other Fabaceae [13, 15].
Two molecular-systematic studies have included Apodanthaceae together with representative samples of other flowering plants [2, 16]. Both also included the parasites' host species to check for possible horizontal gene transfer between parasite and host. Nickrent et al. [16] sequenced the mitochondrial (mt) gene atp1 for Apodanthes caseariae, Berlinianche aethiopica, and Pilostyles thurberi, mt matR for A. caseariae and P. thurberi, and nuclear ribosomal 18S for P. thurberi. The atp1 gene placed the three genera together, but showed Pisum (Fabaceae) and Polemonium (Ericales) embedded among them, albeit without statistical support. The matR gene placed Apodanthes and Pilostyles together (posterior probability 100) and both as the sister clade to Begonia and Cucurbita (Cucurbitales) with a posterior probability of 98. The nuclear 18S region, finally, placed P. thurberi as sister to Gossypium and Pavonia (Malvales), albeit again without statistical support.
Barkman et al. [2] included only P. thurberi, but sequenced three mt genes, atp1, matR, and cox1, for a data set that covered at least one family from 44 of the 45 orders of angiosperm. A maximum likelihood tree from the combined data grouped P. thurberi as sister to Begonia and Echinocystis (the two representatives of the order Cucurbitales), although with low bootstrap support. The relationship received support from a seemingly uniquely shared 3 bp (one codon) insertion in the matR genes of P. thurberi, Begonia and Echinocystis.
Both studies [2, 16] explained the contradictory placements of Apodanthaceae in the atp1, matR, and 18S gene trees as resulting from horizontal gene transfer (HGT). This is a plausible explanation because plant mitochondrial genes are sometimes exchanged between species [17–20], and most known cases of HGT come from parasitic plants, which have permanent tissue contact with their hosts [8, 16, 21–24]. Even nuclear DNA can be transferred between parasites and their hosts [25]. Specifically, Nickrent et al. [16] regarded the matR and atp1 tree topologies as resulting from HGT. The only placement accepted as trustworthy was that of P. thurberi near Malvales in the 18S tree. By contrast, Barkman et al. [2] suggested that matR was not affected by HGT, but that atp1 had recently been transferred from a Fabaceae host species to P. thurberi.
Barkman et al.'s [2] evidence for this interpretation was twofold. First, analyses of atp1 suggested that four endoparasite lineages, Cytinus, Mitrastema, Pilostyles, and Rafflesia, all were closely related to their respective hosts, which appeared extremely unlikely and suggested that atp1 might be prone to HGT. Second, when comparing the atp1 sequences of two accessions of P. thurberi, one from Arizona (AZ), one from Texas (TX), Barkman et al. noted that the sequences differed at eight sites in the gene's central region. In contrast, comparisons of atp1 from Pilostyles-AZ to atp1 from its host revealed identical sequences for 804 bp in the central region, but 15 differences at the 5' and 3' ends. Based on these and other comparisons, Barkman et al. concluded that Pilostyles-AZ and Pilostyles-TX atp1 represent chimeric xenologs that result from multiple horizontal gene transfers. Chimeric atp1 genes in Apodanthaceae were also inferred from the transfer of a 79-nt track of chloroplast (cp) DNA to the mitochondria in Apodanthes caseariae, but not Pilostyles thurberi [26]. The only functional cp sequences so far recovered from any Apodanthaceae come from the small subunit of the plastid ribosomal RNA 16S gene [14, 27]. These results on chimeric sequences indicate that atp1 is unsuited for investigating the phylogenetic relationships of Apodanthaceae.
Other mitochondrial genes that might be explored for the purpose of phylogenetically placing the Apodanthaceae are the mt cox1 gene [2] and the nad1 B/C exons, e.g. [20]. The cox1 gene, however, has an extremely low substitution rate, limiting its usefulness for large-scale phylogenetics [28], and the nad1 B/C region is prone to lateral gene transfer [8, 20]. Nevertheless, the present study also explored these genes for Apodanthaceae phylogenetics.
The most promising genetic markers, however, clearly are the nuclear 18S region and the mt matR gene. The highly conserved 18S gene codes for RNA, a major subunit of the ribosome, and large 18S alignments have been found to contain sufficient signal to recover many known angiosperm family relationships [29–31]. Also, Apodanthaceae 18S trees are required to test the surprising Apodanthaceae/Malvales clade discovered by Nickrent et al. [16]. The maturase-related (matR) gene is retained regardless of photosynthetic ability, making it useful for parasite phylogenetics [2, 6, 9, 16]. Different from the cox1 intron [28], no case of matR loss has come to light [32]. MatR sequences are now available for 700+ flowering plants, and the gene's phylogenetic utility in rosids has been shown in a critical study [33].
We therefore generated 18S and matR sequences from multiple accessions and species of Apodanthaceae from the United States, Chile, Iran, and Australia. The sequences were aligned with representatives of all seven families of Cucurbitales, an order comprising 111 genera with about 2450 species [34]: Anisophylleaceae (29-40 species in 4 gen.), Begoniaceae (1400 spp. in 2 gen.), Coriariaceae (15 spp. in 1 genus), Corynocarpaceae (6 spp. in 1 genus), Cucurbitaceae (950 spp. in c. 100 gen.), Datiscaceae (2 spp. in 1 genus), and Tetramelaceae (2 spp. in 2 gen.). Following Zhu et al. [33], we performed amino acid-level analyses and constructed the DNA alignment according to the amino acid alignment. Such codon-based alignments have been shown to result in better statistical support by reducing misalignment of non-homologous regions [33, 35, 36]. As explained above, we also explored the utility of cox1 and nad1 B/C as potential markers for placing Apodanthaceae within angiosperms.
Results
Phylogenetics
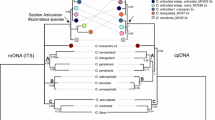
Figure 1 shows a maximum likelihood (ML) tree from the codon alignment of 64 of the 190 matR sequences analyzed in this study (see Additional File 1 for the full 190-taxon tree and Additional File 2 for a tree from the amino acid alignment). The included sequences represent 95 families from 39 of the c. 60 orders of flowering plants, plus 16 accessions from six species of Apodanthaceae from the United States, Chile, and Australia. The rosid orders Fagales, Rosales, and Cucurbitales are each recovered as monophyletic (the Fagales and Cucurbitales with 100% bootstrap support, the Rosales with 80% for codon alignment). The inclusion of Apodanthaceae in Cucurbitales has 100% bootstrap support at the nucleotide level (Figure 1) and 95% on amino acid level (Additional File 2). Family relationships remain unresolved. Apodanthes is sister to Pilostyles, and accessions of the West Australian species P. coccoidea and P. hamiltonii form the sister clade to the North American P. thurberi (represented by four accessions) and the Chilean P. berteroi. We were unable to amplify the matR gene from several samples of the Iranian P. haussknechtii, even with four sets of universal primers.

Maximum likelihood phylogeny obtained from the matR codon (nucleotide) alignment for 64 Cucurbitales, Rosales, Fagales, and Fabales. Numbers above branches indicate ML bootstrap support >75%.
Figure 2 shows an ML tree from the 18S matrix of 197 angiosperm species from 101 families in 16 orders of rosids, including representatives of all families of Cucurbitales and four species of Apodanthaceae from the United States, Iran, Chile, and Australia. Within Cucurbitales, the Apodanthaceae form the sister clade to Coriariaceae and Corynocarpaceae, albeit with low bootstrap support. Within the family, Iranian P. haussknechtii is sister to North American P. thurberi, while South American P. berteroi is sister to the Australian P. hamiltonii. The two 18S sequences of P. thurberi from GenBank [16] fall within Malvales as a sister clade to Tilia, as found in the original study.

Maximum likelihood phylogeny obtained from the 18S nucleotide matrix for 197 rosid taxa. Numbers above branches indicate ML bootstrap support >75%. Numbers in parentheses following the orders' names indicate numbers of genera included in the analysis.
An alignment of 143 cox1 exon sequences (from Cusimano et al. [28]) that included "Berlinianche" aethiopica and Pilostyles thurberi sequences from GenBank [2] placed the latter on an extremely long branch near Viola, Linum, and Malpighia, albeit without statistical support (data not shown). A single Apodanthaceae nad1 B/C sequence could not be successfully aligned with the 70-taxon nad1 B/C data set of Won and Renner [20] nor with newly generated Cucurbitales nad1 B/C sequences.
Codon substitutions
All Apodanthaceae and Cucurbitales share three unique codon changes: at positions 256, a C changed to a T, and at positions 505 and 700, a G to an A (coordinates from Nicotiana tabacum NC006581). Two of these changes are non-synonymous, and the change at position 700 also results in changing the polarity of the encoded amino acid (alanine to threonine). In addition, there is synonymous and variable substitution at position 532; Apodanthes caseariae and the Australian Apodanthaceae (P. hamiltonii and P. coccoidea) here have a codon CUG, while the remaining Cucurbitales have CUC. Both codons encode leucine. American Pilostyles species at this position have AUG, encoding methionine, while Fagales, Fabales and Rosales have AUC, coding for isoleucine. We also found the codon insertion uniquely shared by Cucurbitales and Apodanthaceae (at position 288, coordinates from Nicotiana tabacum NC006581), previously reported for P. thurberi, Begonia, and Echinocystis (at position 341, coordinates from Arabidopsis thaliana NC001284; [2]). Australian Apodanthaceae and A. caseariae moreover share a 12-nt insertion (corresponding to 4 amino acids) at position 1531.
Discussion
Phylogenetic placement of the Apodanthaceae in the Cucurbitales
The mitochondrial and nuclear data analyzed here show that Apodanthaceae belong in the Cucurbitales, an order here represented by 22 or 29 species in 16 or 22 genera from its seven families (Figures 1 and 2). In both trees, all sequences of Apodanthaceae form a clade, and all families of Cucurbitales are recovered as monophyletic. The 18S tree moreover recovers the core cucurbit clade of Datiscaceae, Begoniaceae, Tetramelaceae, and Cucurbitaceae, the Corynocarpaceae and Coriariaceae clade, and the placement of Anisophylleaceae also found with 12,000 nucleotides of nuclear, plastid and mitochondrial gene sequences [34]. While not resolving within-Cucurbitales family relationships, matR is sufficiently variable to recover angiosperm ordinal relationships that agree with those found with other kinds of data [1, 37–39]. An example is that Cucurbitales are sister to Fagales as found by Zhang et al. [34] and Moore et al. [39].
Nickrent et al. [16] were the first to find a relationship between Pilostyles and Cucurbitales using matR, but distrusted this grouping (as well as their atp1 tree) because of possible HGT. Our data reject this explanation because Apodanthaceae accessions from the different hosts and continents group together, which is implausible under the assumption of HGT. Another argument against HGT affecting our data sets is that the nuclear and mitochondrial trees agree in their placement of Apodanthaceae. It was only when Barkman et al. [2] found a codon insertion in the matR gene that was uniquely shared by Pilostyles, Echinocystis and Begonia that a possible phylogenetic relationship among these taxa became a strong hypothesis. The wider taxon sampling used here confirms the presence of a synapomorphic codon insertion in all Cucurbitales (including Apodanthaceae) at position 288 in Nicotiana tabacum NC006581. Additionally, we found three other codon changes shared by Apodanthaceae and Cucurbitales, two of them non-synonymous. This fits with Zhu et al.'s [33] finding that the matR non-synonymous substitution rate is relatively high and close to the gene's synonymous substitution rate, indicating relaxed selection.
Morphological traits supporting the Cucurbitales/Apodanthaceae clade and possible relationships with Coriariaceae and Corynocarpaceae
The Cucurbitales are a large order (7 families, 111 genera, 2450 species), characterized by ancestrally inferior ovaries and parietal placentation [34, 40], traits than can now be interpreted as a morphological synapomorphy with Apodanthaceae, especially since parietal placentation is a rare feature. Other traits characterizing most Cucurbitales are unisexual flowers, and a dioecious mating system [34].
Apodanthaceae also have strictly unisexual flowers, with dioecy very common [14, 41]. However, unisexual flowers and dioecy are strongly overrepresented among heterotrophic angiosperms [42], cautioning against overconfidence in this trait as evolutionarily conservative.
The 18S tree (Figure 2) shows the Apodanthaceae in a polytomy with Coriariaceae (a family of one genus and 15 species) and Corynocarpaceae (one genus with 6 species), albeit without statistical support. These families are sister clades [34] and differ from other Cucurbitales in having superior ovaries with apical placentation and mostly bisexual flowers ([34] for a tabulation of Cucurbitales morphological traits). Within Cucurbitales, they are therefore morphologically distant from Apodanthaceae.
Conclusions
The fossil record of Cucurbitales goes back to the Uppermost Paleocene and Lower Eocene London Clay [34], indicating the deep evolutionary history of the order. So far, no Cucurbitales are known to be hosts of Apodanthaceae, and irrespective of the latter's precise placement in Cucurbitales, it is clear that Apodanthaceae have been on their own evolutionary trajectory for a very long time. The family's presence in both, South America and Africa, might point to a history going back to a time when some of the fragments of the super-continent Gondwana were still close to each other. However, analysis of the biogeography of Apodanthaceae will require inclusion of the two African species, one only known from the type collection destroyed during World War II. The genetically and morphologically well-supported inclusion of Apodanthaceae in the Cucurbitales solves one of the last family-level "problems" in flowering plant phylogenetics [1] and finally allows assessing which genetic material these holoparasites may have acquired from host species.
Methods
Taxon sampling
The matR matrix included 190 sequences, representing 95 families from 39 orders of angiosperms; 22 of the sequences newly generated for this study. The 18S matrix included 197 sequences representing 101 families from 16 orders of rosids; four new Apodanthaceae sequences were generated. For detailed information on voucher material, host species and GenBank accession numbers see additional file 3.
Molecular methods
Molecular methods, including DNA extraction, PCR amplification, and DNA sequencing were performed as described in Zhang et al. [34]. MatR primers used where those of Barkman et al. [9], and 18S primers were those of Nickrent and Starr [43].
Alignments and phylogenetic analyses
We downloaded the matR alignment of Zhu et al. [33] (TreeBase accession number M3533), extracted 30 core rosid sequences from it and added 34 sequences relevant to this study, namely all available and new sequenced representatives of Apodanthaceae, the seven families of Cucurbitales, five of the eight families of Fagales, five families of the nine families of Rosales, and two of the four families of Fabales, including Apodanthaceae host species. DNA sequences were then translated using the ExPASy Proteomics Server [44] and aligned at the amino acid level using MAFFT [45, 46]. The corresponding DNA codon alignment (reverse-translated) was then produced using the PAL2NAL program [47]. The 64-taxon-matrix created generated by PAL2NAL did not include ambiguously aligned sections.
We also produced a 190-taxon matR matrix, which included additional eudicots, using MAFFT and followed by minor manual adjustments. Regions of uncertain alignment in the 190-taxon-matrix were excluded prior to analysis (161 nt from the beginning,76 nt from the end and 7 nt fragment at position 814 of original alignment).
The 567-taxon three-gene alignment of Soltis et al. [30, 31] was received from P. Soltis, the matrix then reduced to the 18S gene (~1800 bp) of the rosids to which were added the 18S sequences of Cucurbitales from Zhang et al. [34] and the newly generated Apodanthaceae sequences. This resulted in 197-taxon 18S matrix.
Phylogenetic analyses for the matR gene were performed at the nucleotide (codon) and the amino acid level, and for the 18S gene at the nucleotide level, using ML and full ML bootstrapping (100 replicates), with the GTR + GAMMA substitution model for the nucleotide alignment and the JTT model for the AA alignment, as implemented in RAxML [48]. The matR 64-taxon tree and the 18S 197-taxon tree were rooted on Nicotiana tabacum, and the matR 190-taxon tree on Austrobaileyales.
Authors' information
NF is currently a postdoc in SSR's group and has a permanent position in the Department of Biology and Pharmaceutical Botany of the Medical University of Gdansk (Poland), from where she was granted a leave-of-absence to pursue this study.
Acknowledgements
The authors thank H. Schaefer for several Cucurbitales matR sequences, S. Zarre, C. Heibl, and K. Dixon for material from Iran, Chile, and Australia, A. Fleischmann and M. Gottschling for helpful discussions, T. Barkman, C. Davis, and D. Nickrent for their constructive reviews, and the associate editor, S. Mathews, for cross-checking information.
References
APG III: An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Botanical Journal of the Linnean Society. 2009, 161: 105-121. 10.1111/j.1095-8339.2009.00996.x.
Barkman TJ, McNeal JR, Lim S-H, Coat G, Croom HB, Young ND, dePamphilis CW: Mitochondrial DNA suggests at least 11 origins of parasitism in angiosperms and reveals genomic chimerism in parasitic plants. BMC Evolutionary Biology. 2007, 7: 248-10.1186/1471-2148-7-248.
Heide-Jørgensen , Hennig S: Parasitic Flowering Plants. 2008, Leiden: Koninklijke Brill NV
DePamphilis CW, Young ND, Wolfe AD: Evolution of plastid gene rps2 in a lineage of hemiparasitic and holoparasitic plants: many losses of photosynthesis and complex patterns of rate variation. Proceedings of the National Academy of Sciences of the United States of America. 1997, 94: 7367-7372. 10.1073/pnas.94.14.7367.
Nickrent DL, Duff RJ, Colwell AE, Wolfe AD, Young ND, Steiner KE, dePamphilis CW: Molecular phylogenetic and evolutionary studies of parasitic plants. Molecular Systematics of Plants II. DNA Sequencing. Edited by: Soltis DE, Soltis PS, Doyle JJ. 1998, Boston: Kluwer Academic Publishers, 211-241.
Nickrent DL, Blarer A, Qiu Y-L, Soltis DE, Soltis PS, Zanis M: Molecular data place Hydnoraceae with Aristolochiaceae. American Journal of Botany. 2002, 89: 1809-1817. 10.3732/ajb.89.11.1809.
Olmstead RG, dePamphilis CW, Wolfe AD, Young ND, Elisens WJ, Reeves PJ: Disintegration of the Scrophulariaceae. American Journal of Botany. 2000, 88: 348-361. 10.2307/2657024.
Davis CC, Wurdack KJ: Host-to-parasite gene transfer in flowering plants: phylogenetic evidence from Malpighiales. Science. 2004, 575: 676-678. 10.1126/science.1100671.
Barkman TJ, Lim S-H, Salleh KM, Nais J: Mitochondrial DNA sequences reveal the photosynthetic relatives of Rafflesia, the world's largest flower. Proceedings of the National Academy of Sciences of the United States of America. 2004, 101: 787-792. 10.1073/pnas.0305562101.
Nickrent DL, Der JP, Anderson FE: Discovery of the photosynthetic relatives of the "Maltese mushroom" Cynomorium. BMC Evolutionary Biology. 2005, 5: 38-10.1186/1471-2148-5-38.
Angiosperm phylogeny website. (Stevens PF 2001 onwards). [http://www.mobot.org/MOBOT/research/APweb/]
Cronquist A: The evolution and classification of flowering plants. 1968, Boston: Houghton Mifflin Company
Blarer A, Nickrent DL, Endress PK: Comparative floral structure and systematics in Apodanthaceae (Rafflesiales). Plant Systematics and Evolution. 2004, 245: 119-42. 10.1007/s00606-003-0090-2.
Thiele KR, Wylie SJ, Maccarone L, Hollick P, McComb JA: Pilostyles coccoidea (Apodanthaceae), a new species from Western Australia described from morphological and molecular evidence. Nuytsia. 2008, 18: 273-284.
Verdcourt B: Rafflesiaceae. Flora of tropical East Africa. Edited by: Beentje HJ. 1998, Rotterdam: Balkema
Nickrent DL, Blarer A, Qiu YL, Vidal-Russell R, Anderson FE: Phylogenetic inference in Rafflesiales: the influence of rate heterogeneity and horizontal gene transfer. BMC Evolutionary Biology. 2004, 4: 40-10.1186/1471-2148-4-40.
Cho Y, Qiu YL, Kuhlman P, Palmer JD: Explosive invasion of plant mitochondria by a group I intron. Proceedings of the National Academy of Sciences of the United States of America. 1998, 95: 14244-14249. 10.1073/pnas.95.24.14244.
Bergthorsson U, Richardson AO, Young GJ, Goertzen LR, Palmer JD: Massive horizontal transfer of mitochondrial genes from diverse land plant donors to the basal angiosperm Amborella. Proceedings of the National Academy of Sciences of the United States of America. 101: 17747-17752. 10.1073/pnas.0408336102.
Bergthorsson U, Adams KL, Thomason B, Palmer JD: Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature. 2003, 424: 197-201. 10.1038/nature01743.
Won H, Renner SS: Horizontal gene transfer from flowering plants to Gnetum. Proceedings of the National Academy of Sciences of the United States of America. 2003, 100: 10824-10829. 10.1073/pnas.1833775100.
Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD: Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evolutionary Biology. 2007, 7: 135-10.1186/1471-2148-7-135.
Mower JP, Stefanovic S, Young GJ, Palmer JD: Plant genetics: gene transfer from parasitic to host plants. Nature. 2004, 432: 165-166. 10.1038/432165b.
Davis CC, Anderson WR, Wurdack KJ: Gene transfer from a parasitic flowering plant to a fern. Proceedings of the Royal Society B: Biological Sciences. 2005, 272: 2237-2242. 10.1098/rspb.2005.3226.
Roney JK, Khatibi PA, Westwood JH: Cross-species translocation of mRNA from host plants into the parasitic plant dodder. Plant Physiology. 2007, 143: 1037-1043. 10.1104/pp.106.088369.
Yoshida S, Maruyama S, Nozaki H, Shirasu K: Horizontal Gene Transfer by the parasitic plant Striga hermonthica. Science. 2010, 328: 1128-10.1126/science.1187145.
Hao W, Palmer JD: Fine-scale mergers of chloroplast and mitochondrial genes create functional, transcompartmentally chimeric mitochondrial genes. Proceedings of the National Academy of Sciences of the United States of America. 2009, 106: 16728-16733. 10.1073/pnas.0908766106.
Nickrent DL, Duff RJ, Konings DAM: Structural analyses of plastid-derived 16S rRNAs in holoparasitic angiosperms. Plant Molecular Biology. 1997, 34: 731-743. 10.1023/A:1005808615763.
Cusimano N, Zhang L-B, Renner SS: Reevaluation of the cox1 group I intron in Araceae and angiosperms indicates a history dominated by loss rather than horizontal transfer. Molecular Biology and Evolution. 2008, 25: 1-12. 10.1093/molbev/msm241.
Soltis PS, Soltis DE: Molecular evolution of 18S rDNA in angiosperms: implications for character weighting in phylogenetic analysis. Molecular systematics of plants II: DNA sequencing. Edited by: Soltis DE, Soltis PS, Doyle JJ. 1998, Boston: Kluwer, 188-210.
Soltis DE, Soltis PS, Chase MW, Mort ME, Albach DC, Zanis M, Savolainen V, Hahn WH, Hoot SB, Fay MF, Axtell M, Swensen SM, Prince LM, Kress WJ, Nixon KC, Farris JS: Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Biological Journal of Linnean Society. 2000, 133: 381-461.
Soltis DE, Gitzendanner MA, Soltis PS: A 567-taxon data set for angiosperms: the challenges posed by Bayesian analyses of large data sets. International Journal of Plant Sciences. 2007, 168: 137-157. 10.1086/509788.
Adams KL, Qiu YL, Stoutemyer M, Palmer JD: Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer during angiosperm evolution. Proceedings of the National Academy of Sciences of the United States of America. 2002, 99: 9905-9912. 10.1073/pnas.042694899.
Zhu XY, Chase MW, Qiu YL, Kong HZ, Dilcher DL, Li JH, Chen ZD: Mitochondrial matR Sequences help to resolve deep phylogenetic relationships in rosids. BMC Evolutionary Biology. 2008, 7: 217-10.1186/1471-2148-7-217.
Zhang L-B, Simmons MP, Kocyan A, Renner SS: Phylogeny of the Cucurbitales based on DNA sequences of nine loci from three genomes: Implications for morphological and sexual system evolution. Molecular Phylogenetics and Evolution. 2006, 39: 305-322. 10.1016/j.ympev.2005.10.002.
Bininda-Emonds ORP: transAlign: using aminoacids to facilitate the multiple alignment of protein-coding DNA sequences. BMC Bioinformatics. 2005, 6: 156-10.1186/1471-2105-6-156.
Suyama M, Torrents D, Bork P: PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acid Research. 2006, 34: W609-12. 10.1093/nar/gkl315.
Soltis DE, Soltis PS, Morgan DR, Swensen SM, Mullin BC, Dowd JM, Martin PG: Chloroplast gene sequence data suggest a single origin of the predisposition for symbiotic nitrogen fixation in angiosperms. Proceedings of the National Academy of Sciences of the United States of America. 1995, 92: 2647-2651. 10.1073/pnas.92.7.2647.
Endress PK, Matthews ML: First steps towards a floral structural characterization of the major rosid subclades. Plant Systematics and Evolution. 2006, 260: 223-251.
Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE: Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proceedings of the National Academy of Sciences of the United States of America. 2010, 107: 4623-4628. 10.1073/pnas.0907801107.
Matthews ML, Endress PK: Comparative floral structure and systematics in Cucurbitales (Corynocarpaceae, Coriariaceae, Tetramelaceae, Datiscaceae, Begoniaceae, Cucurbitaceae, Anisophylleaceae). Botanical Journal of the Linnean Society. 2004, 145: 129-185. 10.1111/j.1095-8339.2003.00281.x.
Dell B, Burbidge AH: Notes on the biology of Pilostyles (Rafflesiaceae) in Western Australia. Western Australian Herbarium Research Notes. 1981, 5: 1-9.
Renner SS, Ricklefs RE: Dioecy and its correlates in the flowering plants. American Journal of Botany. 1995, 82: 596-606. 10.2307/2445418.
Nickrent DL, Starr EM: High rates of nucleotide substitution in nuclear small-subunit (18S) rDNA from holoparasitic flowering plants. Journal of Molecular Evolution. 1994, 39: 62-70. 10.1007/BF00178250.
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A: ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acid Research. 2003, 31: 3784-3788. 10.1093/nar/gkg563.
Katoh K, Misawa K, Kuma K, Miyata T: MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acid Research. 2002, 30: 3059-66. 10.1093/nar/gkf436.
Katoh K, Asimenos G, Toh H: Multiple Alignment of DNA Sequences with MAFFT. Methods in Molecular Biology. 2009, 537: 39-64. full_text.
Suyama M, Torrents D, Bork P: PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acid Research. 2006, 34: W609-12. 10.1093/nar/gkl315.
Stamatakis A, Hoover P, Rougemont J: A rapid bootstrap algorithm for the RAxML web servers. Systematic Biology. 2008, 75: 758-771. 10.1080/10635150802429642.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
NF generated sequences and alignments, performed data analyses, and worked on the manuscript. SSR conceived the study, obtained material, and drafted the manuscript. Both authors read and approved the final manuscript.
Natalia Filipowicz and Susanne S Renner contributed equally to this work.
Electronic supplementary material
12862_2010_1435_MOESM1_ESM.JPEG
{kind=link}
Additional file 1: Maximum likelihood phylogeny obtained from matRsequences representing 190 species from 95 families of flowering plants. Numbers above branches indicate ML bootstrap support >75%. (JPEG 3 MB)
12862_2010_1435_MOESM2_ESM.PDF
Additional file 2: Maximum likelihood phylogeny obtained from the matRamino acid matrix for 64 Cucurbitales, Rosales, Fagales, and Fabales. (PDF 55 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Filipowicz, N., Renner, S.S. The worldwide holoparasitic Apodanthaceae confidently placed in the Cucurbitales by nuclear and mitochondrial gene trees. BMC Evol Biol 10, 219 (2010). https://doi.org/10.1186/1471-2148-10-219
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-10-219